
An ABCC9 Missense Variant Is Associated with Sudden Cardiac Death and Dilated Cardiomyopathy in Juvenile Dogs

 , , ,
, , ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Population
2.2. SNP Genotyping and Analysis
2.3. Haplotype Analysis and Sequencing
2.4. Variant Selection, Genotyping, and Association with SCDY/DCM
3. Results
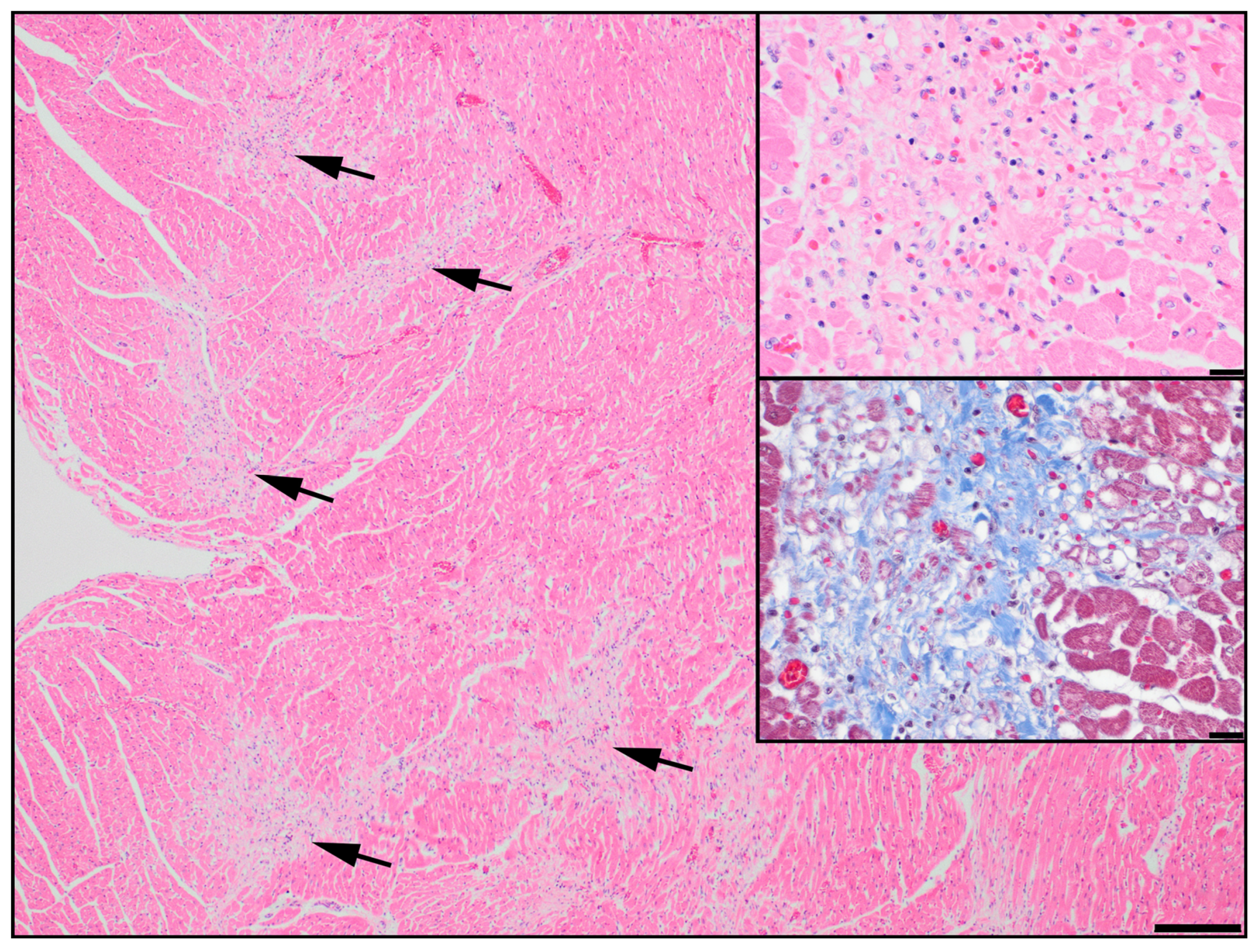
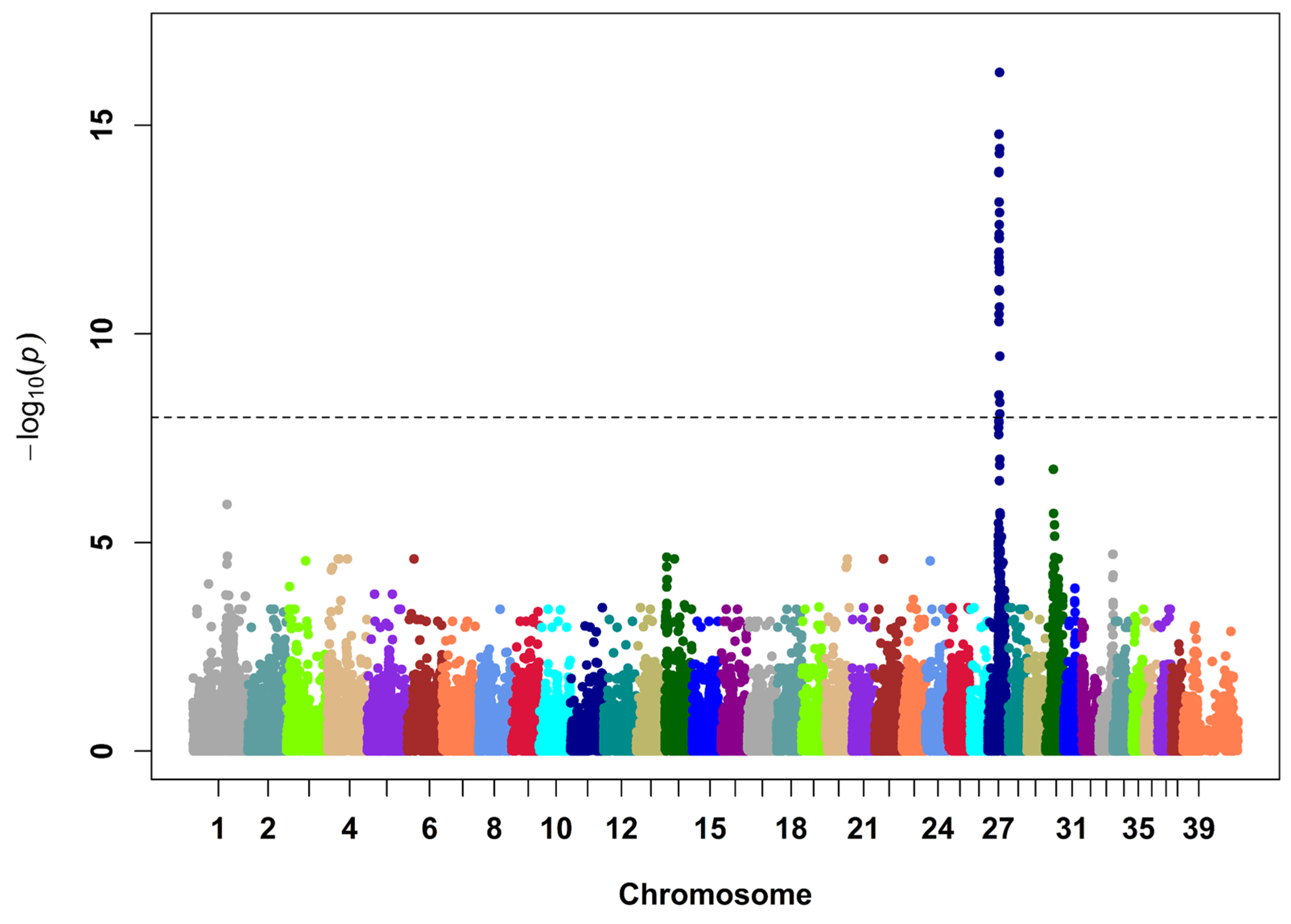
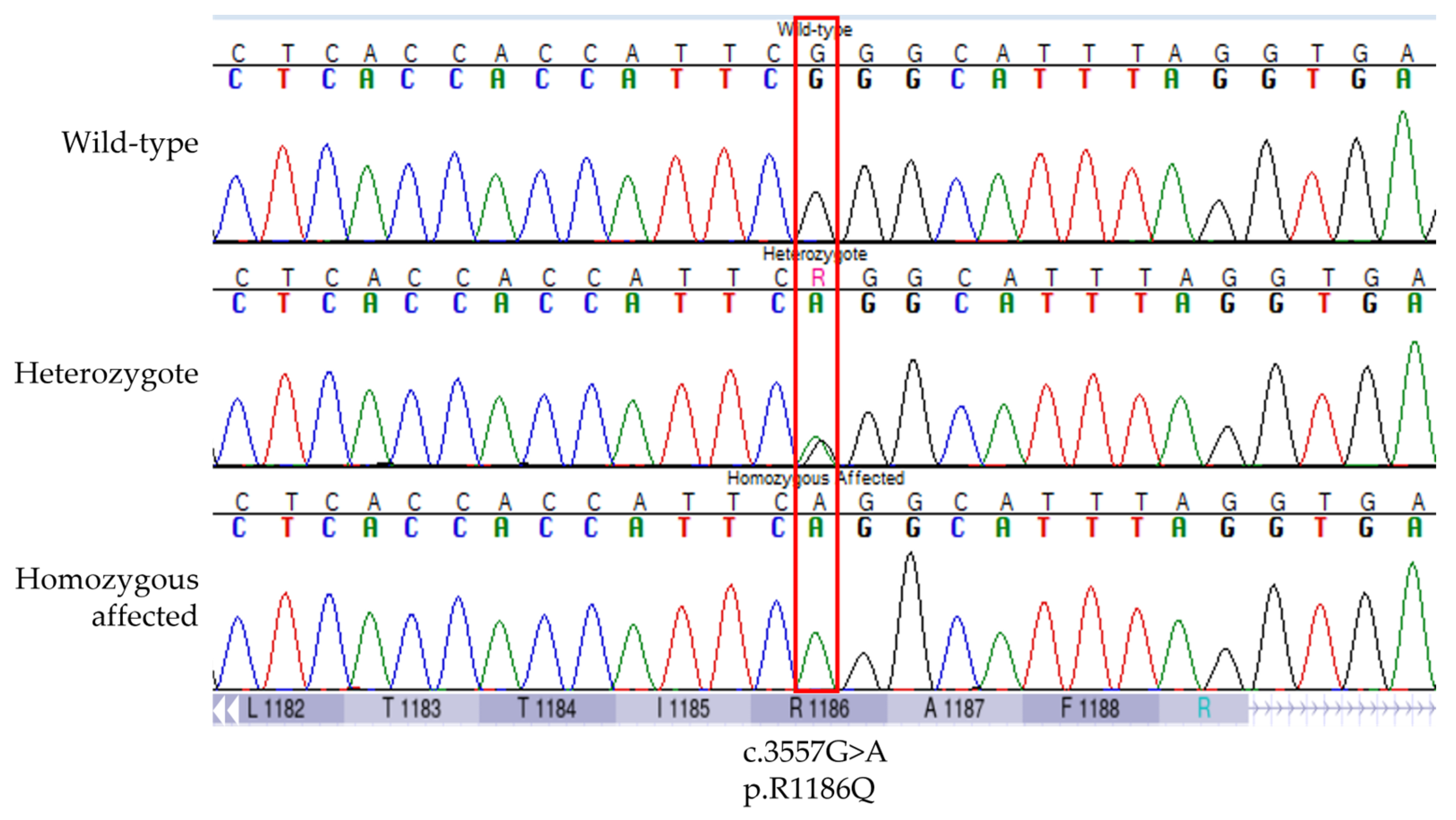
3.1. Discovery of an ABCC9 Missense Variant in a Natural Canine Model of SCDY/DCM
3.2. Validation of the Association between ABCC9 p.R1186Q and SCDY/DCM
3.3. Homologous ABCC9 Variants in Human Databases
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tester, D.J.; Mederios-Domingo, A.; Will, M.L.; Haglund, C.M.; Ackerman, M.J. Cardiac channel molecular autopsy: Insights from 173 consecutive cases of autopsy-negative sudden unexplained death referred for postmortem genetic testing. Mayo Clin. Proc. 2012, 87, 524–539. [Google Scholar] [CrossRef] [PubMed]
- Bagnall, R.D.; Weintraub, R.G.; Ingles, J.; Duflou, J.; Yeates, L.; Lam, L.; Davis, A.M.; Thompson, T.; Connell, V.; Wallace, J.; et al. A prospective study of sudden cardiac death among children and young adults. N. Engl. J. Med. 2016, 374, 2441–2452. [Google Scholar] [CrossRef] [PubMed]
- Hertz, C.L.; Christiansen, S.L.; Ferrero-Miliani, L.; Dahl, M.; Weeke, P.E.; LuCamp; Ottesen, G.L.; Frank-Hansen, R.; Bundgaard, H.; Morling, N. Next-generation sequencing of 100 candidate genes in young victims of suspected sudden cardiac death with structural abnormalities of the heart. Int. J. Legal Med. 2016, 130, 91–102. [Google Scholar] [CrossRef] [PubMed]
- Dewar, L.J.; Alcaide, M.; Fornika, D.; D’Amata, L.; Shafaatalab, S.; Stevens, C.M.; Balachandra, T.; Phillips, S.M.; Sanatani, S.; Morin, R.D.; et al. Investigating the genetic causes of sudden unexpected death in children through targeted next-generation sequencing analysis. Circ. Cardiovasc. Genet. 2017, 10, e001738. [Google Scholar] [CrossRef] [PubMed]
- Larsen, M.K.; Christiansen, S.L.; Hertz, C.L.; Frank-Hansen, R.; Jensen, H.K.; Banner, J.; Morling, N. Targeted molecular genetic testing in young sudden cardiac death victims from Western Denmark. Int. J. Legal Med. 2020, 134, 111–121. [Google Scholar] [CrossRef]
- Mazzarotto, F.; Tayal, U.; Buchan, R.J.; Midwinter, W.; Wilk, A.; Whiffin, N.; Govind, R.; Mazaika, E.; de Marvao, A.; Dawes, T.J.W.; et al. Reevaluating the genetic contribution of monogenic dilated cardiomyopathy. Circulation 2020, 141, 387–398. [Google Scholar] [CrossRef]
- Zhang, L.; Tester, D.J.; Lang, D.; Chen, Y.; Zheng, J.; Gao, R.; Corliss, R.F.; Tang, S.; Kyle, J.W.; Liu, C.; et al. Does sudden unexplained nocturnal death syndrome remain the autopsy negative disorder: A gross, microscopic, and molecular autopsy investigation in Southern China. Mayo Clin. Proc. 2016, 91, 1503–1514. [Google Scholar] [CrossRef]
- Subbotina, E.; Yang, H.Q.; Gando, I.; Williams, N.; Sampson, B.A.; Tang, Y.; Coetzee, W.A. Functional characterization of ABCC9 variants identified in sudden unexpected natural death. Forensic Sci. Int. 2019, 298, 80–87. [Google Scholar] [CrossRef]
- Hershberger, R.E.; Cowan, J.; Jordan, E.; Kinnamon, D.D. The complex and diverse genetic architecture of dilated cardiomyopathy. Circ. Res. 2021, 128, 1514–1532. [Google Scholar] [CrossRef]
- Fan, L.; Yin, P.; Xu, Z. The genetic basis of sudden death in young people—Cardiac and non-cardiac. Gene 2022, 810, 146067. [Google Scholar] [CrossRef]
- Jordan, E.; Peterson, L.; Ai, T.; Asatryan, B.; Bronicki, L.; Brown, E.; Celeghin, R.; Edwards, M.; Fan, J.; Ingles, J.; et al. Evidence-based assessment of genes in dilated cardiomyopathy. Circulation 2021, 144, 7–9. [Google Scholar] [CrossRef]
- Shearin, A.L.; Ostrander, E.A. Leading the way: Canine models of genomics and disease. Dis. Model. Mech. 2010, 3, 27–34. [Google Scholar] [CrossRef]
- Ware, W.A.; Reina-Doreste, Y.; Stern, J.A.; Meurs, K.M. Sudden death associated with QT interval prolongation and KCNQ1 gene mutation in a family of English Springer Spaniels. J. Vet. Intern. Med. 2015, 29, 561–568. [Google Scholar] [CrossRef]
- Meurs, K.M.; Friedenberg, S.G.; Kolb, J.; Saripalli, C.; Tonino, P.; Woodruff, K.; Olby, N.J.; Keene, B.W.; Adin, D.B.; Yost, O.L.; et al. A missense variant in the titin gene in Doberman pinscher dogs with familial dilated cardiomyopathy and sudden cardiac death. Hum. Genet. 2019, 138, 515–524. [Google Scholar] [CrossRef]
- Yost, O.; Friedenberg, S.G.; Jesty, S.A.; Olby, N.J.; Meurs, K.M. The R9H phospholamban mutation is associated with highly penetrant dilated cardiomyopathy and sudden death in a spontaneous canine model. Gene 2019, 20, 118–122. [Google Scholar] [CrossRef]
- Leach, S.B.; Briggs, M.; Hansen, L.; Johnson, G.S. Prevalence, geographic distribution, and impact on lifespan of a dilated cardiomyopathy-associated RNA-binding motif protein 20 variant in genotyped dogs. J. Vet. Cardiol. 2022, 40, 119–125. [Google Scholar] [CrossRef]
- Meurs, K.M.; Mauceli, E.; Lahmers, S.; Acland, G.M.; White, S.N.; Lindblad-Toh, K. Genome-wide association identifies a deletion in the 3′ untranslated region of striatin in a canine model of arrhythmogenic right ventricular cardiomyopathy. Hum. Genet. 2010, 128, 315–324. [Google Scholar] [CrossRef]
- Meurs, K.M.; Lahmers, S.; Keene, B.W.; White, S.N.; Oyama, M.A.; Mauceli, E.; Lindblad-Toh. A splice site mutation in a gene encoding for PDK4, a mitochondrial protein, is associated with the development of dilated cardiomyopathy in the Doberman pinscher. Hum. Genet. 2012, 131, 1319–1325. [Google Scholar] [CrossRef]
- Meurs, K.M.; Friedenberg, S.G.; Olby, N.J.; Condit, J.; Weidman, J.; Rosenthal, S.; Shelton, D. A QIL variant associated with ventricular arrhythmias and sudden cardiac death in the juvenile Rhodesian Ridgeback dog. Genes 2019, 10, 168. [Google Scholar] [CrossRef]
- Legge, C.H.; López, A.; Hanna, P.; Côté, E.; Hare, E.; Martinson, S.A. Histological characterization of dilated cardiomyopathy in the juvenile toy Manchester terrier. Vet. Pathol. 2013, 50, 1043–1052. [Google Scholar] [CrossRef]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A Tool Set for Whole-Genome Association and Population-Based Linkage Analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Stephens, M. Genome-wide efficient mixed-model analysis for association studies. Nat. Genet. 2012, 44, 821–824. [Google Scholar] [CrossRef] [PubMed]
- Scheet, P.; Stephens, M. A fast and flexible statistical model for large-scale population genotype data: Applications to inferring missing genotypes and haplotypic phase. Am. J. Hum. Genet. 2006, 78, 629–644. [Google Scholar] [CrossRef] [PubMed]
- Kent, W.J. BLAT—The BLAST-like alignment tool. Genome Res. 2002, 12, 656–664. [Google Scholar] [CrossRef] [PubMed]
- Jagannathan, V.; Hitte, C.; Kidd, J.M.; Masterson, P.; Murphy, T.D.; Emery, S.; Davis, B.; Buckley, R.M.; Liu, Y.H.; Zhang, X.Q.; et al. Dog10K_Boxer_Tasha_1.0: A long-read assembly of the dog reference genome. Genes 2021, 12, 847. [Google Scholar] [CrossRef]
- Sherry, S.T.; Ward, M.; Sirotkin, K. dbSNP-database for single nucleotide polymorphisms and other classes of minor genetic variation. Genome Res. 1999, 9, 677–679. [Google Scholar] [CrossRef]
- Jagannathan, V.; Drögemüller, C.; Leeb, T.; Dog Biomedical Variant Database Consortium (DBVDC). A comprehensive biomedical variant catalogue based on whole genome sequences of 582 dogs and eight wolves. Anim. Genet. 2019, 50, 695–704. [Google Scholar] [CrossRef]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef]
- Pejaver, V.; Urresti, J.; Lugo-Martinez, J.; Pagel, K.A.; Lin, G.N.; Nam, H.J.; Mort, M.; Cooper, D.N.; Sebat, J.; Iakoucheva, L.M.; et al. Inferring the molecular and phenotypic impact of amino acid variants with MutPred2. Nat. Commun. 2020, 11, 5918. [Google Scholar] [CrossRef]
- Kent, W.J.; Sugnet, C.W.; Furey, T.S.; Roskin, K.M.; Pringle, T.H.; Zahler, A.M.; Haussler, D. The human genome browser at UCSC. Genome Res. 2002, 12, 996–1006. [Google Scholar] [CrossRef]
- Pollard, K.S.; Hubisz, M.J.; Rosenbloom, K.R.; Siepel, A. Detection of nonneutral substitution rates on mammalian phylogenies. Genome Res. 2010, 20, 110–121. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Varadi, M.; Anyango, S.; Deshpande, M.; Nair, S.; Natassia, C.; Yoranova, G.; Yuan, D.; Stroe, O.; Wood, G.; Laydon, A.; et al. AlphaFold Protein Structure Database: Massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 2022, 50, D439–D444. [Google Scholar] [CrossRef]
- The PyMOL Molecular Graphics System, Version 2.5 Schrödinger, LLC. Available online: https://pymol.org/2/ (accessed on 4 April 2023).
- Ittisoponpisan, S.; Islam, S.A.; Khanna, T.; Alhuzimi, E.; David, A.; Sternberg, M.J.E. Can predicted protein 3D structures provide reliable insights into whether missense variants are disease associated? J. Mol. Biol. 2019, 431, 2197–2212. [Google Scholar] [CrossRef]
- Inagaki, N.; Tsuura, Y.; Namba, N.; Masuda, K.; Gonoi, T.; Horie, M.; Seino, Y.; Mizuta, M.; Seino, S. Cloning and functional characterization of a novel ATP-sensitive potassium channel ubiquitously expressed in rat tissues, including pancreatic islets, pituitary, skeletal muscle and heart. J. Biol. Chem. 1995, 270, 5691–5694. [Google Scholar] [CrossRef]
- Inagaki, N.; Gonoi, T.; Clement, J.P.; Wang, C.Z.; Aguilar-Bryan, L.; Bryan, J.; Seinoet, S. A family of sulfonylurea receptors determines the pharmacological properties of ATP-sensitive K+ channels. Neuron 1996, 16, 1011–1017. [Google Scholar] [CrossRef]
- Bienengraeber, M.; Olson, T.M.; Selivanov, V.A.; Kathmann, E.C.; O’Cochlain, F.; Gao, F.; Karger, A.B.; Ballew, J.D.; Hodgson, D.M.; Zingman, L.V.; et al. ABCC9 mutations identified in human dilated cardiomyopathy disrupt catalytic KATP channel gating. Nat. Genet. 2004, 36, 382–387. [Google Scholar] [CrossRef]
- Olson, T.M.; Alekseev, A.E.; Moreau, C.; Liu, X.K.; Zingman, L.V.; Miki, T.; Seino, S.; Asirvatham, S.J.; Jahangir, A.; Terzicet, A. KATP channel mutation confers risk for vein of Marshall adrenergic atrial fibrillation. Nat. Clin. Pract. Cardiovasc. Med. 2007, 4, 110–116. [Google Scholar] [CrossRef]
- Mederios-Domingo, A.; Tan, B.H.; Crotti, L.; Tester, D.J.; Eckhardt, L.; Cuoretti, A.; Kroboth, S.L.; Song, C.; Zhou, Q.; Kopp, D.; et al. Gain-of-function mutation S422L in the KCNJ8-encoded cardiac K(ATP) channel Kir6.1 as a pathogenic substrate for J-wave syndromes. Heart Rhythm. 2010, 7, 1466–1471. [Google Scholar] [CrossRef]
- Tester, D.J.; Tan, B.H.; Medeiros-Domingo, A.; Song, C.; Makielski, J.C.; Ackerman, M.J. Loss-of-function mutations in the KCNJ8-encoded Kir6.1 K(ATP) channel and sudden infant death syndrome. Circ. Cardiovasc. Genet. 2011, 4, 510–515. [Google Scholar] [CrossRef]
- Harakalova, M.; van Harssel, J.J.T.; Terhal, P.A.; van Lieshout, S.; Duran, K.; Renkens, I.; Amor, D.J.; Wilson, L.C.; Kirk, E.P.; Turner, C.L.S.; et al. Dominant missense mutations in ABCC9 cause Cantú syndrome. Nat. Genet. 2012, 44, 793–796. [Google Scholar] [CrossRef] [PubMed]
- van Bon, B.W.M.; Gilissen, C.; Grange, D.K.; Hennekam, R.C.M.; Kayserili, H.; Engels, H.; Reutter, H.; Ostergaard, J.R.; Morava, E.; Tsiakas, K.; et al. Cantú syndrome is caused by mutations in ABCC9. Am. J. Hum. Genet. 2012, 90, 1094–1101. [Google Scholar] [CrossRef] [PubMed]
- Delaney, J.T.; Muhammad, R.; Blair, M.A.; Kor, K.; Fish, F.A.; Roden, D.M.; Darbar, D. A KCNJ8 mutation associated with early repolarization and atrial fibrillation. Europace 2012, 14, 1428–1432. [Google Scholar] [CrossRef]
- Hu, D.; Barajas-Martínez, H.; Terzic, A.; Park, S.; Pfeiffer, R.; Burashnikov, E.; Wu, Y.; Borggrefe, M.; Veltmann, C.; Schimpf, R.; et al. ABCC9 is a novel Brugada and early repolarization syndrome susceptibility gene. Int. J. Cardiol. 2014, 171, 431–442. [Google Scholar] [CrossRef] [PubMed]
- Carnevale, A.; Rosas-Madrigal, S.; Rosendo-Gutiérrez, R.; López-Mora, E.; Romero-Hidalgo, S.; Avila-Vazzini, N.; Jacobo-Albavera, L.; Domínguez-Pérez, M.; Vargas-Alarcón, G.; Pérez-Villatoro, F.; et al. Genomic study of dilated cardiomyopathy in a group of Mexican patients using site-directed next generation sequencing. Mol. Genet. Genomic Med. 2020, 8, e1504. [Google Scholar] [CrossRef]
- Shen, C.; Xu, L.; Sun, X.; Sun, A.; Ge, J. Genetic variants in Chinese patients with sporadic dilated cardiomyopathy: A cross-sectional study. Ann. Transl. Med. 2022, 10, 129. [Google Scholar] [CrossRef]
- Neagoe, O.; Ciobanu, A.; Diaconu, R.; Mirea, O.; Donoiu, I.; Militaru, C. A rare case of familial restrictive cardiomyopathy, with mutations in MYH7 and ABCC9 genes. Discoveries 2019, 30, e99. [Google Scholar] [CrossRef]
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.R.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Jang, W.; et al. ClinVar: Improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018, 46, D1062–D1067. [Google Scholar] [CrossRef]
- Filiano, J.J.; Kinney, H.C. A perspective on neuropathologic findings in victims of the sudden infant death syndrome: The triple-risk model. Biol. Neonate 1994, 65, 194–197. [Google Scholar] [CrossRef]
- Fahrenbach, J.P.; Stoller, D.; Kim, G.; Aggarwal, N.; Yerokun, B.; Earley, J.U.; Hadhazy, M.; Shi, N.Q.; Makielski, J.C.; McNally, E.M. Abcc9 is required for the transition to oxidative metabolism in the newborn heart. FASEB J. 2014, 28, 2804–2815. [Google Scholar] [CrossRef]
- Zingman, L.V.; Hodgson, D.M.; Bast, P.H.; Kane, G.C.; Perez-Terzic, C.; Gumina, R.J.; Pucar, D.; Bienengraeber, M.; Dzeja, P.P.; Miki, T.; et al. Kir6.2 is required for adaptation to stress. Proc. Natl. Acad. Sci. USA 2022, 99, 13278–13283. [Google Scholar] [CrossRef]
- Orphanou, N.; Papatheodorou, E.; Anastasakis, A. Dilated cardiomyopathy in the era of precision medicine: Latest concepts and developments. Heart Fail. Rev. 2022, 27, 1173–1191. [Google Scholar] [CrossRef]
- Nichols, C.G.; Singh, G.K.; Grange, D.K. KATP channels and cardiovascular disease: Suddenly a syndrome. Circ. Res. 2013, 112, 1059–1072. [Google Scholar] [CrossRef]
- Brodehl, A.; Gerull, B. Genetic insights into primary restrictive cardiomyopathy. J. Clin. Med. 2022, 11, 2094. [Google Scholar] [CrossRef]
- Waldmüller, S.; Schroeder, C.; Sturm, M.; Scheffold, T.; Imbrich, K.; Junker, S.; Frische, C.; Hofbeck, M.; Bauer, P.; Bonin, M.; et al. Targeted 46-gene and clinical exome sequencing for mutations causing cardiomyopathies. Mol. Cell. Probes 2015, 29, 308–314. [Google Scholar] [CrossRef]
- Digilio, M.C.; Lepri, F.; Baban, A.; Dentici, M.L.; Versacci, P.; Capolino, R.; Ferese, R.; De Luca, A.; Tartaglia, M.; Marino, B.; et al. RASopathies: Clinical diagnosis in the first year of life. Mol. Syndromol. 2011, 1, 282–289. [Google Scholar] [CrossRef]
- Cannistraci, C.V.; Ogorevc, J.; Zorc, M.; Ravasi, T.; Dovc, P.; Kunej, T. Pivotal role of the muscle-contraction pathway in cryptorchidism and evidence for genomic connections with cardiomyopathy pathways in RASopathies. BMC Med. Genom. 2013, 6, 5. [Google Scholar] [CrossRef]
- Gaar-Humphreys, K.R.; Spanjersberg, T.C.F.; Santarelli, G.; Grinwis, G.C.M.; Szatmári, V.; Roelen, B.A.; Vink, A.; van Tinelen, J.P.; Asselbergs, F.W.; Fieten, H.; et al. Genetic basis of dilated cardiomyopathy in dogs and its potential as a bidirectional model. Animals 2022, 12, 1679. [Google Scholar] [CrossRef]
- Wallace, E.; Howard, L.; Liu, M.; O’Brien, T.; Ward, D.; Shen, S.; Prendiville, T. Long QT syndrome: Genetics and future perspective. Pediatr. Cardiol. 2019, 40, 1419–1430. [Google Scholar] [CrossRef]
- Haghighi, K.; Kolokathis, F.; Gramolini, A.O.; Waggoner, J.R.; Pater, L.; Lynch, R.A.; Fan, G.C.; Tsiapras, D.; Parekh, R.R.; Dorn, G.W., 2nd; et al. A mutation in the human phospholamban gene, deleting arginine 14, results in lethal, hereditary cardiomyopathy. Proc. Natl. Acad. Sci. USA 2006, 103, 1388–1393. [Google Scholar] [CrossRef]
- Schatzberg, S.J.; Olby, N.J.; Breen, M.; Anderson, L.V.B.; Langford, C.F.; Dickens, H.F.; Wilton, S.D.; Zeiss, C.J.; Binns, M.M.; Kornegay, J.N.; et al. Molecular analysis of a spontaneous dystrophin ‘knockout’ dog. Neuromuscul. Disord. 1999, 9, 289–295. [Google Scholar] [CrossRef] [PubMed]
- Nigro, G.; Comi, L.I.; Politano, L.; Bain, R.J. The incidence and evolution of cardiomyopathy in Duchenne muscular dystrophy. Int. J. Cardiol. 1990, 26, 271–277. [Google Scholar] [CrossRef] [PubMed]
- Mazzaccara, C.; Lombardi, R.; Mirra, B.; Barretta, F.; Esposito, M.V.; Uomo, F.; Caiazza, M.; Monda, E.; Losi, M.A.; Limongelli, G.; et al. Next-generation sequencing gene panels in inheritable cardiomyopathies and channelopathies: Prevalence of pathogenic variants and variants of unknown significance in uncommon genes. Biomolecules 2022, 12, 1417. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genomic Alteration * | Protein Alteration † | PhyloP | PolyPhen-2 HumVar | Mut-Pred2 | Allele Frequency | |
|---|---|---|---|---|---|---|
| dbSNP ‡ | DBVDC | |||||
| CFA27: g.21042635C > T | p.R1186Q | 7.8 | 0.998 | 0.722 § | 0 | 0 |
| CFA27: g.21025527G > A | p.D1316= | 1.7 | NA | NA | 0.34 | 0.30 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Furrow, E.; Tate, N.; Minor, K.; Martinson, S.; Larrabee, S.; Anttila, M.; Sleeper, M.; Henthorn, P. An ABCC9 Missense Variant Is Associated with Sudden Cardiac Death and Dilated Cardiomyopathy in Juvenile Dogs. Genes 2023, 14, 988. https://doi.org/10.3390/genes14050988
Furrow E, Tate N, Minor K, Martinson S, Larrabee S, Anttila M, Sleeper M, Henthorn P. An ABCC9 Missense Variant Is Associated with Sudden Cardiac Death and Dilated Cardiomyopathy in Juvenile Dogs. Genes. 2023; 14(5):988. https://doi.org/10.3390/genes14050988
Chicago/Turabian StyleFurrow, Eva, Nicole Tate, Katie Minor, Shannon Martinson, Shannon Larrabee, Marjukka Anttila, Meg Sleeper, and Paula Henthorn. 2023. "An ABCC9 Missense Variant Is Associated with Sudden Cardiac Death and Dilated Cardiomyopathy in Juvenile Dogs" Genes 14, no. 5: 988. https://doi.org/10.3390/genes14050988
APA StyleFurrow, E., Tate, N., Minor, K., Martinson, S., Larrabee, S., Anttila, M., Sleeper, M., & Henthorn, P. (2023). An ABCC9 Missense Variant Is Associated with Sudden Cardiac Death and Dilated Cardiomyopathy in Juvenile Dogs. Genes, 14(5), 988. https://doi.org/10.3390/genes14050988