
Split Hand-Foot and Deafness in a Patient with 7q21.13-q21.3 Deletion Not Including the DLX5/6 Genes

 , , , , , , , , and
, , , , , , , , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Karyotype and FISH Analysis
2.2. SNP Array
2.3. Lymphoblastoid Cell Line Establishment
2.4. Expression Study
3. Results
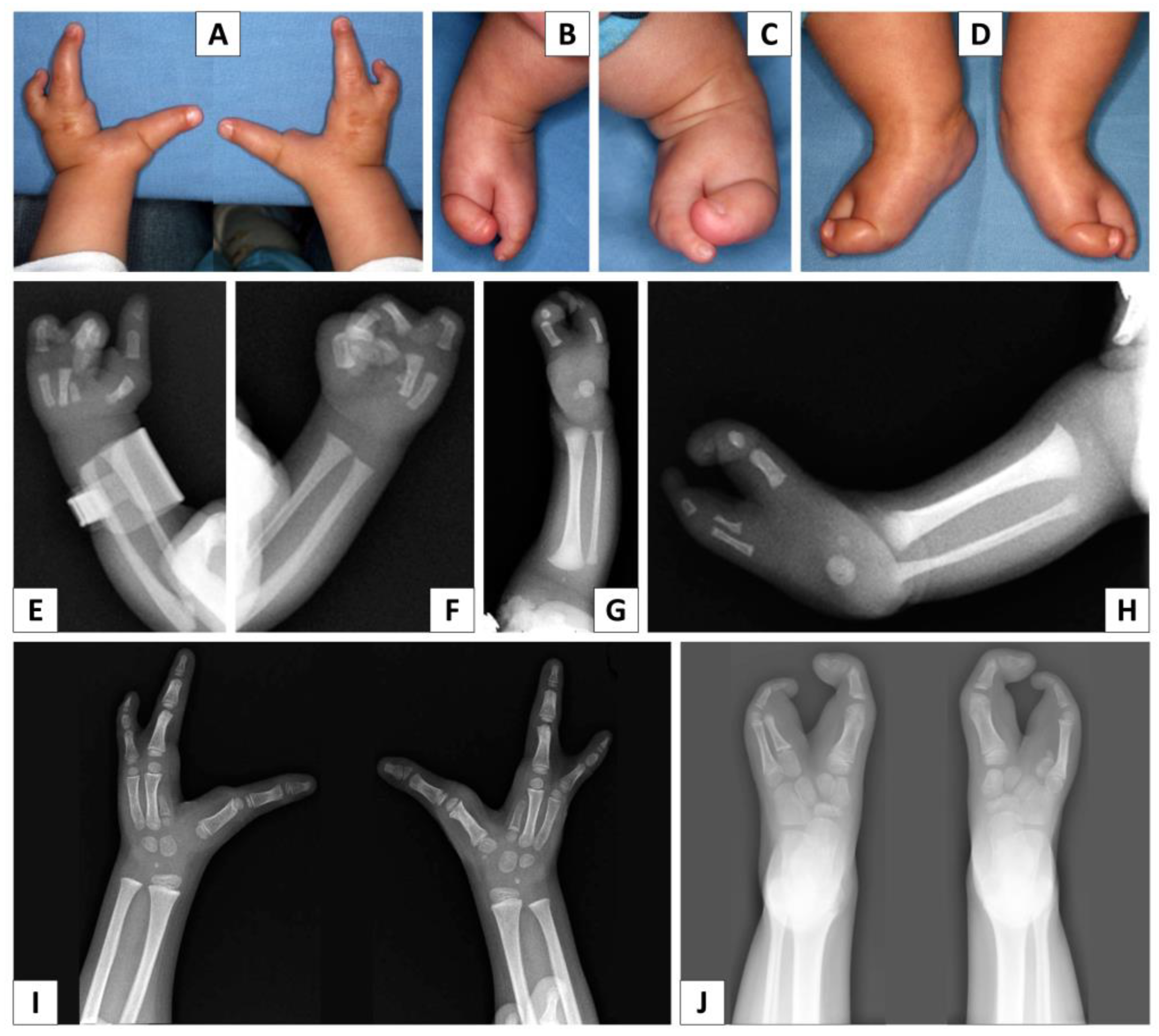
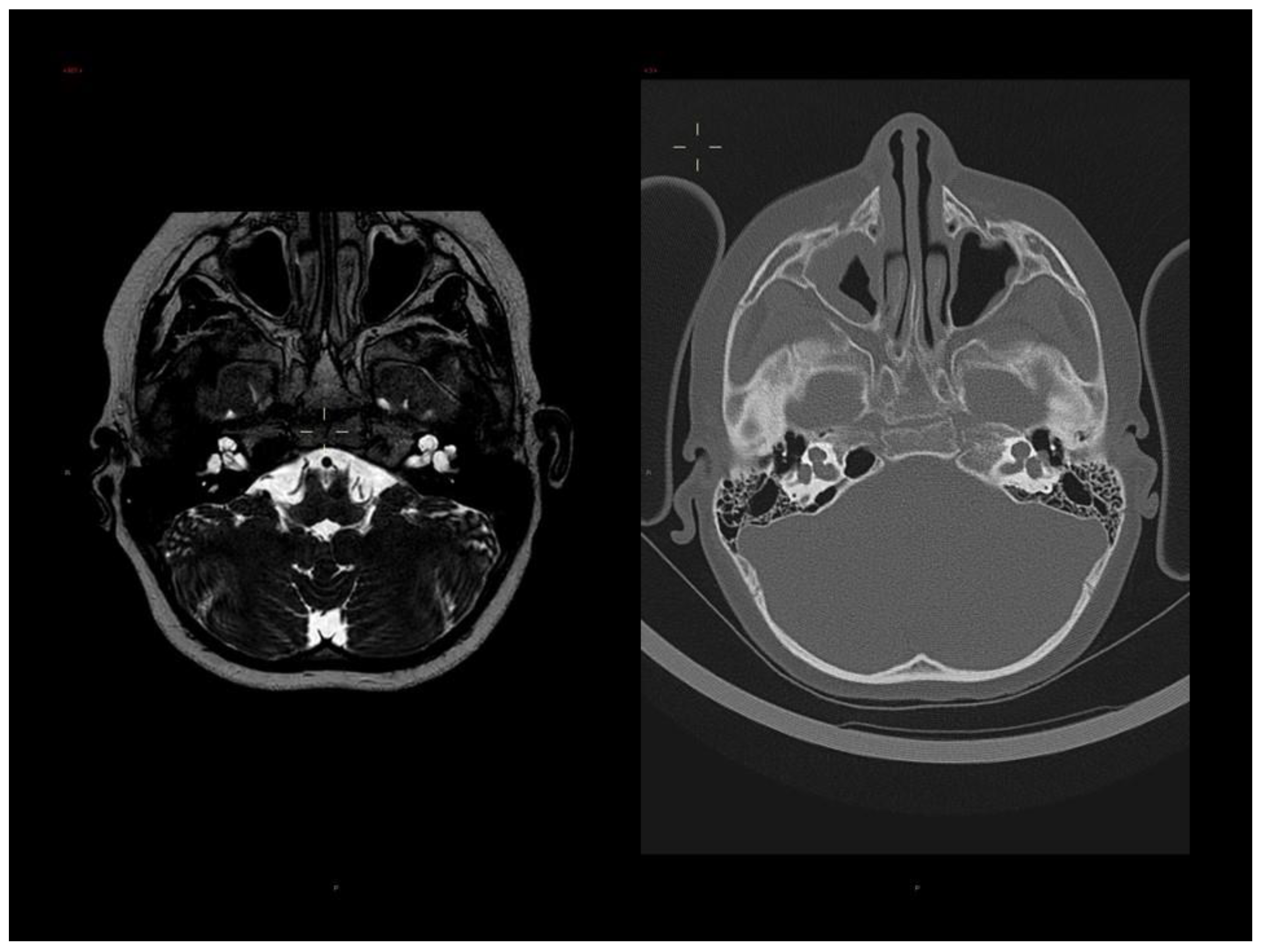
3.1. Clinical Report
3.2. Molecular Analyses
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Duijf, P.H.; van Bokhoven, H.; Brunner, H.G. Pathogenesis of split-hand/split-foot malformation. Hum. Mol. Genet. 2003, 12, 51R–60R. [Google Scholar] [CrossRef] [PubMed]
- Sifakis, S.; Basel, D.; Ianakiev, P.; Kilpatrick, M.; Tsipouras, P. Distal limb malformations: Underlying mechanisms and clinical associations. Clin. Genet. 2001, 60, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Ullah, A.; Gul, A.; Umair, M.; Irfanullah; Ahmad, F.; Aziz, A.; Wali, A.; Ahmad, W. Homozygous sequence variants in the WNT10B gene underlie split hand/foot malformation. Genet. Mol. Biol. 2018, 41, 1–8. [Google Scholar] [CrossRef]
- Sowińska-Seidler, A.; Socha, M.; Jamsheer, A. Split-hand/foot malformation—Molecular cause and implications in genetic counseling. J. Appl. Genet. 2013, 55, 105–115. [Google Scholar] [CrossRef] [PubMed]
- Bernardini, L.; Palka, C.; Ceccarini, C.; Capalbo, A.; Bottillo, I.; Mingarelli, R.; Novelli, A.; Dallapiccola, B. Complex rearrangement of chromosomes 7q21.13-q22.1 confirms the ectrodactyly-deafness locus and suggests new candidate genes. Am. J. Med. Genet. Part A 2007, 146A, 238–244. [Google Scholar] [CrossRef] [PubMed]
- van Silfhout, A.T.; Akker, P.C.v.D.; Dijkhuizen, T.; Verheij, J.B.G.M.; Olderode-Berends, M.J.W.; Kok, K.; Sikkema-Raddatz, B.; A van Ravenswaaij-Arts, C.M. Split hand/foot malformation due to chromosome 7q aberrations (SHFM1): Additional support for functional haploinsufficiency as the causative mechanism. Eur. J. Hum. Genet. 2009, 17, 1432–1438. [Google Scholar] [CrossRef]
- Wieland, I.; Muschke, P.; Jakubiczka, S.; Volleth, M.; Freigang, B.; Wieacker, P.F. Refinement of the deletion in 7q21.3 associated with split hand/foot malformation type 1 and Mondini dysplasia. J. Med. Genet. 2004, 41, e54. [Google Scholar] [CrossRef][Green Version]
- Levi, G.; Narboux-Nême, N.; Cohen-Solal, M. DLX Genes in the Development and Maintenance of the Vertebrate Skeleton: Implications for Human Pathologies. Cells 2022, 11, 3277. [Google Scholar] [CrossRef]
- Kantaputra, P.N.; Carlson, B.M. Genetic regulatory pathways of split-hand/foot malformation. Clin. Genet. 2018, 95, 132–139. [Google Scholar] [CrossRef]
- Robledo, R.F.; Rajan, L.; Li, X.; Lufkin, T. The Dlx5 and Dlx6 homeobox genes are essential for craniofacial, axial, and appendicular skeletal development. Genes Dev. 2002, 16, 1089–1101. [Google Scholar] [CrossRef]
- Merlo, G.R.; Paleari, L.; Mantero, S.; Genova, F.; Beverdam, A.; Palmisano, G.L.; Barbieri, O.; Levi, G. Mouse model of split hand/foot malformation type I. Genes. 2002, 33, 97–101. [Google Scholar] [CrossRef] [PubMed]
- Shamseldin, H.E.; Faden, M.A.; Alashram, W.; Alkuraya, F.S. Identification of a novel DLX5 mutation in a family with autosomal recessive split hand and foot malformation. J. Med. Genet. 2011, 49, 16–20. [Google Scholar] [CrossRef]
- Wang, X.; Xin, Q.; Li, L.; Li, J.; Zhang, C.; Qiu, R.; Qian, C.; Zhao, H.; Liu, Y.; Shan, S.; et al. Exome sequencing reveals a heterozygous DLX5 mutation in a Chinese family with autosomal-dominant split-hand/foot malformation. Eur. J. Hum. Genet. 2014, 22, 1105–1110. [Google Scholar] [CrossRef] [PubMed]
- Sowińska-Seidler, A.; Badura-Stronka, M.; Latos-Bieleńska, A.; Stronka, M.; Jamsheer, A. Heterozygous DLX5 nonsense mutation associated with isolated split-hand/foot malformation with reduced penetrance and variable expressivity in two unrelated families. Birth Defects Res. Part A: Clin. Mol. Teratol. 2014, 100, 764–771. [Google Scholar] [CrossRef] [PubMed]
- Ullah, A.; Hammid, A.; Umair, M.; Ahmad, W. A Novel Heterozygous Intragenic Sequence Variant in DLX6 Probably Underlies First Case of Autosomal Dominant Split-Hand/Foot Malformation Type 1. Mol. Syndr. 2016, 8, 79–84. [Google Scholar] [CrossRef] [PubMed]
- Birnbaum, R.Y.; Clowney, E.J.; Agamy, O.; Kim, M.J.; Zhao, J.; Yamanaka, T.; Pappalardo, Z.; Clarke, S.L.; Wenger, A.M.; Nguyen, L.; et al. Coding exons function as tissue-specific enhancers of nearby genes. Genome Res. 2012, 22, 1059–1068. [Google Scholar] [CrossRef]
- Birnbaum, R.Y.; Everman, D.B.; Murphy, K.K.; Gurrieri, F.; Schwartz, C.E.; Ahituv, N. Functional characterization of tissue-specific enhancers in the DLX5/6 locus. Hum. Mol. Genet. 2012, 21, 4930–4938. [Google Scholar] [CrossRef]
- Qiao, Y.; Tyson, C.; Hrynchak, M.; Lopez-Rangel, E.; Hildebrand, J.; Martell, S.; Fawcett, C.; Kasmara, L.; Calli, K.; Harvard, C.; et al. Clinical application of 2.7M Cytogenetics array for CNV detection in subjects with idiopathic autism and/or intellectual disability. Clin. Genet. 2012, 83, 145–154. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Rasmussen, M.B.; Kreiborg, S.; Jensen, P.; Bak, M.; Mang, Y.; Lodahl, M.; Budtz-Jørgensen, E.; Tommerup, N.; Tranebjærg, L.; Rendtorff, N.D. Phenotypic subregions within the split-hand/foot malformation 1 locus. Hum. Genet. 2016, 135, 345–357. [Google Scholar] [CrossRef]
- Brown, K.K.; Reiss, J.A.; Crow, K.; Ferguson, H.L.; Kelly, C.; Fritzsch, B.; Morton, C.C. Deletion of an enhancer near DLX5 and DLX6 in a family with hearing loss, craniofacial defects, and an inv(7)(q21.3q35). Hum. Genet. 2009, 127, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Gezdirici, A.; Yenigun, A.; Koparir, E.; Yosunkaya, E.; Ulucan, H.; Seven, M.; Yuksel, A.; Ozen, M. A rare case of split hand/foot malformation with sensorineural hearing loss and Mondini dysplasia. Clin. Dysmorphol. 2013, 22, 33–35. [Google Scholar] [CrossRef] [PubMed]
- Katarzyna, M.-M.; Jarosław, S.; Katarzyna, J.-P.; Wojciech, S.; Magdalena, F. Recurrent Streptococcus Pneumoniae Meningitis in a Child with Split Hand and Foot Malformation and Undiagnosed Mondini Dysplasia. J. Dev. Phys. Disabil. 2015, 27, 823–829. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Rowley, M.J.; Corces, V.G. Organizational principles of 3D genome architecture. Nat. Rev. Genet. 2018, 19, 789–800. [Google Scholar] [CrossRef] [PubMed]
- Benko, S.; Fantes, J.A.; Amiel, J.; Kleinjan, D.-J.; Thomas, S.; Ramsay, J.; Jamshidi, N.; Essafi, A.; Heaney, S.; Gordon, C.T.; et al. Highly conserved non-coding elements on either side of SOX9 associated with Pierre Robin sequence. Nat. Genet. 2009, 41, 359–364. [Google Scholar] [CrossRef]
- Klopocki, E.; Lohan, S.; Brancati, F.; Koll, R.; Brehm, A.; Seemann, P.; Dathe, K.; Stricker, S.; Hecht, J.; Bosse, K.; et al. Copy-Number Variations Involving the IHH Locus Are Associated with Syndactyly and Craniosynostosis. Am. J. Hum. Genet. 2011, 88, 70–75. [Google Scholar] [CrossRef]
- Kouwenhoven, E.N.; van Heeringen, S.J.; Tena, J.J.; Oti, M.; Dutilh, B.E.; Alonso, M.E.; de la Calle-Mustienes, E.; Smeenk, L.; Rinne, T.; Parsaulian, L.; et al. Genome-Wide Profiling of p63 DNA–Binding Sites Identifies an Element that Regulates Gene Expression during Limb Development in the 7q21 SHFM1 Locus. PLOS Genet. 2010, 6, e1001065. [Google Scholar] [CrossRef]
- Rattanasopha, S.; Tongkobpetch, S.; Srichomthong, C.; Kitidumrongsook, P.; Suphapeetiporn, K.; Shotelersuk, V. Absent expression of the osteoblast-specific maternally imprinted genes, DLX5 and DLX6, causes split hand/split foot malformation type I. J. Med. Genet. 2014, 51, 817–823. [Google Scholar] [CrossRef]
- Allen, H.L.; Caswell, R.; Xie, W.; Xu, X.; Wragg, C.; Turnpenny, P.D.; Turner, C.L.S.; Weedon, M.N.; Ellard, S. Next generation sequencing of chromosomal rearrangements in patients with split-hand/split-foot malformation provides evidence for DYNC1I1 exonic enhancers of DLX5/6 expression in humans. J. Med. Genet. 2014, 51, 264–267. [Google Scholar] [CrossRef][Green Version]
- Tayebi, N.; Jamsheer, A.; Flöttmann, R.; Sowinska-Seidler, A.; Doelken, S.C.; Oehl-Jaschkowitz, B.; Hülsemann, W.; Habenicht, R.; Klopocki, E.; Mundlos, S.; et al. Deletions of exons with regulatory activity at the DYNC1I1 locus are associated with split-hand/split-foot malformation: Array CGH screening of 134 unrelated families. Orphanet J. Rare Dis. 2014, 9, 108. [Google Scholar] [CrossRef]
- Delgado, S.; Velinov, M. 7q21.3 Deletion involving enhancer sequences within the gene DYNC1I1 presents with intellectual disability and split hand-split foot malformation with decreased penetrance. Mol. Cytogenet. 2015, 8, 37. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Zaldívar, H.M.; Martínez-Irías, D.G.; Espinoza-Moreno, N.A.; Napky-Rajo, J.S.; Bueso-Aguilar, T.A.; Reyes-Perdomo, K.G.; Montes-Gambarelli, J.A.; Euceda, I.M.; Ponce-Barahona, A.F.; Gámez-Fernández, C.A.; et al. A novel description of a syndrome consisting of 7q21.3 deletion including DYNC1I1 with preserved DLX5/6 without ectrodactyly: A case report. J. Med. Case Rep. 2016, 10, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Saitsu, H.; Kurosawa, K.; Kawara, H.; Eguchi, M.; Mizuguchi, T.; Harada, N.; Kaname, T.; Kano, H.; Miyake, N.; Toda, T.; et al. Characterization of the complex 7q21.3 rearrangement in a patient with bilateral split-foot malformation and hearing loss. Am. J. Med. Genet. Part A 2009, 149A, 1224–1230. [Google Scholar] [CrossRef]
- Okita, C.; Meguro, M.; Hoshiya, H.; Haruta, M.; Sakamoto, Y.-K.; Oshimura, M. A new imprinted cluster on the human chromosome 7q21-q31, identified by human-mouse monochromosomal hybrids. Genomics 2003, 81, 556–559. [Google Scholar] [CrossRef]
- Horike, S.-I.; Cai, S.; Miyano, M.; Cheng, J.-F.; Kohwi-Shigematsu, T. Loss of silent-chromatin looping and impaired imprinting of DLX5 in Rett syndrome. Nat. Genet. 2004, 37, 31–40. [Google Scholar] [CrossRef]
- LaSalle, J.M. The Odyssey of MeCP2 and parental imprinting. Epigenetics 2007, 2, 5–10. [Google Scholar] [CrossRef]
- Schüle, B.; Li, H.H.; Fisch-Kohl, C.; Purmann, C.; Francke, U. DLX5 and DLX6 Expression Is Biallelic and Not Modulated by MeCP2 Deficiency. Am. J. Hum. Genet. 2007, 81, 492–506. [Google Scholar] [CrossRef]
- Itaba-Matsumoto, N.; Maegawa, S.; Yamagata, H.; Kondo, I.; Oshimura, M.; Nanba, E. Imprinting status of paternally imprinted DLX5 gene in Japanese patients with Rett syndrome. Brain Dev. 2007, 29, 491–495. [Google Scholar] [CrossRef] [PubMed]
- Miyano, M.; Horike, S.-I.; Cai, S.; Oshimura, M.; Kohwi-Shigematsu, T. DLX5 expression is monoallelic and Dlx5 is up-regulated in the Mecp2-null frontal cortex. J. Cell. Mol. Med. 2008, 12, 1188–1191. [Google Scholar] [CrossRef]
- Hannula-Jouppi, K.; Muurinen, M.; Lipsanen-Nyman, M.; Reinius, L.E.; Ezer, S.; Greco, D.; Kere, J. Differentially methylated regions in maternal and paternal uniparental disomy for chromosome 7. Epigenetics 2013, 9, 351–365. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID | Nr of Affected Relatives | Limb Anomalies | Neurodevelopment | Hearing Loss | Inner Ear Abnormality | Other |
|---|---|---|---|---|---|---|
| Van Silfhout et al. [6] | 1 | 1 (bil SHFM) | 1 (PDD NOS) | 0 | 0 | AVM right hand |
| Kouwenhoven et al. [27] | 1 | 1 (bil SFM) | 0 | 0 | 0 | |
| Brown et al. [21] | 5 | 0 | 2/5 | 1 | 1 | Craniofacial abnormalities |
| Rattanasopha et al. [28] | 6 (and 2 unaffected carriers) | 1 (variable) | 0 | 0 | 0 | Right hand polydactyly (1/8) |
| Allen et al. 1/2 [29] | 1 | 1 (variable) | 0 | 1 (sensorineural deafness) | 0 | |
| Allen et al. 2/2 [29] | 2 | 1 (variable) | 0 | 0 | 0 | |
| Tayebi et al. 1/4 [30] | 3 | 1 (bil SHFM) | 0 | 0 | 0 | |
| Tayebi et al. 2/4 [30] | 5 | 1 | 0 | 1 (severe HL) | 0 | |
| Tayebi et al. 3/4 [30] | 6 | 1 | 0 | 0 | 0 | |
| Tayebi et al. 4/4 [30] | 2 (monozygotic twins) | 1 | 0 | 0 | 0 | |
| Delgado & Velinov [31] | 4 | 1 | 1 (ID/DD) | 0 | 0 | |
| Rasmussen et al. [20] | 5 (2 available for clinical examination) | 1 | 2/2 (autism) | 1 | 1 | Craniofacial abnormalities |
| Ramos-Zaldìva et al. [32] | 1 | 0 | 1 (paranoid personality disorder) | 1 | 0 | Dysmorphic features |
| Saitsu et al. [33] | 1 | 1 | 1 (DD) | 1 (severe HL, mixed type) | 0 | |
| Our case | 1 | 1 | 1 (mild DD) | 1 | 1 | Poor growth |
| ID | Karyotype | Type of Alteration | Deletion Size (kb) | Proximal bp | Distal bp | DLX5/6 Deleted | eExons of DYNC1I1 Deleted |
|---|---|---|---|---|---|---|---|
| van Silfhout et al. [6] | 46,XY,inv(7)(p22q21.3) | Inversion | ~95,530,000 | ~95,700,000 | No | No | |
| Kouwenhoven et al. [27] | Deletion | 880 | ~95,552,000 * | ~96,432,000 * | No | Yes | |
| Brown et al. [21] | 46,XX,inv7(q21.3q35) or 46,X,inv7(q21.3q35) | Inversion | 96,401,902 | 96,407,985 | No | No | |
| Rattanasopha et al. [28] | Deletion | 104 | 95,694,099 | 95,797,866 | No | Yes | |
| Allen et al. 1/2 [29] | t(2;7)(p25.1;q21.3) | Translocation | 96,229,309 | 96,229,309 | No | No | |
| Allen et al. 2/2 [29] | Deletion | 106 | 95,704,812 | 95,810,747 | No | Yes | |
| Tayebi et al. 1/4 [30] | Deletion | 167 | 95,615,187 | 95,783,313 | No | Yes | |
| Tayebi et al. 2/4 [30] | Deletion | 510 | 95,624,825 | 96,135,521 | No | Yes | |
| Tayebi et al. 3/4 [30] | Deletion | 205 | 95,667,046 | 95,872,044 | No | Yes | |
| Tayebi et al. 4/4 [30] | Deletion | 169 | 95,693,341 | 95,862,369 | No | Yes | |
| Delgado & Velinov [31] | Deletion | 1032 | 94,769,383 | 95,801,045 | No | Yes | |
| Rasmussen et al. [20] | 46,XX,inv(7)(q22q33) | Inversion | 96,378,046 | 96,378,047 | No | No | |
| Ramos-Zaldìva et al. [32] | 46,XY | Deletion | 3190 | 93,389,222 | 96,579,845 | No | Yes |
| Saitsu et al. [33] | 46,XX,t(7;15)(q21;q15),t(9;14)(q21;q11.2) | Translocation | 96,097,195 | 96,276,197 | No | No | |
| Our case | 46,XY | Deletion | 6300 | 89,831,744 | 96,116,907 | No | Yes |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ambrosetti, I.; Bernardini, L.; Pollazzon, M.; Giuffrida, M.G.; Guida, V.; Peluso, F.; Baroni, M.C.; Polizzi, V.; Napoli, M.; Rosato, S.; et al. Split Hand-Foot and Deafness in a Patient with 7q21.13-q21.3 Deletion Not Including the DLX5/6 Genes. Genes 2023, 14, 1526. https://doi.org/10.3390/genes14081526
Ambrosetti I, Bernardini L, Pollazzon M, Giuffrida MG, Guida V, Peluso F, Baroni MC, Polizzi V, Napoli M, Rosato S, et al. Split Hand-Foot and Deafness in a Patient with 7q21.13-q21.3 Deletion Not Including the DLX5/6 Genes. Genes. 2023; 14(8):1526. https://doi.org/10.3390/genes14081526
Chicago/Turabian StyleAmbrosetti, Irene, Laura Bernardini, Marzia Pollazzon, Maria Grazia Giuffrida, Valentina Guida, Francesca Peluso, Maria Chiara Baroni, Valeria Polizzi, Manuela Napoli, Simonetta Rosato, and et al. 2023. "Split Hand-Foot and Deafness in a Patient with 7q21.13-q21.3 Deletion Not Including the DLX5/6 Genes" Genes 14, no. 8: 1526. https://doi.org/10.3390/genes14081526
APA StyleAmbrosetti, I., Bernardini, L., Pollazzon, M., Giuffrida, M. G., Guida, V., Peluso, F., Baroni, M. C., Polizzi, V., Napoli, M., Rosato, S., Trimarchi, G., Gelmini, C., Caraffi, S. G., Wischmeijer, A., Frattini, D., Novelli, A., & Garavelli, L. (2023). Split Hand-Foot and Deafness in a Patient with 7q21.13-q21.3 Deletion Not Including the DLX5/6 Genes. Genes, 14(8), 1526. https://doi.org/10.3390/genes14081526