

The First Complete Mitochondrial Genome of Genus Isocapnia (Plecoptera: Capniidae) and Phylogenetic Assignment of Superfamily Nemouroidea

Abstract
1. Introduction
2. Materials and Methods
2.1. Sampling Collection and DNA Extraction
2.2. PCR Amplification and Sequencing
2.3. Mitogenome Assembly and Annotation
2.4. Phylogenetic Analysis
3. Results and Discussion
3.1. Characteristics of the Mitogenome
3.2. Base Composition
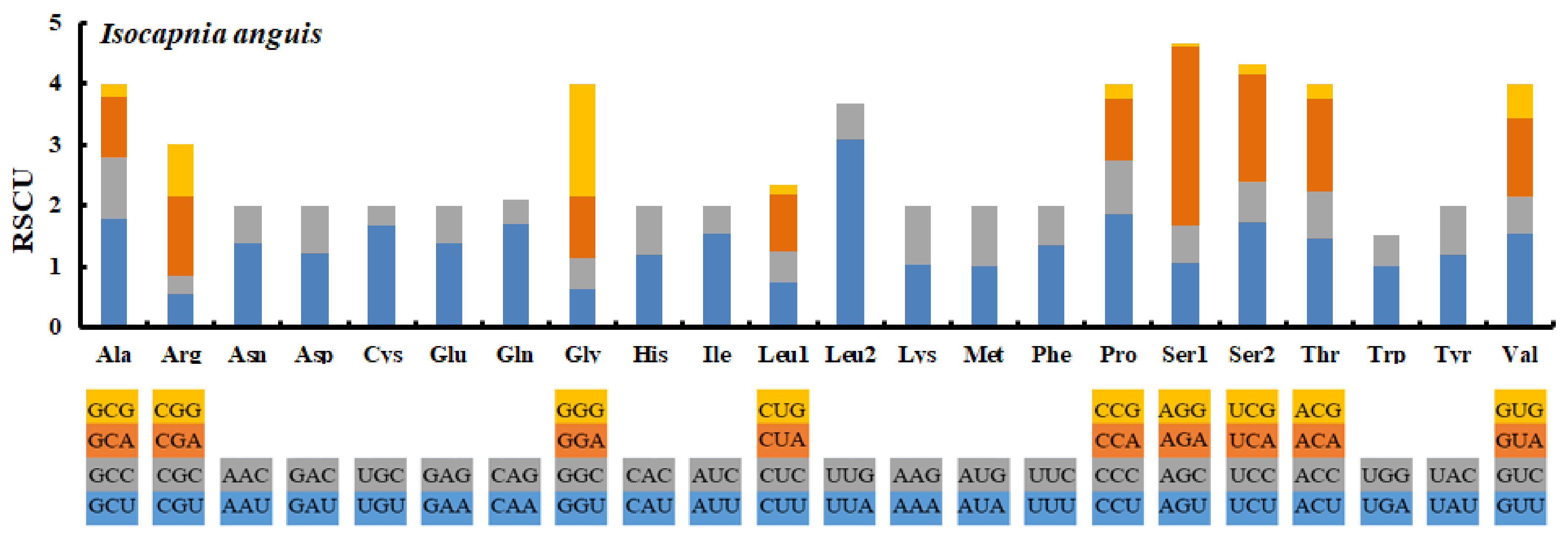
3.3. Protein Coding Genes
3.4. Transfer RNA Genes
3.5. The Control Region
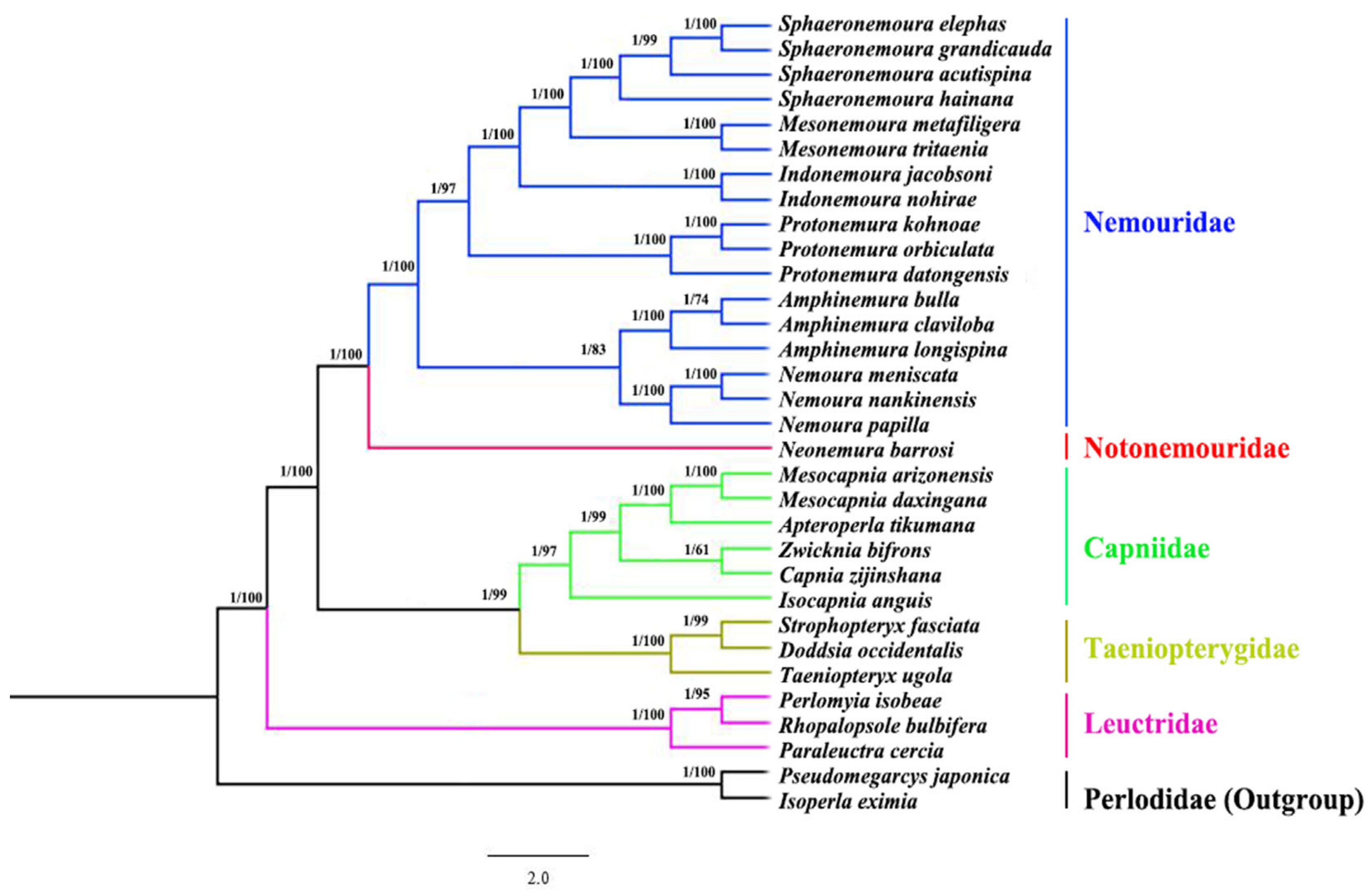
3.6. Phylogenetic Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Simon, C.; Buckley, T.R.; Frati, F.; Stewart, J.B.; Beckenbach, A.T. Incorporating molecular evolution into phylogenetic analysis, and a new compilation of conserved polymerase chain reaction primers for animal mitochondrial DNA. Annu. Rev. Ecol. Evol. Syst. 2006, 37, 545–579. [Google Scholar] [CrossRef]
- Cameron, S.L. Insect mitochondrial genomics: Implications for evolution and phylogeny. Annu. Rev. Entomol. 2014, 59, 95–117. [Google Scholar] [CrossRef]
- Yuan, M.L.; Zhang, Q.L.; Zhang, L.; Guo, Z.L.; Liu, Y.J.; Shen, Y.Y.; Shao, R. High-level phylogeny of the Coleoptera inferred with mitochondrial genome sequences. Mol. Phylogenet. Evol. 2016, 104, 99–111. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Leavengood, J.M.; Chapman, E.G.; Burkhardt, D.; Song, F.; Jiang, P.; Liu, J.; Zhou, X.; Cai, W. Mitochondrial phylogenomics of Hemiptera reveals adaptive innovations driving the diversification of true bugs. Proc. R. Soc. B 2017, 284, 20171223. [Google Scholar] [CrossRef]
- Wolstenholme, D.R. Animal mitochondrial DNA: Structure and evolution. Int. Rev. Cytol. 1992, 141, 173–216. [Google Scholar] [PubMed]
- Simon, C.; Frati, F.; Beckenbach, A.; Crespi, B.; Liu, H.; Flook, P. Evolution, weighting, and phylogenetic utility of mitochondrial gene sequences and a compilation of conserved polymerase chain reaction primers. Ann. Entomol. Soc. Am. 1994, 87, 651–701. [Google Scholar] [CrossRef]
- Gissi, C.; Iannelli, F.; Pesole, G. Evolution of the mitochondrial genome of metazoa as exemplified by comparison of congeneric species. Heredity 2008, 101, 301–320. [Google Scholar] [CrossRef]
- DeWalt, R.E.; Maehr, M.D.; Hopkins, H.; Neu-Becker, U.; Stueber, G. Plecoptera Species File Online. Version 5.0/5.0. Available online: http://Plecoptera.SpeciesFile.org (accessed on 13 February 2023).
- Stewart, K.W.; Stark, B.P. Nymphs of North American Stonefly Genera (Plecoptera), 2nd ed.; The Caddis Press: Columbus, OH, USA, 2002. [Google Scholar]
- Stewart, K.W.; Stark, B.P. Plecoptera. In An Introduction to the Aquatic Insects of North America, 4th ed.; Merritt, R.W., Cummins, K.W., Berg, M.B., Eds.; Kendall/Hunt Publishing Company: Dubuque, IA, USA, 2008; Chapter 14; pp. 311–384. [Google Scholar]
- Fochetti, R.; Figueroa, J.M.T. Global diversity of stoneflies (Plecoptera, Insecta) in freshwater. Hydrobiologia 2008, 595, 365–377. [Google Scholar] [CrossRef]
- Banks, N. New native neuropteroid insects. Psyche J. Entomol. 1938, 45, 72–79. [Google Scholar] [CrossRef]
- Zhiltzova, L.A. Plecoptera, Gruppe Euholognatha. Fauna Russ. Neighboring Ctries New Ser. 2003, 145, 1–538. [Google Scholar]
- Chen, Z.T.; Du, Y.Z. First record of Isocapnia (Plecoptera: Capniidae) from China with description of a new species. Ann. Soc. Entomol. Fr. 2017, 53, 42–46. [Google Scholar] [CrossRef]
- Judson, S.W.; Nelson, C.R. A guide to Mongolian stoneflies (Insecta: Plecoptera). Zootaxa 2012, 3541, 1–118. [Google Scholar] [CrossRef]
- Zwick, P. Phylogenetic System and Zoogeography of the Plecoptera. Annu. Rev. Entomol. 2000, 45, 709–746. [Google Scholar] [CrossRef] [PubMed]
- Ding, S.M.; Li, W.H.; Wang, Y.; Cameron, S.L.; Murányi, D.; Yang, D. The phylogeny and evolutionary timescale of stoneflies (Insecta: Plecoptera) inferred from mitochondrial genomes. Mol. Phylogenet. Evol. 2019, 135, 123–135. [Google Scholar] [CrossRef] [PubMed]
- Davis, N.G. Application of Next-Generation Transcriptomic Tools for Non-Model Organisms: Gene Discovery and Marker Development within Plecoptera (Insecta). Master’s Thesis, Brigham Young University, Provo, UT, USA, 2013; pp. 1–39. [Google Scholar]
- Chen, Z.T.; Du, Y.Z. The first two mitochondrial genomes from Taeniopterygidae (Insecta: Plecoptera): Structural features and phylogenetic implications. Int. J. Biol. Macromol. 2018, 111, 70–76. [Google Scholar] [CrossRef]
- Cao, J.J.; Wang, Y.; Li, W.H. Comparative mitogenomic analysis of species in the subfamily Amphinemurinae (Plecoptera: Nemouridae) reveal conserved mitochondrial genome organization. Int. J. Biol. Macromol. 2019, 138, 292–301. [Google Scholar] [CrossRef]
- Cao, J.J.; Wang, Y.; Guo, X.; Wang, G.Q.; Li, W.H.; Murányi, D. Two complete mitochondrial genomes from Leuctridae (Plecoptera: Nemouroidea): Implications for the phylogenetic relationships among stoneflies. J. Insect Sci. 2021, 21, 16. [Google Scholar] [CrossRef]
- Shen, Y.; Du, Y.Z. The mitochondrial genome of Leuctra sp. (Plecoptera: Leuctridae) and its performance in phylogenetic analyses. Zootaxa 2019, 4671, 571–580. [Google Scholar] [CrossRef]
- Zhao, M.Y.; Huo, Q.B.; Du, Y.Z. Molecular phylogeny inferred from the mitochondrial genomes of Plecoptera with Oyamia nigribasis (Plecoptera: Perlidae). Sci. Rep. 2020, 10, 20955. [Google Scholar] [CrossRef]
- Guo, X.; Guo, C.; Dong, X.; Zhang, H.; Murányi, D.; Li, W.; Wang, Y. Mitochondrial Genome of Strophopteryx fasciata (Plecoptera: Taeniopterygidae), with a Phylogenetic Analysis of Nemouroidea. Genes 2022, 13, 1116. [Google Scholar] [CrossRef]
- Wu, H.Y.; Ji, X.Y.; Yu, W.W.; Du, Y.Z. Complete mitochondrial genome of the stonefly Cryptoperla stilifera Sivec (Plecoptera: Peltoperlidae) and the phylogeny of Polyneopteran insects. Gene 2014, 537, 177–183. [Google Scholar] [CrossRef] [PubMed]
- Mo, R.R.; Wang, Y.; Cao, J.J.; Wang, G.Q.; Li, W.H.; Murányi, D. Two complete mitochondrial genomes of the subfamily Chloroperlinae (Plecoptera: Chloroperlidae) and their phylogenetic implications. Arthropod Syst. Phylogeny 2022, 80, 155–168. [Google Scholar] [CrossRef]
- Wang, Y.; Cao, J.; Li, W.; Chen, X. The mitochondrial genome of Mesocapnia daxingana (Plecoptera: Capniidae). Conserv. Genet. Resour. 2017, 9, 639–642. [Google Scholar] [CrossRef]
- Chen, Z.T.; Du, Y.Z. Complete mitochondrial genome of Capnia zijinshana (Plecoptera: Capniidae) and phylogenetic analysis among stoneflies. J. Asia-Pac. Entomol. 2017, 20, 305–312. [Google Scholar] [CrossRef]
- Grant, J.R.; Stothard, P. The CGView server: A comparative genomics tool for circular genomes. Nucl. Acids Res. 2008, 36, 181–184. [Google Scholar] [CrossRef]
- Bernt, M.; Donath, A.; Jühling, F.; Externbrinka, F.; Florentz, C.; Fritzsch, G.; Pützh, J.; Middendorf, M.; Stadler, P.F. MITOS: Improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenet. Evol. 2013, 69, 313–319. [Google Scholar] [CrossRef]
- Laslett, D.; Canbäck, B. ARWEN, a program to detect tRNA genes in metazoan mitochondrial nucleotide sequences. Bioinformatics 2008, 24, 172–175. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA 7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef]
- Perna, N.T.; Kocher, T.D. Patterns of nucleotide composition at fourfold degenerate sites of animal mitogenome. J. Mol. Evol. 1995, 41, 353–358. [Google Scholar] [CrossRef]
- Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef]
- National Center for Biotechnology Information (NCBI). Available online: https://www.ncbi.nlm.nih.gov (accessed on 5 February 2023).
- Wei, X.; Cao, J.; Kong, F.; Wang, Y. The mitochondrial genome analysis of Sphaeronemoura elephas (Plecoptera: Nemouridae) from Jiangxi Province of southeastern China. Mitochondrial DNA Part B Resour. 2020, 5, 1107–1108. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.J.; Wang, Y.; Huang, Y.R.; Li, W.H. Mitochondrial genomes of the stoneflies Mesonemoura metafiligera and Mesonemoura tritaenia (Plecoptera, Nemouridae), with a phylogenetic analysis of Nemouroidea. ZooKeys 2019, 835, 43–63. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Tian, T.; Zhou, M.; Li, W.; Cao, J. The complete mitochondrial genome of a stonefly species, Protonemura datongensis (Plecoptera: Nemouridae). Mitochondrial DNA Part B 2020, 5, 2006–2007. [Google Scholar] [CrossRef]
- Chen, J.; Cao, J.; Chen, M.; Chen, S.; Li, W.; Wang, Y. The complete mitochondrial genome of Amphinemura claviloba (Wu, 1973)(Plecoptera: Nemouridae). Zootaxa 2020, 4732, 4732. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Cao, J.; Murányi, D.; Chen, X.; Yan, F. The complete mitochondrial genome of Amphinemura bulla Shimizu, 1997 (Plecoptera: Nemouridae) from Japan. Mitochondrial DNA Part B 2021, 6, 846–847. [Google Scholar] [CrossRef]
- Cao, J.; Wang, Y.; Ma, G.; Li, W. Complete mitochondrial genome of a stonefly, Nemoura papilla (Plecoptera: Nemouridae). Mitochondrial DNA Part B 2019, 4, 806–807. [Google Scholar] [CrossRef]
- Chen, Z.T.; Du, Y.Z. First Mitochondrial Genome from Nemouridae (Plecoptera) Reveals novel features of the elongated control region and phylogenetic implications. Int. J. Mol. Sci. 2017, 18, 996. [Google Scholar] [CrossRef]
- Chen, S.; Cao, J.; Li, W.; Wang, Y. Characterization of the complete mitochondrial genome of Nemoura meniscata Li & yang (Plecoptera: Nemouridae) from China. Mitochondrial DNA Part B Resour. 2020, 5, 1052–1053. [Google Scholar]
- Elbrecht, V.; Leese, F. The mitochondrial genome of the Arizona Snowfly Mesocapnia arizonensis (Plecoptera, Capniidae). Mitochondrial DNA Part A 2016, 27, 3365–3366. [Google Scholar] [CrossRef]
- Wang, Y.; Cao, J.; Murányi, D.; Li, W. Comparison of two complete mitochondrial genomes from Perlodidae (Plecoptera: Perloidea) and the family-level phylogenetic implications of Perloidea. Gene 2018, 675, 254–264. [Google Scholar] [CrossRef]
- Vaidya, G.; Lohman, D.J.; Meier, R. SequenceMatrix: Concatenation software for the fast assembly of multi-gene datasets with character set and codon information. Cladistics 2010, 27, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Ronquist, F.; Huelsenbeck, J.P. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 2003, 19, 1572–1574. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Y.; Zhao, M.; Huo, Q.; Du, Y. Mitochondrial Genomes of the Genus Claassenia (Plecoptera: Perlidae) and Phylogenetic Assignment to Subfamily Perlinae. Genes 2021, 12, 1986. [Google Scholar] [CrossRef]
- Nguyen, L.T.; Schmidt, H.A.; Haeseler, A.V.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Stewart, J.B.; Beckenbach, A.T. Insect mitochondrial genomics: The complete mitogenome sequence of the meadow spittlebug Philaenus spumarius (Hemiptera: Auchenorrhyncha: Cercopoidae). Genome 2005, 48, 46–54. [Google Scholar] [CrossRef]
- Clary, D.O.; Wolstenholme, D.R. The ribosomal RNA genes of Drosophila mitochondrial DNA. Nucleic Acids Res. 1985, 13, 4029–4045. [Google Scholar] [CrossRef]
- Hassanin, A.; Léger, N.; Deutsch, J. Evidence for multiple reversals of asymmetric mutational constraints during the evolution of the mitochondrial genome of Metazoa, and consequences for phylogenetic inferences. Syst. Biol. 2005, 54, 277–298. [Google Scholar] [CrossRef]
- Hassanin, A. Phylogeny of Arthropoda inferred from mitochondrial sequences: Strategies for limiting the misleading effects of multiple changes in pattern and rates of substitution. Mol. Phylogenet. Evol. 2006, 38, 100–116. [Google Scholar] [CrossRef]
- Wei, S.J.; Shi, M.; Chen, X.X.; Sharkey, M.J.; van Achterberg, C.; Ye, G.Y.; He, J.H. New views on strand asymmetry in insect mitochondrial genomes. PLoS ONE 2010, 5, e12708. [Google Scholar] [CrossRef]
- Lindahl, T. Instability and decay of the primary structure of DNA. Nature 1993, 362, 709–715. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Ding, S.; Yang, D. The complete mitochondrial genome of a stonefly species, Kamimuria chungnanshana Wu, 1948 (Plecoptera: Perlidae). Mitochondrial DNA Part A 2016, 27, 3810–3811. [Google Scholar] [CrossRef] [PubMed]
- Boore, J.L. Complete mitochondrial genome sequence of Urechis caupo, a representative of the phylum Echiura. BMC Genome 2004, 5, e67. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Guo, X.; Guo, C.; Wang, X.; Wang, Y.; Yan, F. Complete Mitochondrial Genome of Malenka flexura (Plecoptera: Nemouridae) and Phylogenetic Analysis. Genes 2022, 13, 911. [Google Scholar] [CrossRef]
- Garey, J.R.; Wolstenholme, D.R. Platyhelminth mitochondrial DNA: Evidence for early evolutionary origin of a tRNAserAGN that contains a dihydrouridine arm replacement loop, and of serine-specifying AGA and AGG codons. J. Mol. Evol. 1989, 28, 374–387. [Google Scholar] [CrossRef]
- Gutell, R.R.; Lee, J.C.; Cannone, J.J. The accuracy of ribosomal RNA comparative structure models. Curr. Opin. Struct. Biol. 2002, 12, 301–310. [Google Scholar] [CrossRef]
- Zhang, D.X.; Szymura, J.M.; Hewitt, G.M. Evolution and structure conservation of the control region of insect mitochondrial DNA. J. Mol. Evol. 1995, 40, 382–391. [Google Scholar] [CrossRef]
- Taanman, J.W. The mitochondrial genome: Structure, transcription, translation and replication. BBA Bioenerg. 1999, 1410, 103–123. [Google Scholar] [CrossRef]
- Zhang, D.X.; Hewitt, G.M. Insect mitochondrial control region: A review of its structure, evolution and usefulness in evolutionary studies. Biochem. Syst. Ecol. 1997, 25, 99–120. [Google Scholar] [CrossRef]
- Yang, F.; Du, Y.Z.; Wang, L.P.; Cao, J.M.; Yu, W.W. The complete mitochondrial genome of the leafminer Liriomyza sativae (Diptera: Agromyzidae): Great difference in the A + T-rich region compared to Liriomyza trifolii. Gene 2011, 485, 7–15. [Google Scholar] [CrossRef]
- Crozier, R.H.; Crozier, Y.C. The mitochondrial genome of the honeybee Apis mellifera: Complete sequence and genome organization. Genetics 1993, 133, 97–117. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.A.; Walsh, K.A.; Wolf, M.R.; McPheron, B.A.; Marden, J.H. Molecular phylogenetic analysis of evolutionary trends in stonefly wing structure and locomotor behavior. Proc. Natl. Acad. Sci. USA 2000, 97, 13178–13183. [Google Scholar] [CrossRef] [PubMed]
- Terry, M.D. Phylogeny of the Polyneopterous Insects with Emphasis on Plecoptera: Molecular and Morpological Evidence. Ph.D. Thesis, Brigham Young University, Provo, UT, USA, 2003; pp. 1–118. [Google Scholar]
- Muranyi, D.; Gamboa, M.; Orci, K.M. Zwicknia Gen. n., a New Genus for the Capnia Bifrons Species Group, with Descriptions of Three New Species Based on Morphology, Drumming Signals and Molecular Genetics, and a Synopsis of the West Palaearctic and Nearctic Genera of Capniidae (Plecoptera). Zootaxa 2014, 3812, 1–82. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Family | Species | Number (bp) | GenBank Number | References |
|---|---|---|---|---|
| Nemouridae | Sphaeronemoura grandicauda | 15,661 | MH085454 | [20] |
| Sphaeronemoura acutispina | 15,016 | MH085455 * | [35] | |
| Sphaeronemoura hainana | 15,260 | MK111420 * | [17] | |
| Sphaeronemoura elephas | 15,846 | MN944385 | [36] | |
| Mesonemoura tritaenia | 15,778 | MH085451 | [37] | |
| Mesonemoura metafiligera | 15,739 | MH085450 | [37] | |
| Protonemura kohnoae | 15,707 | MH085452 | [20] | |
| Protonemura datongensis | 15,756 | MT276842 | [38] | |
| Protonemura orbiculata | 15,758 | MH085453 | [20] | |
| Amphinemura claviloba | 15,707 | MN720741 | [39] | |
| Amphinemura bulla | 15,827 | MW339348 | [40] | |
| Amphinemura longispina | 15,709 | MH085446 | [20] | |
| Indonemoura nohirae | 15,738 | MH085449 | [20] | |
| Indonemoura jacobsoni | 15,642 | MH085448 | [20] | |
| Nemoura papilla | 15,774 | MK290826 | [41] | |
| Nemoura nankinensis | 16,602 | KY940360 | [42] | |
| Nemoura meniscata | 15,895 | MN944386 | [43] | |
| Notonemouridae | Neonemura barrosi | 14,852 | MK111418 * | [17] |
| Capniidae | Mesocapnia daxingana | 15,524 | KY568983 * | [27] |
| Mesocapnia arizonensis | 14,921 | KP642637 * | [44] | |
| Capnia zijinshana | 16,310 | KX094942 | [28] | |
| Apteroperla tikumana | 15,564 | NC_027698 | [35] | |
| Zwicknia bifrons | 15,380 | MT872688* | [35] | |
| Isocapnia anguis | 16,200 | OQ735414 | This study | |
| Taeniopterygidae | Doddsia occidentalis | 16,020 | MG589787 | [19] |
| Strophopteryx fasciata | 15,527 | ON500674 | [24] | |
| Taeniopteryx ugola | 15,353 | MG589786 | [19] | |
| Leuctridae | Paraleuctra cercia | 15,625 | MK492251 | [21] |
| Rhopalopsole bulbifera | 15,599 | MK111419 * | [17] | |
| Perlomyia isobeae | 15,795 | MK492252 | [21] | |
| Perlodidae(Outgroup) | Isoperla eximia | 16,034 | MG910457 | [45] |
| Pseudomegarcys japonica | 16,067 | MG910458 | [45] |
| Feature | Proportion of Nucleotides | No. of Nucleotides | |||||||
|---|---|---|---|---|---|---|---|---|---|
| T% | C% | A% | G% | AT% | GC% | AT Skew | GC Skew | ||
| Genome | 31.28 | 20.64 | 34.91 | 13.17 | 66.19 | 33.81 | 0.05 | −0.22 | 16,200 |
| Protein coding genes | 37.63 | 18.62 | 27.15 | 16.60 | 64.78 | 35.22 | −0.16 | −0.06 | 11,219 |
| First position | 36.58 | 18.61 | 27.38 | 17.41 | 63.96 | 36.02 | −0.14 | −0.03 | 3739 |
| Second position | 37.94 | 18.00 | 27.14 | 16.90 | 65.08 | 34.90 | −0.17 | 0.03 | 3739 |
| Third position | 38.37 | 19.25 | 26.90 | 15.46 | 65.27 | 34.71 | −0.18 | −0.11 | 3739 |
| PCG−J | 34.34 | 22.55 | 28.61 | 14.50 | 62.95 | 37.05 | −0.09 | −0.22 | 6904 |
| First codon position | 28.67 | 22.42 | 34.01 | 14.90 | 62.68 | 37.32 | 0.09 | −0.20 | 2302 |
| Second codon position | 35.16 | 22.56 | 24.12 | 18.17 | 59.28 | 40.72 | −0.19 | −0.11 | 2301 |
| Third codon position | 39.20 | 22.69 | 27.68 | 10.43 | 66.88 | 33.12 | −0.17 | −0.37 | 2301 |
| PCG−N | 40.88 | 14.82 | 25.64 | 18.67 | 66.52 | 33.48 | −0.23 | 0.12 | 5609 |
| First codon position | 40.91 | 12.62 | 26.68 | 19.79 | 67.59 | 32.41 | −0.21 | 0.22 | 1870 |
| Second codon position | 40.77 | 17.71 | 20.65 | 20.87 | 61.42 | 38.58 | −0.33 | 0.08 | 1869 |
| Third codon position | 40.96 | 14.12 | 29.57 | 15.35 | 70.53 | 29.47 | −0.16 | 0.04 | 1870 |
| tRNA genes | 34.24 | 13.69 | 34.78 | 17.29 | 69.02 | 30.98 | 0.01 | 0.12 | 1475 |
| rRNA genes | 37.51 | 10.45 | 33.69 | 18.35 | 71.20 | 28.80 | −0.05 | 0.27 | 2125 |
| lrRNA | 37.66 | 9.08 | 34.81 | 18.45 | 72.47 | 27.53 | −0.04 | 0.34 | 1333 |
| srRNA | 37.25 | 12.75 | 31.82 | 18.18 | 69.07 | 30.93 | −0.08 | 0.18 | 792 |
| CR | 30.08 | 20.67 | 36.54 | 12.71 | 66.62 | 33.38 | 0.10 | −0.24 | 1393 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rehman, A.; Huo, Q.-B.; Du, Y.-Z. The First Complete Mitochondrial Genome of Genus Isocapnia (Plecoptera: Capniidae) and Phylogenetic Assignment of Superfamily Nemouroidea. Genes 2023, 14, 965. https://doi.org/10.3390/genes14050965
Rehman A, Huo Q-B, Du Y-Z. The First Complete Mitochondrial Genome of Genus Isocapnia (Plecoptera: Capniidae) and Phylogenetic Assignment of Superfamily Nemouroidea. Genes. 2023; 14(5):965. https://doi.org/10.3390/genes14050965
Chicago/Turabian StyleRehman, Abdur, Qing-Bo Huo, and Yu-Zhou Du. 2023. "The First Complete Mitochondrial Genome of Genus Isocapnia (Plecoptera: Capniidae) and Phylogenetic Assignment of Superfamily Nemouroidea" Genes 14, no. 5: 965. https://doi.org/10.3390/genes14050965
APA StyleRehman, A., Huo, Q.-B., & Du, Y.-Z. (2023). The First Complete Mitochondrial Genome of Genus Isocapnia (Plecoptera: Capniidae) and Phylogenetic Assignment of Superfamily Nemouroidea. Genes, 14(5), 965. https://doi.org/10.3390/genes14050965