

Comparative Analysis of CRISPR-Cas Systems in Pseudomonas Genomes
 ,
,  , and
, and 
Abstract
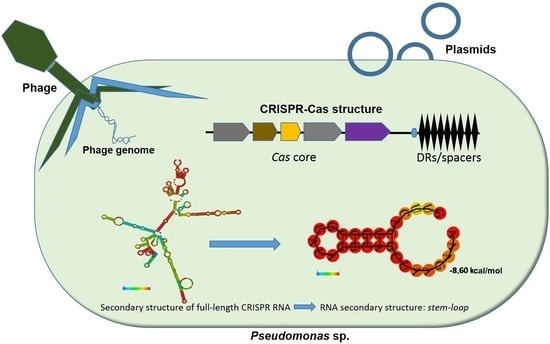
1. Introduction
2. Materials and Methods
2.1. Obtaining Genomic Sequences of Pseudomonas
2.2. Identification of CRISPR Structures
2.3. Identification and Comparison of CRISPR-Associated Genes (CAS) and CAS Proteins
2.4. Determination of the Origin and Diversity of Spacer Sequences
2.5. Identification of Protospacer Adjacent Motifs (PAMs) and Self-Targeting
2.6. Determination of the Conservation of Direct Repeats (DRs) and the Prediction of the RNA Secondary Structure
2.7. Statistical Analysis
3. Results and Discussions
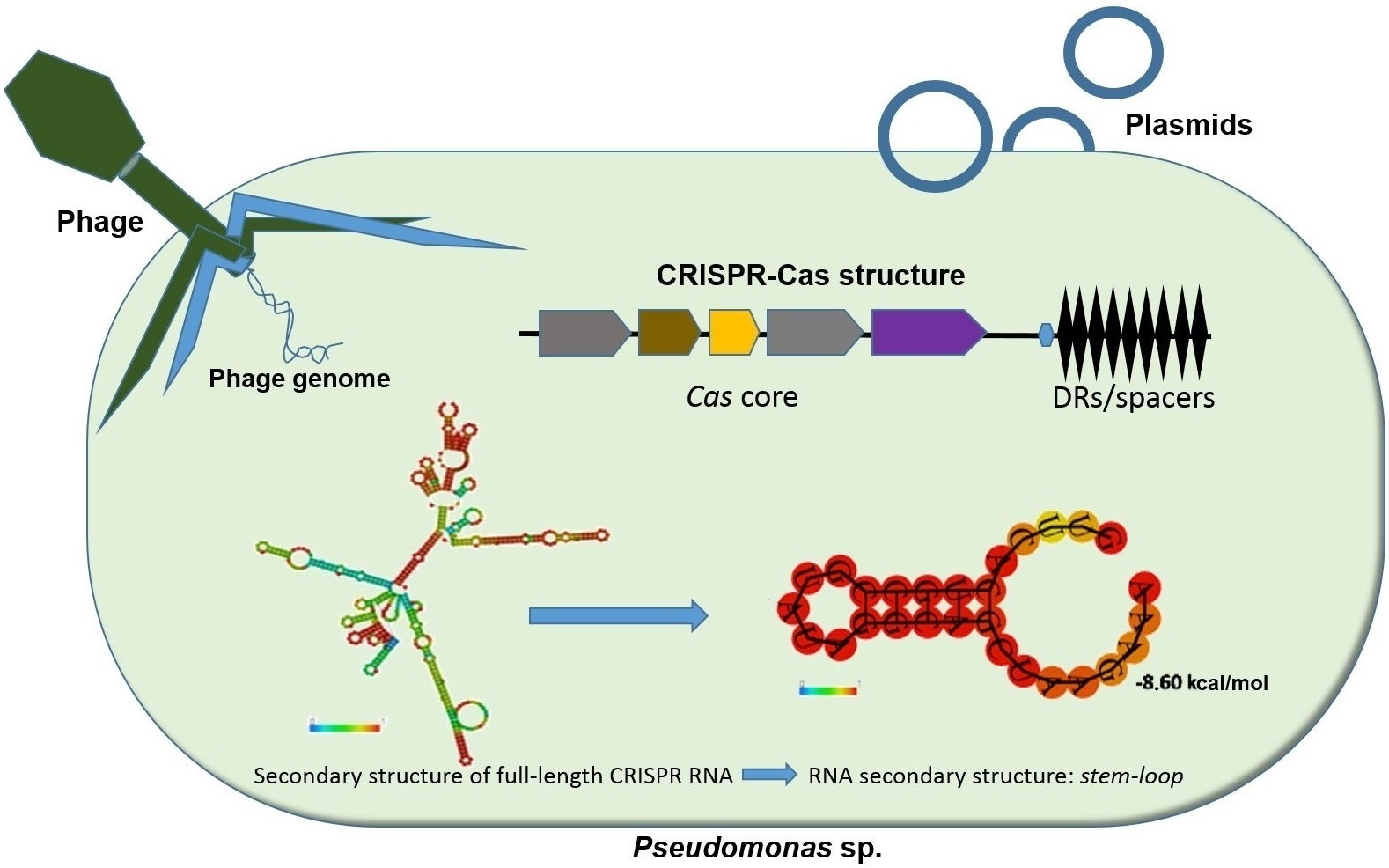
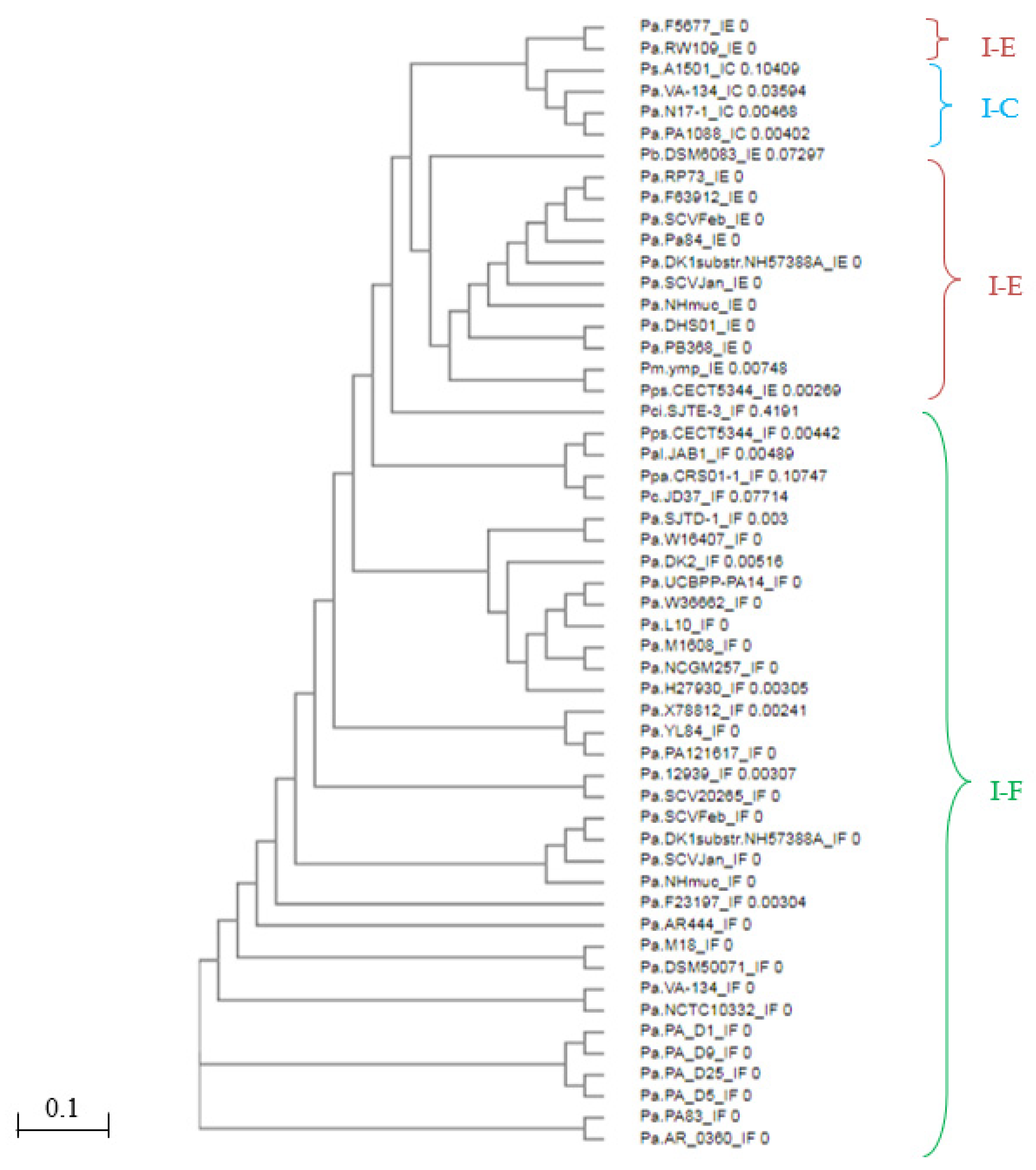
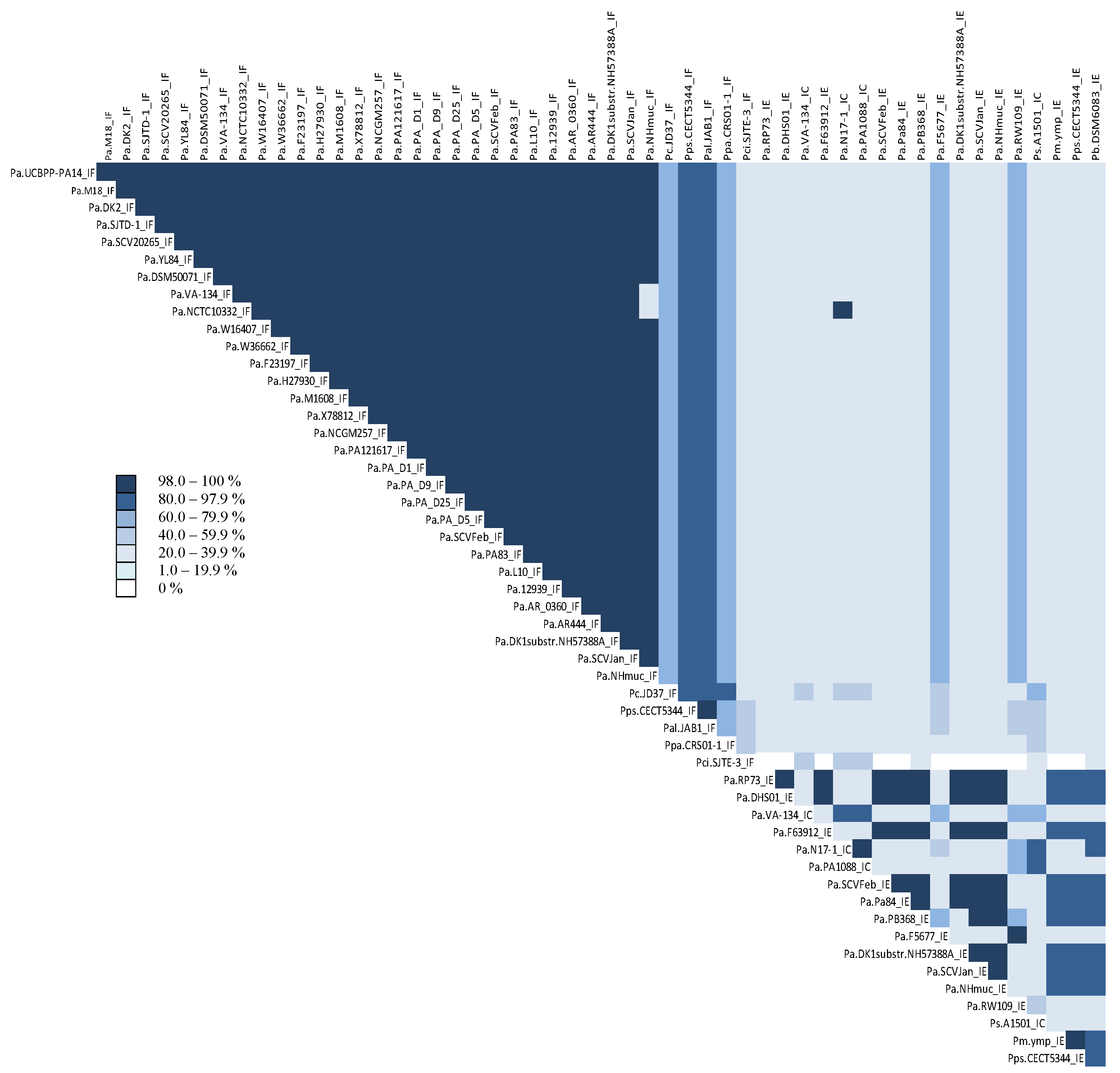
3.1. Identification of CRISPR Structures
3.2. Identification and Comparison of CRISPR-Associated Genes (cas) and CAS Proteins
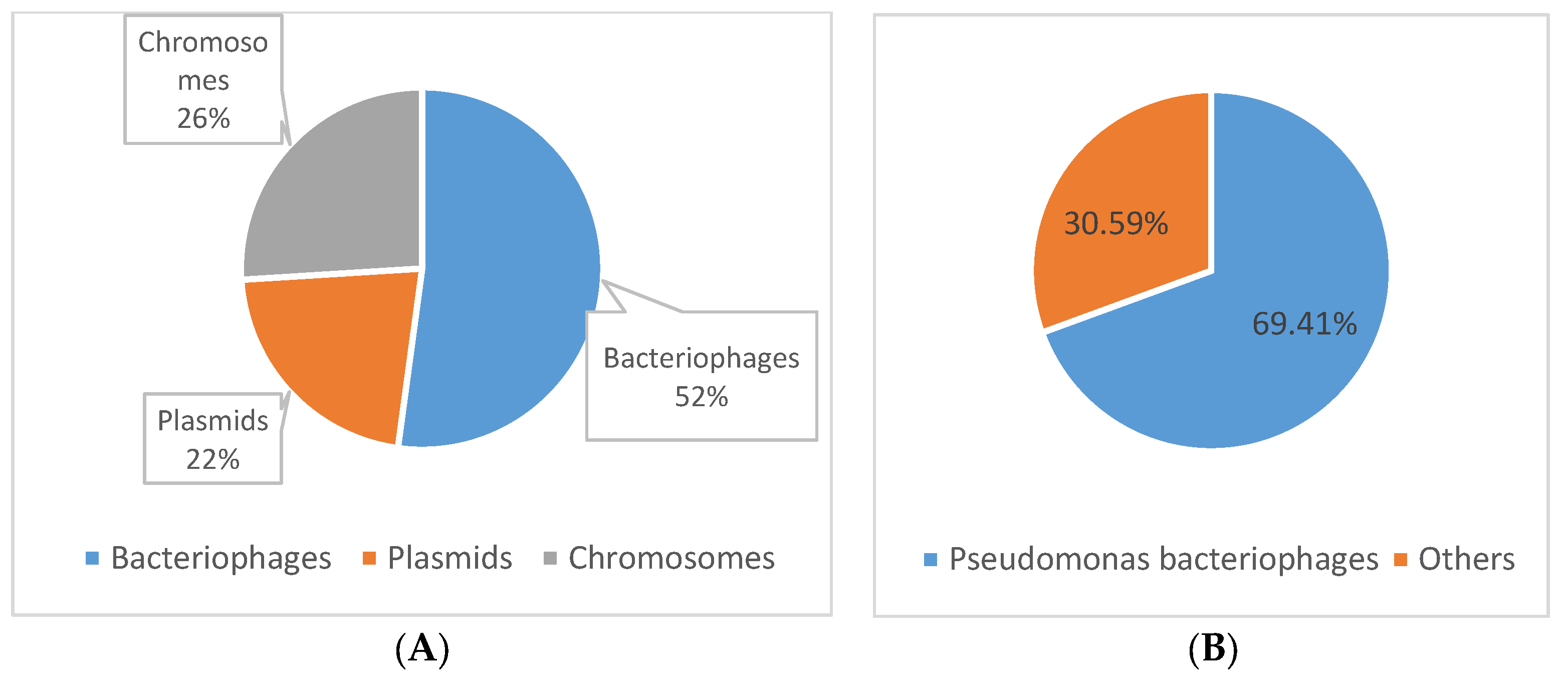
3.3. Determining the Origin of the Spacer Sequences
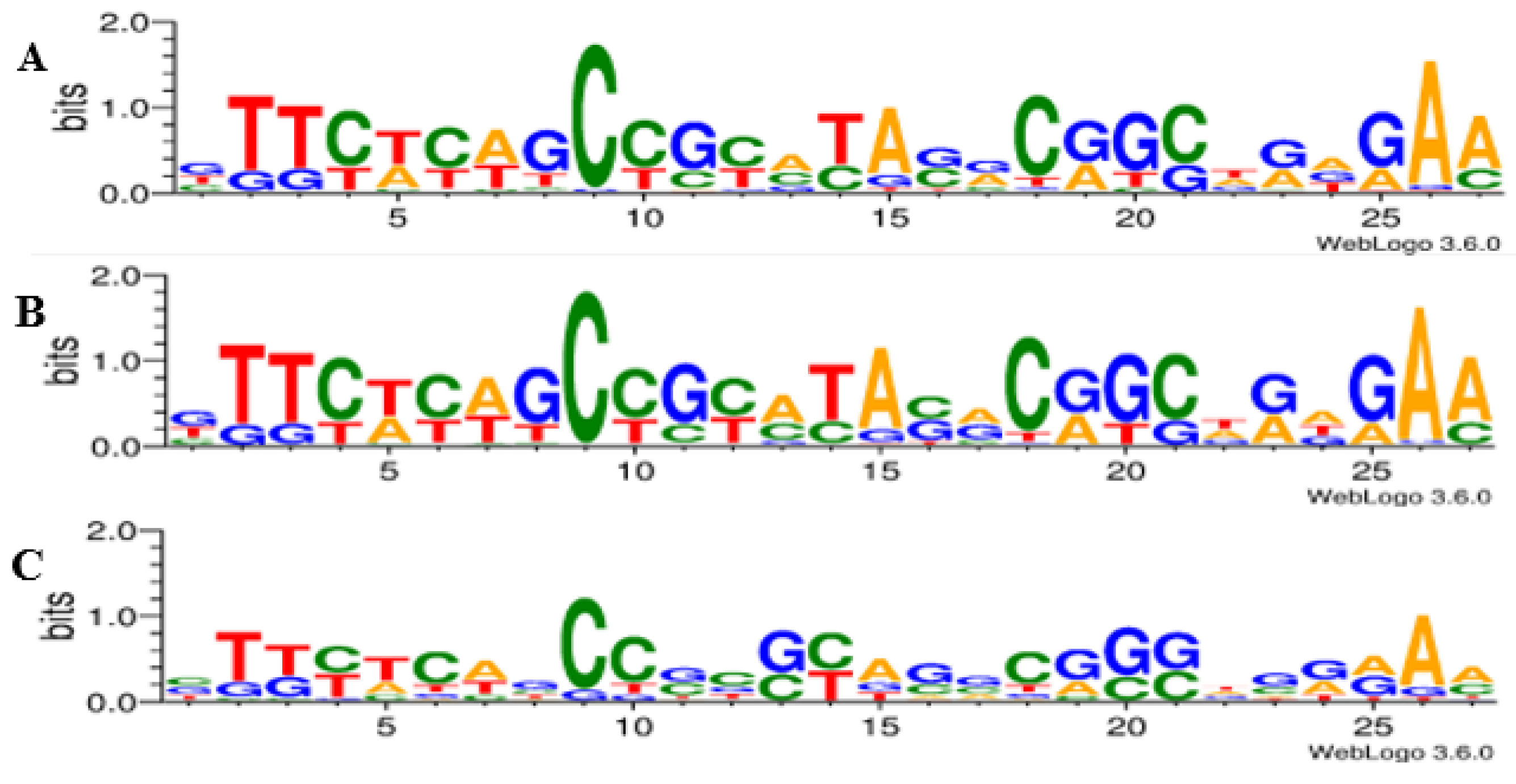
3.4. Identification of Protospacer Adjacent Motifs (PAMs) and Self-Targeting
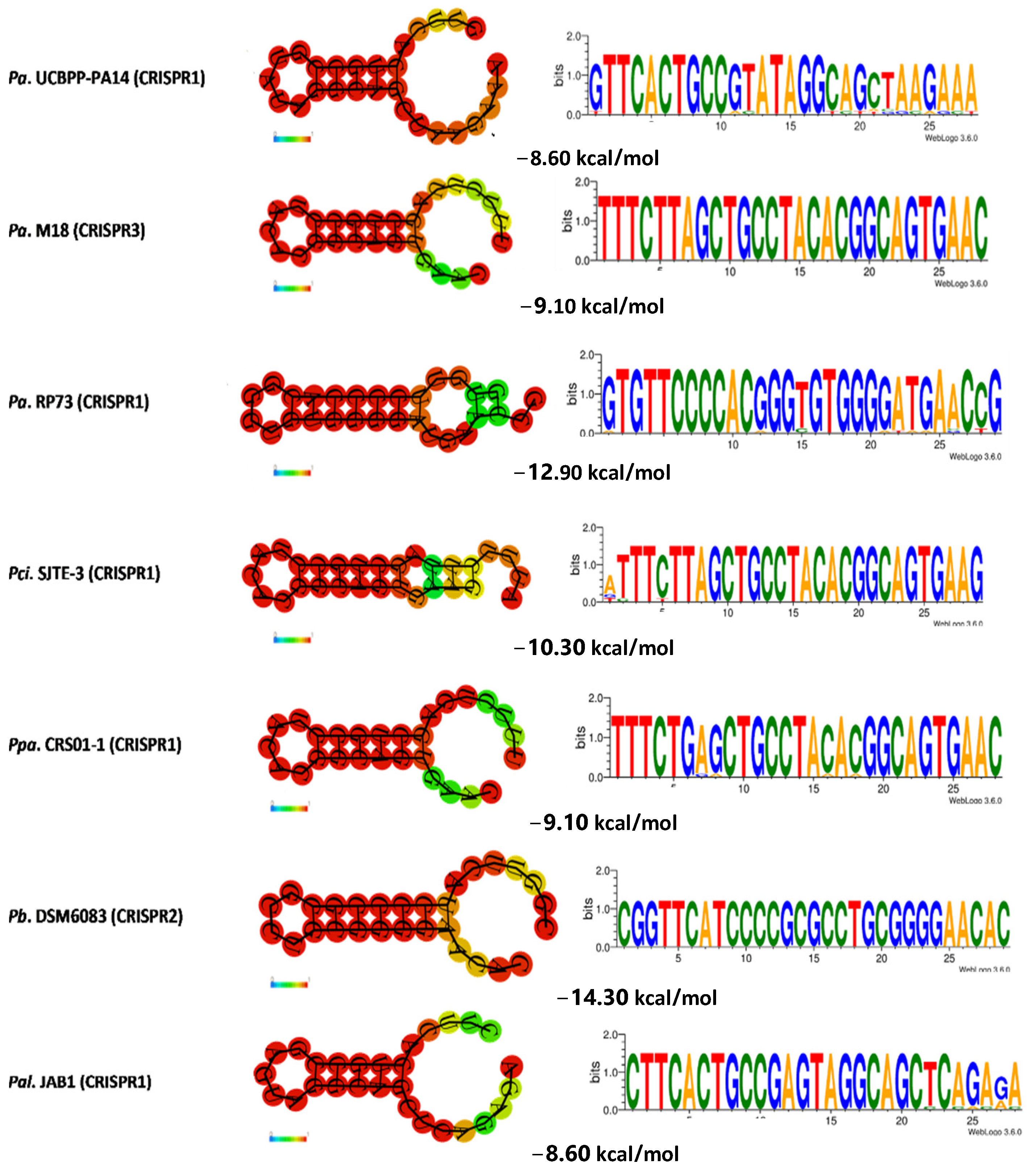
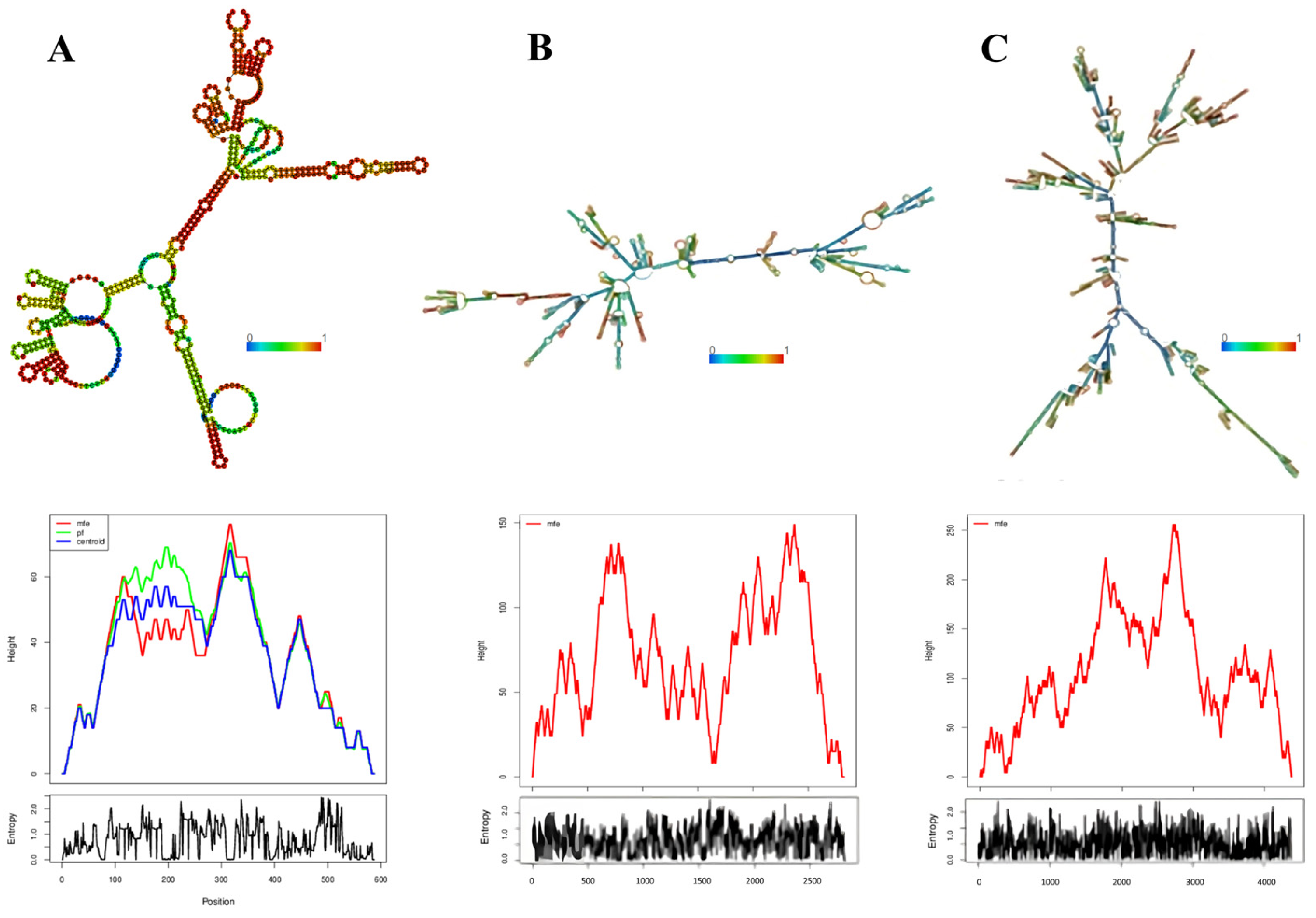
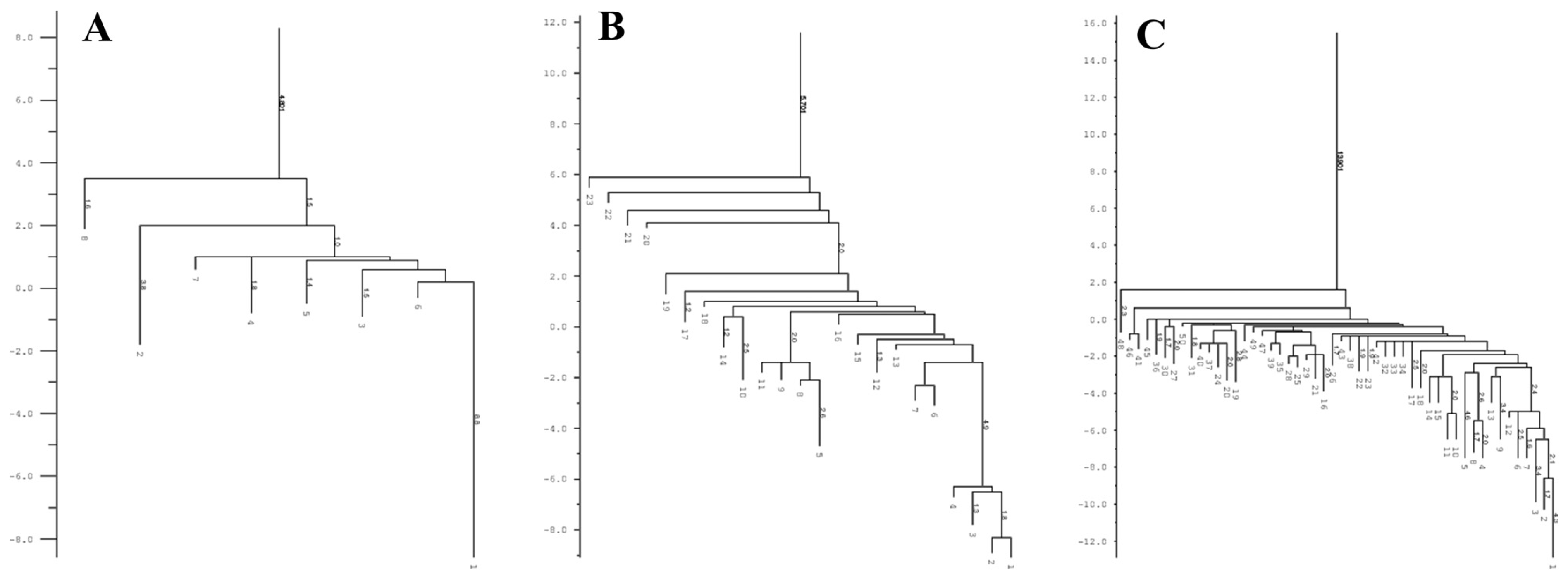
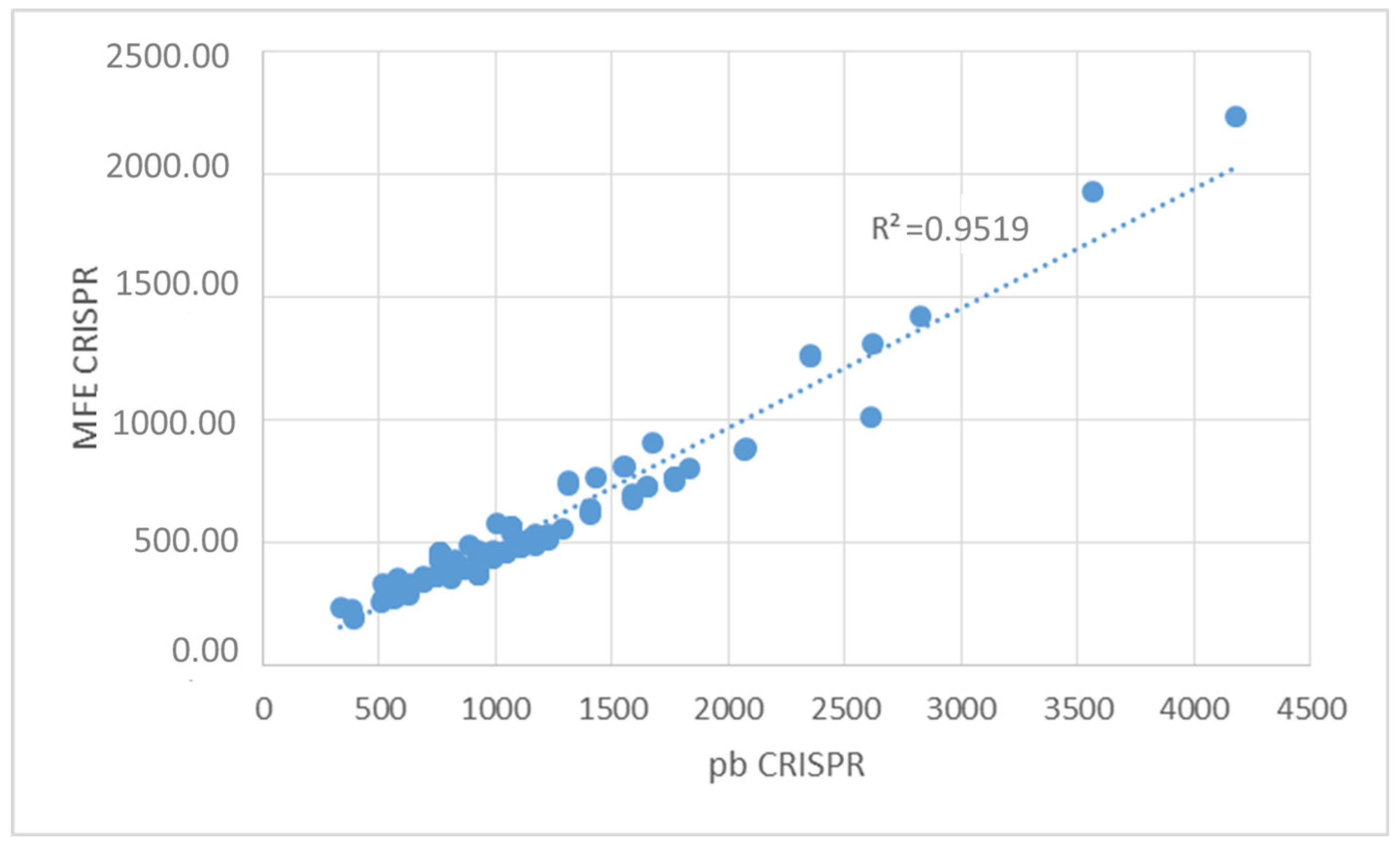
3.5. Determination of the Conservation of Direct Repeats (DRs) and the Prediction of the RNA Secondary Structure
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- NCBI. Pseudomonas Taxonomy. Documento en Línea. 2017. Available online: https://www.ncbi.nlm.nih.gov/Taxonomy/Browser/wwwtax.cgi?id=286 (accessed on 30 January 2020).
- Iglewski, B. Pseudomonas. In Medical Microbiology, 4th ed.; Baron, S., Ed.; University of Texas Medical Branch at Galveston: Galveston, TX, USA, 1996. [Google Scholar]
- Lau, G.; Hassett, D.; Ran, H.; Kong, F. The role of pyocyanin in Pseudomonas aeruginosa infection. Trends Mol. Med. 2004, 10, 599–606. [Google Scholar] [CrossRef] [PubMed]
- Nain, Z.; Minnatul, M. Whole-genome sequence, functional annotation, and comparative genomics of the high biofilm-producing multidrug-resistant Pseudomonas aeruginosa MZ4A isolated from clinical waste. Gene Rep. 2021, 22, 10999. [Google Scholar] [CrossRef]
- Davies, J. Pseudomonas aeruginosa in cystic fibrosis: Pathogenesis and persistence. Pediatr. Respir. Rev. 2002, 3, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Terry, E.; Ruíz, J.; Tejeda, T. Efecto de un bioproducto a base de Pseudomonas aeruginosa en el cultivo del tomate (Solanum licopersicumMill). Rev. Colomb. Biotecnol. 2010, 12, 32–38. [Google Scholar]
- Piqueres, M. Actividad Antimicrobiana de Compuestos de Origen Natural y su Aplicación en la Industria Cosmética; Trabajo de Fin de Grado; Universidad Miguel Hernández de Elche: Elche, Spain, 2019. [Google Scholar]
- Kwok, W.C.; Ho, J.C.M.; Tam, T.C.C.; Ip, M.S.M.; Lam, D.C.L. Risk factors for Pseudomonas aeruginosa colonization in non-cystic fibrosis bronchiectasis and clinical implications. Respir. Res. 2021, 22, 1–8. [Google Scholar] [CrossRef]
- Lyons, C.; Raustad, N.; Bustos, M.; Shiaris, M. Incidence of Type II CRISPR1-Cas Systems in Enterococcus Is Species-Dependent. PLoS ONE 2015, 10, 1–11. [Google Scholar] [CrossRef]
- Rodriguez, M.; Hullahalli, K.; Palmer, K. CRISPR mediatedremoval of antibiotic resistance genes in Enterococcus faecalis populations. FASEB J. 2017, 31, 909.3. [Google Scholar]
- Smith, L.M.; Jackson, S.A.; Malone, L.M.; Ussher, J.E.; Gardner, P.P.; Fineran, P.C. The Rcs stress response inversely controls surface and CRISPR–Cas adaptive immunity to discriminate plasmids and phages. Nat. Microbiol. 2021, 6, 162–172. [Google Scholar] [CrossRef]
- Zegans, M.; Wagner, J.; Cady, K.; Murphy, D.; Hammond, J.; O’Toole, G. Interaction between Bacteriophage DMS3 and Host CRISPR Region Inhibits Group Behaviors of Pseudomonas aeruginosa. J. Bacteriol. 2009, 191, 210–219. [Google Scholar] [CrossRef]
- Cady, K.; O’Toole, G. Non-Identity-Mediated CRISPR-Bacteriophage Interaction Mediated via the Csy and Cas3 Proteins. J. Bacteriol. 2011, 193, 3433–3445. [Google Scholar] [CrossRef]
- Cady, K.; White, A.; Hammond, J.; Abendroth, M.; Karthikeyan, R.; Lalitha, P.; Zegans, M.; O’Toole, G. Prevalence, conservation and functional analysis of Yersinia and Escherichia CRISPR regions in clinical Pseudomonas aeruginosa isolates. Microbioloy 2011, 157, 430–437. [Google Scholar] [CrossRef]
- Grissa, I.; Vergnaud, G.; Pourcel, C. The CRISPRdb database and tools todisplay CRISPRs and to generate dictionaries of spacers and repeats. BMC Bioinform. 2007, 23, 172. [Google Scholar]
- Luz, A.C.d.O.; da Silva, J.M.A.; Rezende, A.M.; de Barros, M.P.S.; Leal-Balbino, T.C. Analysis of direct repeats and spacers of CRISPR/Cas systems type I-F in Brazilian clinical strains of Pseudomonas aeruginosa. Mol. Genet. Genom. 2019, 294, 1095–1105. [Google Scholar] [CrossRef] [PubMed]
- Wheatley, R.M.; MacLean, R.C. CRISPR-Cas systems restrict horizontal gene transfer in Pseudomonas aeruginosa. ISME J. 2021, 15, 1420–1433. [Google Scholar] [CrossRef] [PubMed]
- León, L.M.; Park, A.E.; Borges, A.L.; Zhang, J.Y.; Bondy-Denomy, J. Mobile element warfare via CRISPR and anti-CRISPR in Pseudomonas aeruginosa. Nucleic Acids Res. 2021, 49, 2114–2125. [Google Scholar] [CrossRef]
- Gagaletsios, L.A.; Papagiannitsis, C.C.; Petinaki, E. Prevalence and analysis of CRISPR/Cas systems in Pseudomonas aeruginosa isolates from Greece. Mol. Genet. Genom. 2022, 297, 1767–1776. [Google Scholar] [CrossRef] [PubMed]
- Dela Ahator, S.; Liu, Y.; Wang, J.; Zhang, L.H. The virulence factor regulator and quorum sensing regulate the type I-F CRISPR-Cas mediated horizontal gene transfer in Pseudomonas aeruginosa. Front. Microbiol. 2022, 13, 987656. [Google Scholar] [CrossRef]
- Heussler, G.E.; Miller, J.L.; Price, C.E.; Collins, A.J.; O’Toole, G.A. Requirements for Pseudomonas aeruginosa Type I-F CRISPR-Cas Adaptation Determined Using a Biofilm Enrichment Assay. J. Bacteriol. 2016, 198, 3080–3090. [Google Scholar] [CrossRef]
- Høyland-Kroghsbo, N.M.; Paczkowski, J.; Mukherjee, S.; Broniewski, J.; Westra, E.; Bondy-Denomy, J.; Bassler, B.L. Quorum sensing controls thePseudomonas aeruginosaCRISPR-Cas adaptive immune system. Proc. Natl. Acad. Sci. USA 2017, 114, 131–135. [Google Scholar] [CrossRef]
- Palmer, K.L.; Whiteley, M. DMS3-42: The Secret to CRISPR-Dependent Biofilm Inhibition in Pseudomonas aeruginosa. J. Bacteriol. 2011, 193, 3431–3432. [Google Scholar] [CrossRef]
- Benson, D.; Karsch, I.; Lipman, D.; Ostell, J.; Wheeler, D. GenBank. Nucleic Acids Res. 2008, 36, 25–30. [Google Scholar] [CrossRef] [PubMed]
- Bernal, A.; Ear, U.; Kyrpides, N. Genomes OnLine Database (GOLD): A monitor of genome projects world-wide. Nucleic Acids Res. 2001, 29, 126–127. [Google Scholar] [CrossRef]
- Hyatt, D.; Chen, G.; LoCascio, F.; Land, M.; Larimer, F.; Hauser, L. Prodigal: Prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 2010, 11, 119. [Google Scholar] [CrossRef] [PubMed]
- Abby, S.; Néron, B.; Ménager, H.; Touchon, M.; Rocha, E. MacSyFinder: A program to mine genomes for molecular systems with an application to CRISPR-Cas systems. PLoS ONE 2014, 9, e110726. [Google Scholar] [CrossRef] [PubMed]
- Horvath, P.; Coûté-Monvoisin, A.-C.; Romero, D.A.; Boyaval, P.; Fremaux, C.; Barrangou, R. Comparative analysis of CRISPR loci in lactic acid bacteria genomes. Int. J. Food Microbiol. 2009, 131, 62–70. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular Evolutionary Genetics Analysis Version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef]
- Larkin, M.; Blackshields, G.; Brown, N.; Chenna, R.; McGettigan, P.; McWilliam, H.; Valentin, F.; Wallace, I.; Wilm, A.; Lopez, R.; et al. Clustal W and Clustal X version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef]
- Shen, J.; Lv, L.; Wang, X.; Xiu, Z.; Chen, G. Comparative analysis of CRISPR-Cas systems in Klebsiella genomes. J. Basic Microbiol. 2017, 57, 325–336. [Google Scholar] [CrossRef]
- Li, W.; Bian, X.; Evivie, S.E.; Huo, G.-C. Comparative Analysis of Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR) of Streptococcus thermophilus St-I and its Bacteriophage-Insensitive Mutants (BIM) Derivatives. Curr. Microbiol. 2016, 73, 393–400. [Google Scholar] [CrossRef]
- Crooks, G.; Hon, G.; Chandonia, J.; Brenner, S. WebLogo: A sequence Logo Generator. Genome Res. 2004, 14, 1188–1190. [Google Scholar] [CrossRef]
- Hofacker, I. Vienna RNA secondary structure server. Nucleic Acids Res. 2003, 31, 3429–3431. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, S.; Zaved, T.; Hassan, F.; Kabir, Y.; Smith, M.; Arif, M. Assessment of the Evolutionary Origin and Possibility of CRISPR-Cas (CASS) Interference Pathway in Vibrio cholerae O395. Silico Biol. 2009, 9, 245–254. [Google Scholar] [CrossRef]
- Mohanraju, P.; Makarova, K.S.; Zetsche, B.; Zhang, F.; Koonin, E.V.; van der Oost, J. Diverse evolutionary roots and mechanistic variations of the CRISPR-Cas systems. Science 2016, 353, aad5147. [Google Scholar] [CrossRef] [PubMed]
- Makarova, K.S.; Wolf, Y.I.; Iranzo, J.; Shmakov, S.A.; Alkhnbashi, O.S.; Brouns, S.J.J.; Charpentier, E.; Cheng, D.; Haft, D.H.; Horvath, P.; et al. Evolutionary classification of CRISPR–Cas systems: A burst of class 2 and derived variants. Nat. Rev. Microbiol. 2020, 18, 67–83. [Google Scholar] [CrossRef]
- Hidalgo-Cantabrana, B.; Barrangou, R. Characterization and applications of Type I CRISPR-Cas systems. Biochem. Soc. Trans. 2020, 48, 15–23. [Google Scholar] [CrossRef]
- Burstein, D.; Sun, C.; Brown, C.; Sharon, I.; Anantharaman, K.; Probst, A.; Thomas, B.; Banfield, J. Major bacterial lineages are essentially devoid of CRISPR-Cas viral defence systems. Nat. Commun. 2016, 7, 1–8. [Google Scholar] [CrossRef]
- Osakabe, K.; Wada, N.; Miyaji, T.; Murakami, E.; Marui, K.; Ueta, R.; Hashimoto, R.; Abe-Hara, C.; Kong, B.; Yano, K.; et al. Genome editing in plants using CRISPR type I-D nuclease. Commun. Biol. 2020, 3, 1–10. [Google Scholar] [CrossRef]
- Van der Oost, J.; Westra, E.; Jackson, R.; Wiedenheft, B. Unravelling the structural and mechanistic basis of CRISPR-Cas systems. Nat. Rev. Microbiol. 2014, 12, 479–492. [Google Scholar] [CrossRef]
- Wright, A.; Nunez, J.; Doudna, J. Biology and Applications of CRISPR Systems: Harnessing Nature’s Toolbox for Genome Engineering. Cell 2016, 164, 29–44. [Google Scholar] [CrossRef]
- Parra, Á.; Antequera, L.A.; Lossada, C.; Fernández-Materán, F.V.; Parra, M.D.; Centanaro, P.; Arcos, D.; Castro, A.; Moncayo, L.; Freddy Romero Paz, J.L.; et al. Comparative Analysis of CRISPR-Cas Systems in Vibrio and Photobacterium Genomes of High Influence in Aquaculture Production. Biointerface Res. Appl. Chem. 2021, 11, 9513–9529. [Google Scholar]
- Tuminauskaite, D.; Norkunaite, D.; Fiodorovaite, M.; Tumas, S.; Songailiene, I.; Tamulaitiene, G.; Sinkunas, T. DNA interference is controlled by R-loop length in a type I-F1 CRISPR-Cas system. BMC Biol. 2020, 18, 1–16. [Google Scholar] [CrossRef]
- Ozcan, A.; Pausch, P.; Linden, A.; Wulf, A.; Schuhle, K.; Heider, J.; Urlaub, H.; Heimerl, T.; Bange, G.; Randau, L. Procesamiento de ARN CRISPR tipo IV y formación de complejo efector en Aromatoleum aromaticum. Nat. Microbiol. 2019, 4, 89–96. [Google Scholar] [CrossRef] [PubMed]
- Mendoza, S.D.; Nieweglowska, E.S.; Govindarajan, S.; Leon, L.M.; Berry, J.D.; Tiwari, A.; Chaikeeratisak, V.; Pogliano, J.; Agard, D.A.; Bondy-Denomy, J. A bacteriophage nucleus-like compartment shields DNA from CRISPR nucleases. Nature 2019, 577, 244–248. [Google Scholar] [CrossRef] [PubMed]
- Makarova, K.S.; Aravind, L.; I Wolf, Y.; Koonin, E.V. Unification of Cas protein families and a simple scenario for the origin and evolution of CRISPR-Cas systems. Biol. Direct 2011, 6, 1–27. [Google Scholar] [CrossRef]
- Makarova, K.S.; Wolf, Y.I.; Alkhnbashi, O.S.; Costa, F.; Shah, S.A.; Saunders, S.J.; Barrangou, R.; Brouns, S.J.J.; Charpentier, E.; Haft, D.H.; et al. An updated evolutionary classification of CRISPR–Cas systems. Nat. Rev. Microbiol. 2015, 13, 722–736. [Google Scholar] [CrossRef]
- Takeuchi, N.; Wolf, Y.I.; Makarova, K.S.; Koonin, E.V. Nature and Intensity of Selection Pressure on CRISPR-Associated Genes. J. Bacteriol. 2012, 194, 1216–1225. [Google Scholar] [CrossRef]
- Makarova, K.S.; Haft, D.H.; Barrangou, R.; Brouns, S.J.J.; Charpentier, E.; Horvath, P.; Moineau, S.; Mojica, F.J.M.; Wolf, Y.I.; Yakunin, A.F.; et al. Evolution and classification of the CRISPR–Cas systems. Nat. Rev. Genet. 2011, 9, 467–477. [Google Scholar] [CrossRef] [PubMed]
- Makarova, K.S.; Grishin, N.V.; Shabalina, S.A.; Wolf, Y.I.; Koonin, E.V. A putative RNA-interference-based immune system in prokaryotes: Computational analysis of the predicted enzymatic machinery, functional analogies with eukaryotic RNAi, and hypothetical mechanisms of action. Biol. Direct 2006, 1, 7. [Google Scholar] [CrossRef]
- Nuñez, J.K.; Kranzusch, P.J.; Noeske, J.; Wright, A.V.; Davies, C.W.; Doudna, J.A. Cas1–Cas2 complex formation mediates spacer acquisition during CRISPR–Cas adaptive immunity. Nat. Struct. Mol. Biol. 2014, 21, 528–534. [Google Scholar] [CrossRef]
- Nuñez, J.K.; Lee, A.S.Y.; Engelman, A.; Doudna, J.A. Integrase-mediated spacer acquisition during CRISPR–Cas adaptive immunity. Nature 2015, 519, 193–198. [Google Scholar] [CrossRef]
- Shah, S.A.; Garrett, R.A. CRISPR/Cas and Cmr modules, mobility and evolution of adaptive immune systems. Res. Microbiol. 2011, 162, 27–38. [Google Scholar] [CrossRef] [PubMed]
- Garrett, R.A.; Vestergaard, G.; Shah, S. Archaeal CRISPR-based immune systems: Exchangeable functional modules. Trends Microbiol. 2011, 19, 549–556. [Google Scholar] [CrossRef] [PubMed]
- Munck, C.; Sheth, R.U.; Freedberg, D.E.; Wang, H.H. Recording mobile DNA in the gut microbiota using an Escherichia coli CRISPR-Cas spacer acquisition platform. Nat. Commun. 2020, 11, 95. [Google Scholar] [CrossRef] [PubMed]
- Mojica, F.; Díez, C.; García, J.; Soria, E. Intervening sequences of regularly spaced prokaryotic repeats derive from foreign genetic elements. J. Mol. Evol. 2005, 60, 174–182. [Google Scholar] [CrossRef]
- Javalkote, V.S.; Kancharla, N.; Bhadra, B.; Shukla, M.; Soni, B.; Sapre, A.; Goodin, M.; Bandyopadhyay, A.; Dasgupta, S. CRISPR-based assays for rapid detection of SARS-CoV-2. Methods 2020, 203, 594–603. [Google Scholar] [CrossRef]
- Bolotin, A.; Quinquis, B.; Sorokin, A.; Ehrlich, S.D. Clustered regularly interspaced short palindrome repeats (CRISPRs) have spacers of extrachromosomal origin. Microbiology 2005, 151, 2551–2561. [Google Scholar] [CrossRef]
- Pourcel, C.; Salvignol, G.; Vergnaud, G. CRISPR elements in Yersinia pestis acquire new repeats by preferential uptake of bacteriophage DNA, and provide additional tools for evolutionary studies. Microbiology 2005, 151, 653–663. [Google Scholar] [CrossRef]
- Singh, A.; Gaur, M.; Sharma, V.; Khanna, P.; Bothra, A.; Bhaduri, A.; Mondal, A.K.; Dash, D.; Singh, Y.; Misra, R. Comparative Genomic Analysis of Mycobacteriaceae Reveals Horizontal Gene Transfer-Mediated Evolution of the CRISPR-Cas System in the Mycobacterium tuberculosis Complex. Msystems 2021, 6, e00934-20. [Google Scholar] [CrossRef]
- Godde, J.S.; Bickerton, A. The Repetitive DNA Elements Called CRISPRs and Their Associated Genes: Evidence of Horizontal Transfer Among Prokaryotes. J. Mol. Evol. 2006, 62, 718–729. [Google Scholar] [CrossRef]
- Haft, D.; Selengut, J.; Mongodin, E.; Nelson, K. A guild of 45 CRISPR-associated (Cas) protein families and multiple CRISPR/Cas subtypes exist in prokaryotic genomes. PLoS Comput. Biol. 2005, 1, e60. [Google Scholar] [CrossRef]
- Rollins, M.F.; Schuman, J.T.; Paulus, K.; Bukhari, H.S.; Wiedenheft, B. Mechanism of foreign DNA recognition by a CRISPR RNA-guided surveillance complex from Pseudomonas aeruginosa. Nucleic Acids Res. 2015, 43, 2216–2222. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.-H.; Lee, G.; Park, C.; Koo, J.; Kim, E.-H.; Bae, E.; Suh, J.-Y. The structure of AcrIE4-F7 reveals a common strategy for dual CRISPR inhibition by targeting PAM recognition sites. Nucleic Acids Res. 2022, 50, 2363–2376. [Google Scholar] [CrossRef]
- Marraffini, L.A.; Sontheimer, E.J. Self versus non-self discrimination during CRISPR RNA-directed immunity. Nature 2010, 463, 568–571. [Google Scholar] [CrossRef]
- Wimmer, F.; Beisel, C.L. CRISPR-Cas Systems and the Paradox of Self-Targeting Spacers. Front. Microbiol. 2020, 10, 3078. [Google Scholar] [CrossRef] [PubMed]
- Paez-Espino, D.; Morovic, W.; Sun, C.L.; Thomas, B.C.; Ueda, K.-I.; Stahl, B.; Barrangou, R.; Banfield, J.F. Strong bias in the bacterial CRISPR elements that confer immunity to phage. Nat. Commun. 2013, 4, 1430. [Google Scholar] [CrossRef]
- Stringer, A.; Cooper, L.; Kadaba, S.; Shrestha, S.; Wade, J. Characterization of Primed Adaptation in the Escherichia coli type I-E CRISPR-Cas System. bioRxiv 2020. [Google Scholar] [CrossRef]
- Bhaya, D.; Davison, M.; Barrangou, R. Los sistemas CRISPR-Cas en bacterias y arqueas: RNAs pequeños y versátiles para la regulación y defensa. Rev. Genet. 2011, 45, 273–297. [Google Scholar] [CrossRef]
- Shechner, D.M. Targeting Noncoding RNA Domains to Genomic Loci with CRISPR-Display: Guidelines for Designing, Building, and Testing sgRNA–ncRNA Expression Constructs. In CRISPR Guide RNA Design. Methods in Molecular Biology; Fulga, T.A., Knapp, D.J.H.F., Ferry, Q.R.V., Eds.; Humana: New York, NY, USA, 2021; Volume 2162. [Google Scholar] [CrossRef]
- Kunin, V.; Sorek, R.; Hugenholtz, P. Evolutionary conservation of sequence and secondary structures in CRISPR repeats. Genome Biol. 2007, 8, R61–R67. [Google Scholar] [CrossRef]
- Qu, D.; Lu, S.; Wang, P.; Mengxue, J.; Yi, S.; Jianzhong, H. Análisis del sistema CRISPR-Cas de Proteus y los factores que afectan el mecanismo funcional. Life Sci. 2019, 231, 1–9. [Google Scholar] [CrossRef]
- Lossada, C.; Fernández, F.; Parra, M.; Moncayo, L.; Romero, F.; Paz, J.; Vera, J.; Pérez, A.; Portillo, E.; Alvarado, Y.; et al. Biological Significance of the Thermodynamic Stability of CRISPR Structures Associated with Unconventional Functions. Biointerface Res. Appl. Chem. 2021, 3, 10381–10392. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Subtype | Cas Genes | Species |
|---|---|---|
| IF | cas1, cas2-cas3, cas6, csy1, csy2, csy3 | P. aeruginosa UCBPP-PA14, M18, DK2, SJTD-1, SCV20265, YL84, DSM50071, NCTC10332, W16407, W36662, F23197, H27930, M1608, X78812, NCGM2572, PA121617, PA_D1, PA_D9, PA_D25, PA_D5, PA83, L10, 12939, AR_0360, AR444. P. chlororaphis JD37 P. alcaliphila JAB1 P. parafulva CRS01-1 P. citronellolis SJTE-3 |
| IE | cas1, cas2, cas3, cas5, cas6, cse1, cse2 | P. aeruginosa RP73, DHS01, F63912, Pa84, PB368, F5677, RW109. P. balearica DSM6083 P. mendocina ymp |
| IC | cas1, cas2, cas3, cas4, cas5, cas7, cas8c | P. aeruginosa N17-1, PA1088, P. stutzeri A1501 |
| IV | csf1, csf2, csf3, csf4, csf5 | P. putida KF715 |
| IF/IE | cas1, cas2-cas3, cas6, csy1, csy2, csy3 cas1, cas2, cas3, cas5, cas6, cse1, cse2 | P. aeruginosa SCVfeb, SCVJan, Nhmuc, DK1 substr. NH57388A. P. pseudoalcaligenes CECT5344 |
| IF/IC | cas1, cas2-cas3, cas6, csy1, csy2, csy3 cas1, cas2, cas3, cas4, cas5, cas7, cas8c | P. aeruginosa VA-134 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Parra-Sánchez, Á.; Antequera-Zambrano, L.; Martínez-Navarrete, G.; Zorrilla-Muñoz, V.; Paz, J.L.; Alvarado, Y.J.; González-Paz, L.; Fernández, E. Comparative Analysis of CRISPR-Cas Systems in Pseudomonas Genomes. Genes 2023, 14, 1337. https://doi.org/10.3390/genes14071337
Parra-Sánchez Á, Antequera-Zambrano L, Martínez-Navarrete G, Zorrilla-Muñoz V, Paz JL, Alvarado YJ, González-Paz L, Fernández E. Comparative Analysis of CRISPR-Cas Systems in Pseudomonas Genomes. Genes. 2023; 14(7):1337. https://doi.org/10.3390/genes14071337
Chicago/Turabian StyleParra-Sánchez, Ángel, Laura Antequera-Zambrano, Gema Martínez-Navarrete, Vanessa Zorrilla-Muñoz, José Luis Paz, Ysaias J. Alvarado, Lenin González-Paz, and Eduardo Fernández. 2023. "Comparative Analysis of CRISPR-Cas Systems in Pseudomonas Genomes" Genes 14, no. 7: 1337. https://doi.org/10.3390/genes14071337
APA StyleParra-Sánchez, Á., Antequera-Zambrano, L., Martínez-Navarrete, G., Zorrilla-Muñoz, V., Paz, J. L., Alvarado, Y. J., González-Paz, L., & Fernández, E. (2023). Comparative Analysis of CRISPR-Cas Systems in Pseudomonas Genomes. Genes, 14(7), 1337. https://doi.org/10.3390/genes14071337