3.1. Chromosome Genome Assembly of M. chilensis Using Proximity Ligation
With two HiFi single-molecule real-time cells in the PacBio Sequel platform, we generated 53.8 Gb of high-quality DNA genome information. This data comprised 63 million reads with a total length of 882 Gbp (
Table 1). These long reads were assembled with the Hifiasm package using default parameters [
45], yielding a primary assembly of 13,762 contigs equivalent to 2.19 Gb, with an N50 of 206 Mb. The genome size assembly made by Hifiasm was comparable with the previous genome size described for closely related species: 1.28 Gb for
M. galloprovincialis [
32], 1.57 Gb for
M. coruscus [
28], and 1.79 Gb for
Dreissena polymorpha [
27].
In vivo Hi-C is a technique that maps physical DNA–DNA proximity across the entire genome [
68,
69]. The method was introduced as a genome-wide version of its predecessor, 3C (chromosome conformation capture). It has been a powerful tool in chromosome-scale genome assembly of many animals in recent years [
70,
71]. In this study, Hi-C experiments and data analysis of hemocyte cells were used for the chromosome assembly of the blue mussel
M. chilensis. Here, Phase Genomics (Seattle, WA, USA) prepared and sequenced two Hi-C libraries, resulting in ~20× coverage and ~253 million 150 bp paired-end reads (
Table 1). The Hi-C analysis evidenced that 44.68% of high-quality reads showed intercontig signals or Cis-close position (<10 kbp on the same contig), and an additional 4.09% of sequence reads revealed a Cis-far conformation (>10 kbp on the same contig) (
Table 2).
Hi-C reads were aligned using Bowtie version 1.3.1 [
72] to order and orient the 13,762 contigs, and scaffolding was performed using Proximo (Phase Genomics, Seattle, WA, USA). We then applied Juicebox for visual inspection and manual correction [
73]. We also manually removed 1894 scaffolds that were microbe-sized and disconnected from the rest of the assembly. Then, 11,868 contigs were used for the first chromosome-level high-quality
M. chilensis assembly (
Table 3).
The N50 and total genome length were calculated in 134 Mbp and 1938 Gbps, respectively. The
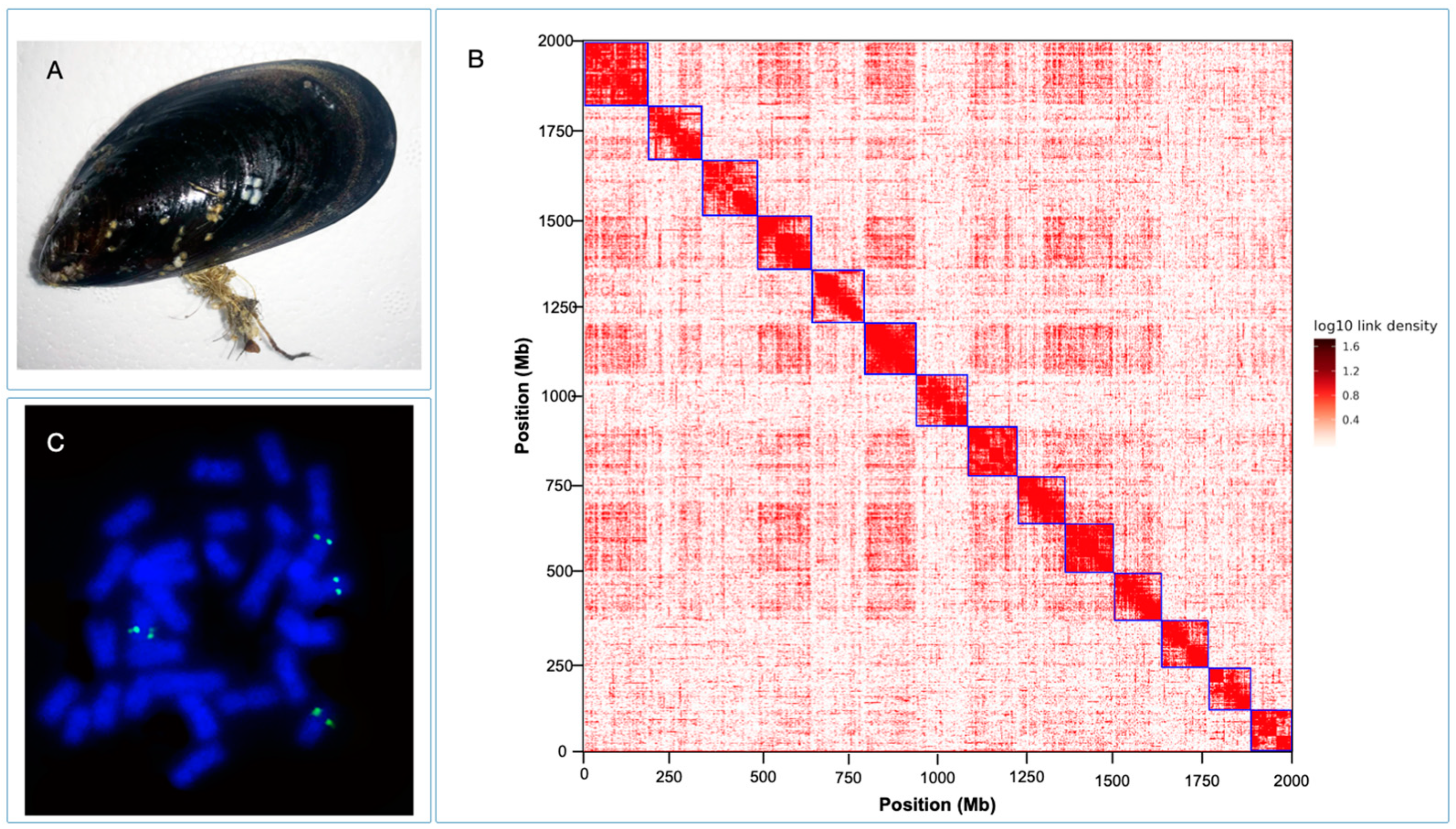
M. chilensis genome provides a valuable genomic resource for research in mussel biology and for developing novel sustainable strategies in mussel aquaculture. The Hi-C data generated 14 chromosomes assembled with HiFi consensus long DNA reads (
Figure 1B). The cytogenetic analysis performed for
M. chilensis revealed a conservative karyotype for the
Mytilus genus composed of 2n = 14 [
21]. Physical localization of 28S-rRNA revealed two loci mapped in different submetacentric/subtelocentric chromosome pairs (
Figure 1C), confirming the presence of major rDNA clusters subterminal to the long arms of two chromosome pairs reported in
M. edulis and
M. galloprovincialis [
74]. Concerning genome assembly, the largest scaffold was assembled from 998 contigs with a total size of 173.3 Mb. Meanwhile, the smallest scaffold was 117.3 Mb, consisting of 744 contigs (
Table 3). Notably, the number of contigs in the scaffolds was 11,868 (100% of all contigs in chromosome clusters, 86.24% of all contigs) and accounted for 1.93 Gbps of genome size (100% of all length in chromosome clusters, 88.43% of all sequence length). The completeness of genome assembly was assessed by the single-copy ortholog set (BUSCO, V5.3.2) [
75]. The results showed the following BUSCO scores: (i) Eukaryota Odb10; C:94.1% (S:72.9%, D:21.2%), F:3.1%, M:2.8%, and n:255. (ii) Metazoa Odb10; C:95.1% (S:75.5%, D:19.6%), F:2.5%, M:2.4%, and n:954. (iii) Mollusca Odb10, C:85% (S:70.1%, D:14.9%), F:3.6%, M:11.4%, and n:5295.
3.3. Protein-Coding Gene Prediction and Functional Annotation in the M. chilensis Genome
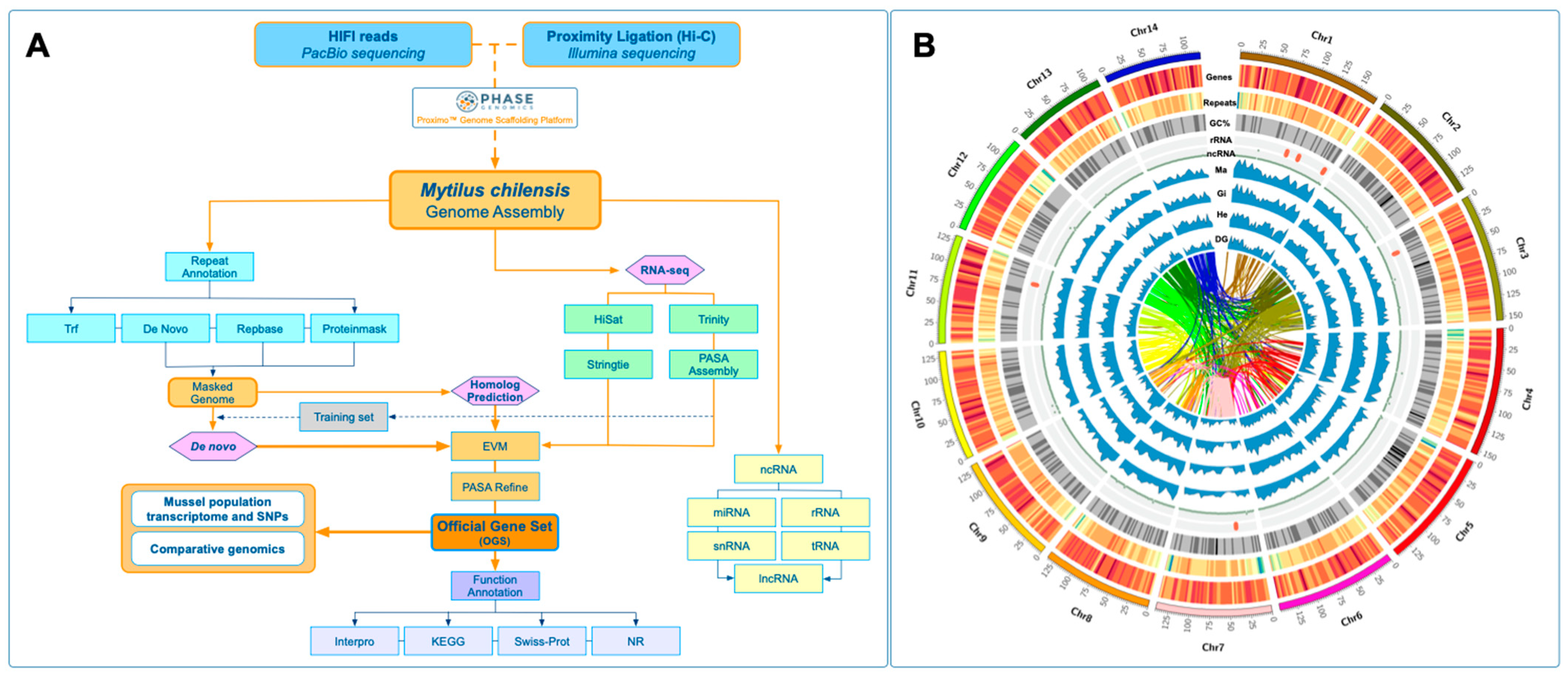
For the identification of protein-coding genes, de novo, homolog prediction, and RNA-seq evidence were used as the training set (
Figure 2A). For homologous predictions, the protein sequences from
Crassostrea gigas,
Mytilus galloprovincialis,
M. coruscus, and
Dreissena polymorpha genomes were extracted using the respectively published references and aligned against the blue mussel genome using TBLASTN (E-value < 1 × 10
−5) (
Table 5). The gene sequence structure of each candidate gene and previously mentioned tools were used to predict protein-coding genes. Finally, a non-redundant reference gene set was generated using the EvidenceModeler (v.2.0) (EVM) and PASA2 tools (v.2.5.2) (
Figure 2A).
Taken together, 34,530 protein-coding genes were identified with a 6531 bp average transcript length, 1377 bp average CDS length, 4.92 average of exons per gene, and 1377 and 1316 average length of exons and introns, respectively (
Table 6). Additionally, 516 tRNAs were predicted using tRNAscan-SE, and 143 rRNA genes were annotated using RNAmmer. For non-coding RNAs with putative regulatory roles, 1365 miRNAs and 43,011 long non-coding RNAs were identified and annotated within the
M. chilensis genome (
Table 7). For functional annotation, the predicted proteins within the blue mussel genome were searched by homology against seven databases: Swiss-Prot, Nr, Nt, KEGG, eggnog, GO, and Pfam (
Figure 2A). Overall, 70.45%, 73.01%, 8.98%, 64.94%, 80.57%, 33.61%, and 96.33% of genes matched entries in these databases, respectively. A total of 34,530 genes (100%) were successfully annotated by gene function and conserved protein motifs (
Table 8). The genomic features annotated for the native blue mussel
M. chilensis were displayed using a Circos plot [
64]. Herein, this graphical representation shows the primary genomic features for the 14 chromosomes. Specifically, gene density, repeat density, GC content, rRNA localization, and ncRNAs were plotted. The transcriptome expression profiles for the mantle, gills, hemocytes, and digestive gland tissues were also displayed in connection with the syntenic blocks (
Figure 2B).
3.4. Comparative Genomics
Smooth-shelled blue mussels of the genus
Mytilus represent a model group because of their cosmopolitan distribution, socioecological importance, and intriguing evolutionary history. This taxon provides new insights into the process of speciation and how hybridization and introgression can be one of the biggest threats to global mussel biodiversity [
82]. A survey of single nucleotide polymorphisms (SNPs) on southern hemisphere blue mussels has provided a new layer for understanding their biology, taxonomy, and phylogeography [
83,
84]. However, SNP markers cannot be applied as a single tool to evidence chromosome rearrangement events during the
Mytilus evolution. Here, whole-genome sequencing in smooth-shelled blue mussels and relative bivalve species is a priority for global mussel aquaculture, biosecurity, and conservation.
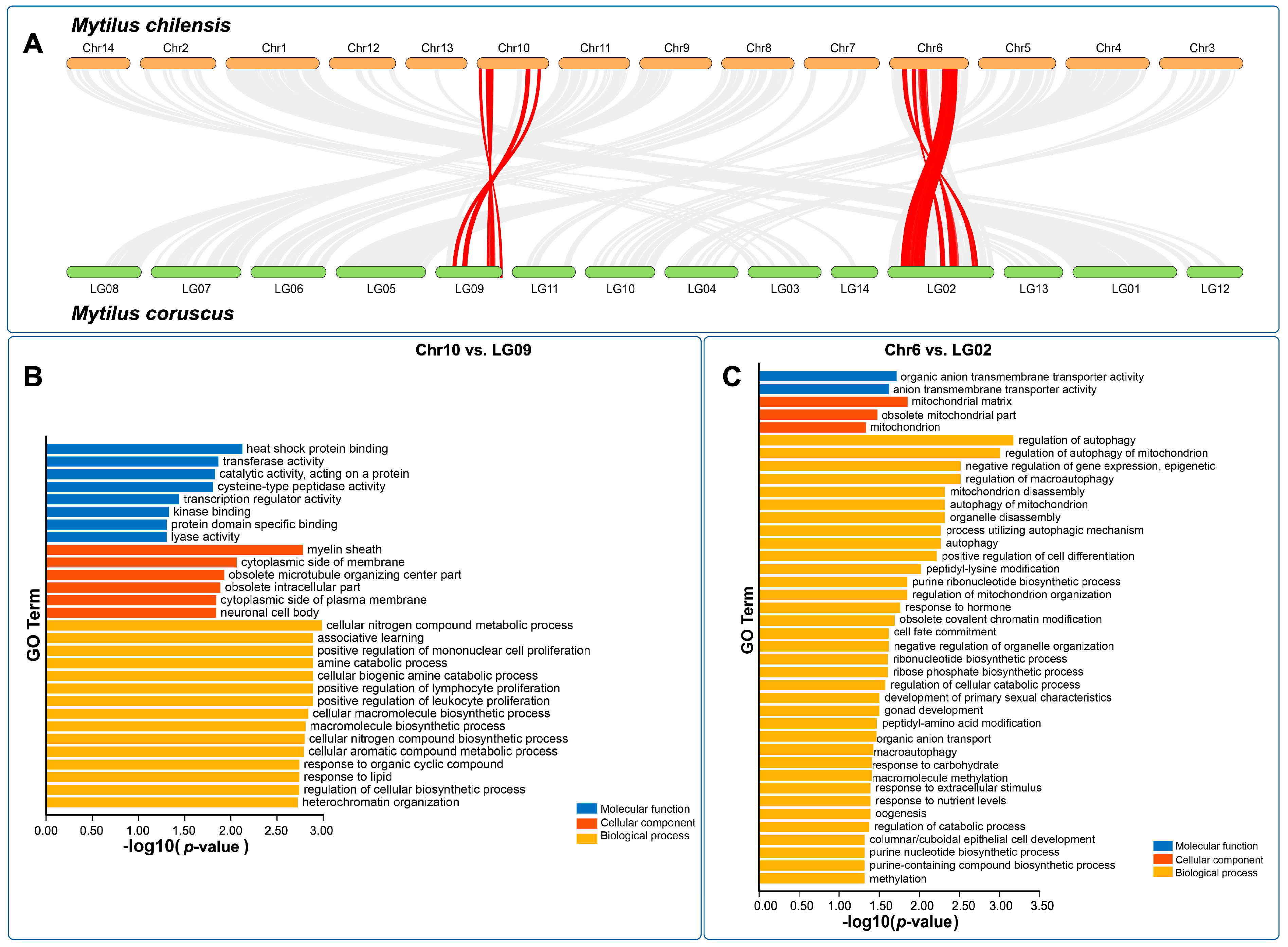
With the aim of exploring genomic rearrangements in
Mytilus, the reported reference genomes for
M. coruscus and
M. chilensis were analyzed. Of the 34,530 predicted genes from the
M. chilensis genome, 18,758 (54.32%) were found in syntenic collinear blocks after being compared with the
M. coruscus genome (
Figure 3A). These syntenic blocks consisted of 671 alignments with a minimum of 5 genes per block. The number of alignments per chromosome ranged from 27 on chromosome 13 to 69 on chromosome 3. Chromosomes with higher genes in collinear blocks were chromosomes 1, 4, and 6, with 1227, 1091, and 1088 genes, respectively. Blocks with less than five genes or E-value < 1 × 10
−5 were discarded from this analysis. Most collinear blocks were located at the same pair of chromosomes between the two genomes. For example,
M. chilensis Chr1 had only syntenic blocks with LG01 from
M. coruscus in the same order. However, chromosomes 6 and 10 from
M. chilensis had collinearity with chromosomes LG09 and LG02 in
M. coruscus but were orientated as two inversed blocks per pair of chromosomes (red lines in
Figure 3A and
Figure S2). The genes in these alignments from inversed blocks were extracted, blasted, and gene ontology terms were identified. Enrichment analyses from GO terms were obtained from Chr10, and LG09 inversed blocks and Chr6 and LG02 pair of chromosomes (
Figure 3B,C). Most molecular function-enriched GO terms in the Chr10 and LG09 pair were associated with heat shock protein (HSP) binding. By contrast, in the Chr6 and LG02 pair, most of the enriched GO terms were associated with the mitochondria and biological processes related to autophagy or regulation of gene expression by epigenetic changes. Notably, chromosome rearrangements have been associated with adaptative genetic traits in marine organisms [
85], where specific architectural proteins such as HSPs may have distinct roles in establishing 3D genome organization [
86].
3.5. Comparative Analysis of Steamer-like Elements in Bivalvia
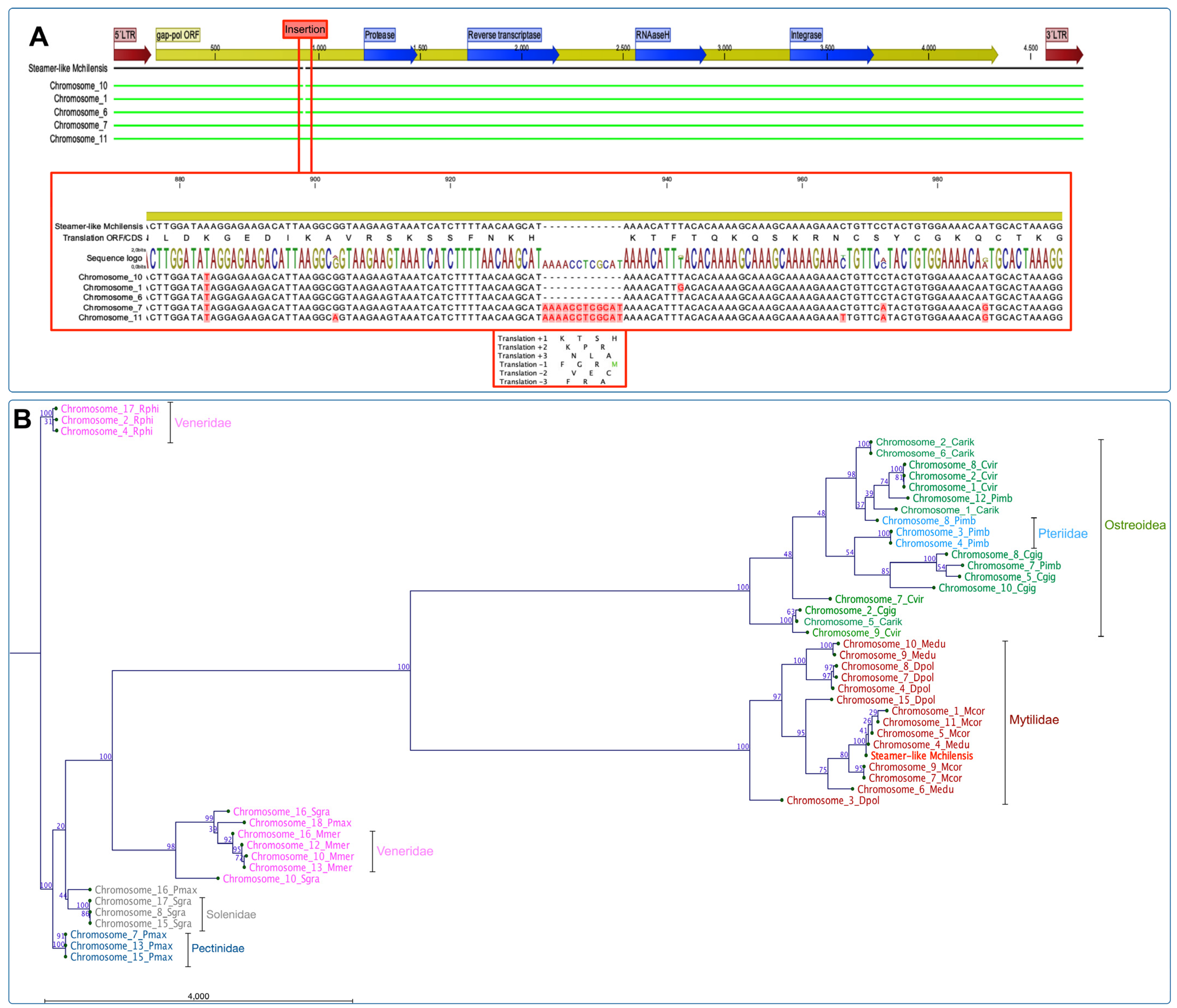
To explore the gene expansion of retrotransposon elements among representative species from Bivalvia, we primarily characterized the Steamer-like elements (SLEs) in
M. chilensis using the approach described by Arriagada et al. [
62]. The analysis evidenced that the genome of
M. chilensis contains five copies of SLEs distributed in chromosomes 1, 6, 7, 10, and 11. The alignment showed that all SLE copies are flanked by two LTRs (5′ and 3′) containing the Gag-Pol ORFs and the domains annotated to protease, reverse transcriptase, RNAaseH, and integrase. Notably, an insertion composed of 12 nucleotides at position 933^934 was exclusively found in chromosomes 7 and 11. The translation for the inserted nucleotides suggests four amino acids, K, T, S, and H, in a positive orientation. However, the translation evaluated in the reading frame (−1) evidenced a methionine localized before the RNAaseH coding gene (
Figure 4A).
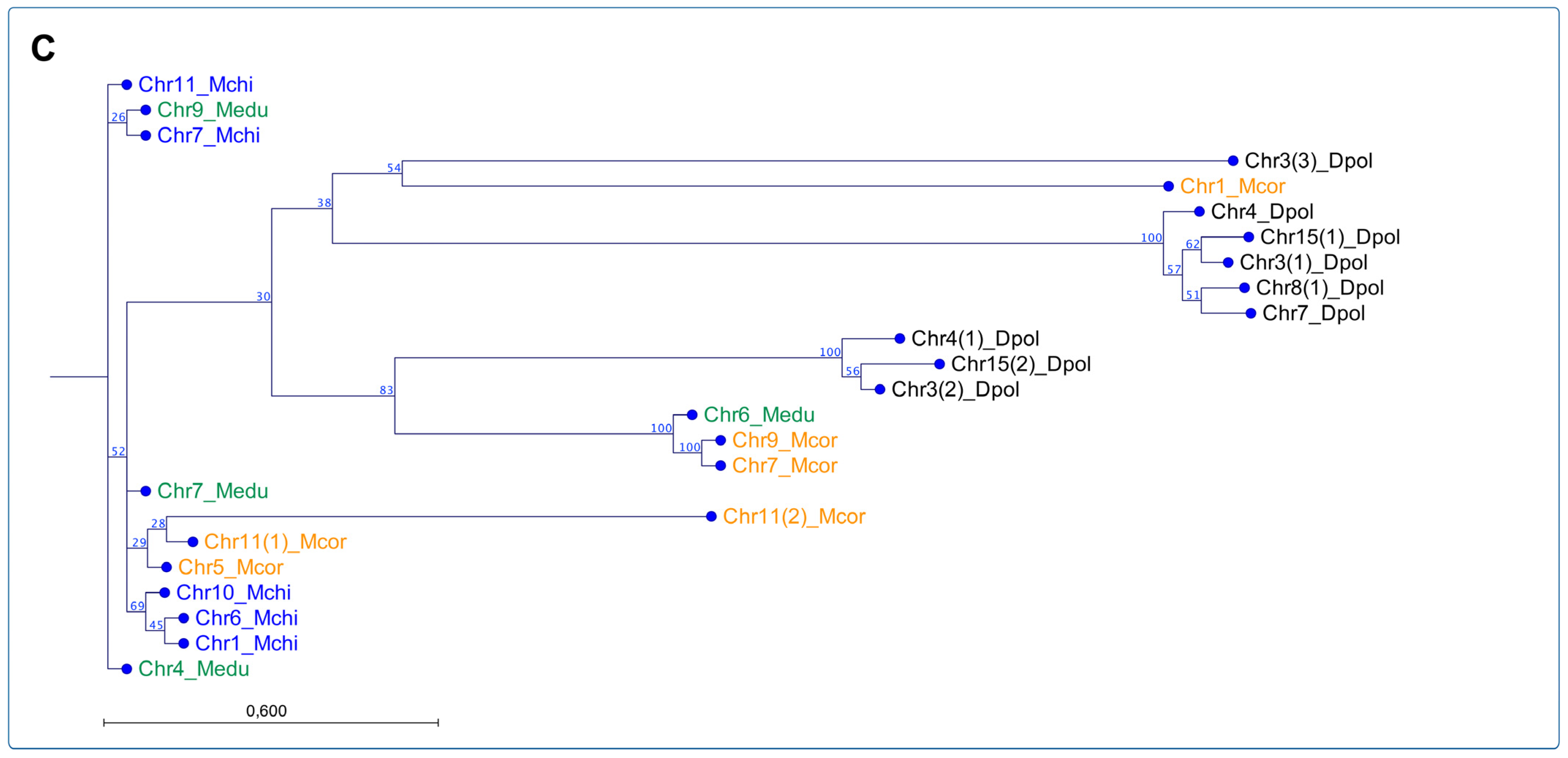
Furthermore, the phylogenetic analysis using publicly available reference genomes assembled at chromosome level for eleven bivalve species using maximum likelihood (ML) revealed a six-chromosome cluster composed of bivalves belonging to the families Veneridae, Solenidae, Pectinidae, Ostreidae, Pteriidae, and Mytilidae (
Figure 4B). The phylogenetic-reconstruction-rooted SLEs were found in three chromosomes (2, 4, and 17) from
R. philippinarum. The other Veneridae member,
M. mercenaria showed a cluster of four chromosomes (10, 12, 13, and 16) and related to two chromosomes of
S. grandis (10 and 16). This last species formed a unique cluster composed of three chromosomes (8, 15, and 17), similar to
P. maximus, with three chromosomes. Concerning the mussel and oyster genomes assembled at the chromosome level, the phylogenic analysis revealed two main clusters composed of species belonging to Ostreida and Mytilidae, where the first taxon was comprised of the Ostreidae and Pteriidae families. Herein, one cluster was rooted with three SLE sequences from
C. virginica,
C. gigas, and
C. ariakensis located on chromosomes 9, 2, and 5, respectively. The second major cluster was composed of SLEs annotated in chromosomes from Ostreidae and Pteriidae, where
C. virginica chromosomes were closely related to
P. imbricata. The third cluster was observed containing three SLE sequences from
C. virginica and
C. ariakensis: chromosomes 1, 2, 8, and 1, 2, and 6, respectively (
Figure 4B). The analysis of the Mytilidae family revealed two primary clusters comprising SLEs located in chromosomes from
M. edulis and
D. polymorpha, and
M. coruscus,
M. chilensis, respectively (
Figure 4B). This last cluster grouped five chromosomes from
M. coruscus (Chr. 1, 4, 5, 7, and 11), and two from
M. edulis (Chr. 4 and 6). The Steamer-like sequence characterized for
M. chilensis was also observed in this cluster. Finally, a detailed analysis of the three mussel species reported with genome assemblies at the chromosome level was conducted (
Figure 4C). Notably, a rooted cluster comprising chromosomes 7, 11, 9 for
M. couscous, and 4 and 6 for
M. edulis were closely related. Herein, two primary clusters of SLEs located in chromosomes from
M. chilensis,
M. edulis, and
M. coruscus were observed. The analysis suggested that the SLEs identified on
M. chilensis chromosomes are closely related to the SLEs annotated on chromosomes 9 and 4 in
M. edulis; meanwhile, the SLEs located in chromosomes 1, 5, and 11 in
M. coruscus were also identified in the same chromosome cluster. The second main cluster observed comprised exclusively SLEs annotated in
D. polymorpha chromosomes, except the SLE copies identified in chromosomes 9 and 10 of
M. edulis. Interestingly, the SLEs annotated in chromosome 9 from
M. edulis are shared among the three primary clusters analyzed, suggesting putative translocation gene events in Mytilidae.
Overall, the phylogenic relationships of SLEs revealed that the reported bivalve genomes comprise between 3 and 6 loci. A lower number of SLEs was found in Solenidae, Pectinidae, and Veneridae, followed by Mytilidae. A higher number of SLE loci was observed in genomes belonging to the Ostreida order. As far as we know, the evolution of the bivalve chromosomes has mainly been studied using cytogenetic techniques combining molecular probes on candidate genes to detect genome rearrangements that drive the speciation process [
87,
88,
89]. However, the availability of reference genomes assembled at the chromosome level opens new perspectives for exploring molecular evolution in several taxonomic orders through gene collinearity analysis. The study by Yang [
28] highlighted putative chromosome rearrangements among the king scallop
Pecten maximus, the blood clam
Scapharca broughtonii, the hard-shelled mussel
Mytilus coruscus, the pearl oyster
Pinctada martensii, and the Pacific oyster
Crassostrea gigas genomes. Notably, the chromosome synteny illustrated that large-scale rearrangements are common events between the scallop and oyster but scarce between the scallop and mussel genomes. The reported evidence suggested that almost all the chromosome rearrangements between the mussel and oyster genomes are different, implicating independent chromosome fusion events. The SLE loci identified in all the genomes analyzed in the current study suggest that SLEs are relatively conserved in chromosome position for some taxa. For instance, the SLE loci in Veneridae, Pectinidae, and Solenidae appear to be associated with chromosomes 10, 13, 12, and 16. This sharing characteristic can reflect common genetic events during the evolution of these taxonomical groups. Similarly, the Ostreidae and Mytilidae families share SLE loci annotated to chromosomes 1, 2, 8, and 10. The detailed analysis of SLEs in Mytilidae shows that the transposon identified in
M. chilensis was shared between
M. edulis and
M. coruscus, where SLEs in
D. polymorpha appear to be more phylogenetically distant than
Mytilus species. Interestingly, the mutation identified on the SLEs localized in the
M. chilensis genome (insertion of twelve nucleotides), specifically on chromosomes 7 and 11, was shared with the SLE annotated on chromosome 9 in
M. edulis. This cumulative evidence reveals diverse chromosome rearrangements, reflecting a complex evolutionary history of bivalve chromosomes.
3.7. Whole-Genome Transcript Expression Analysis in Two M. chilensis Populations
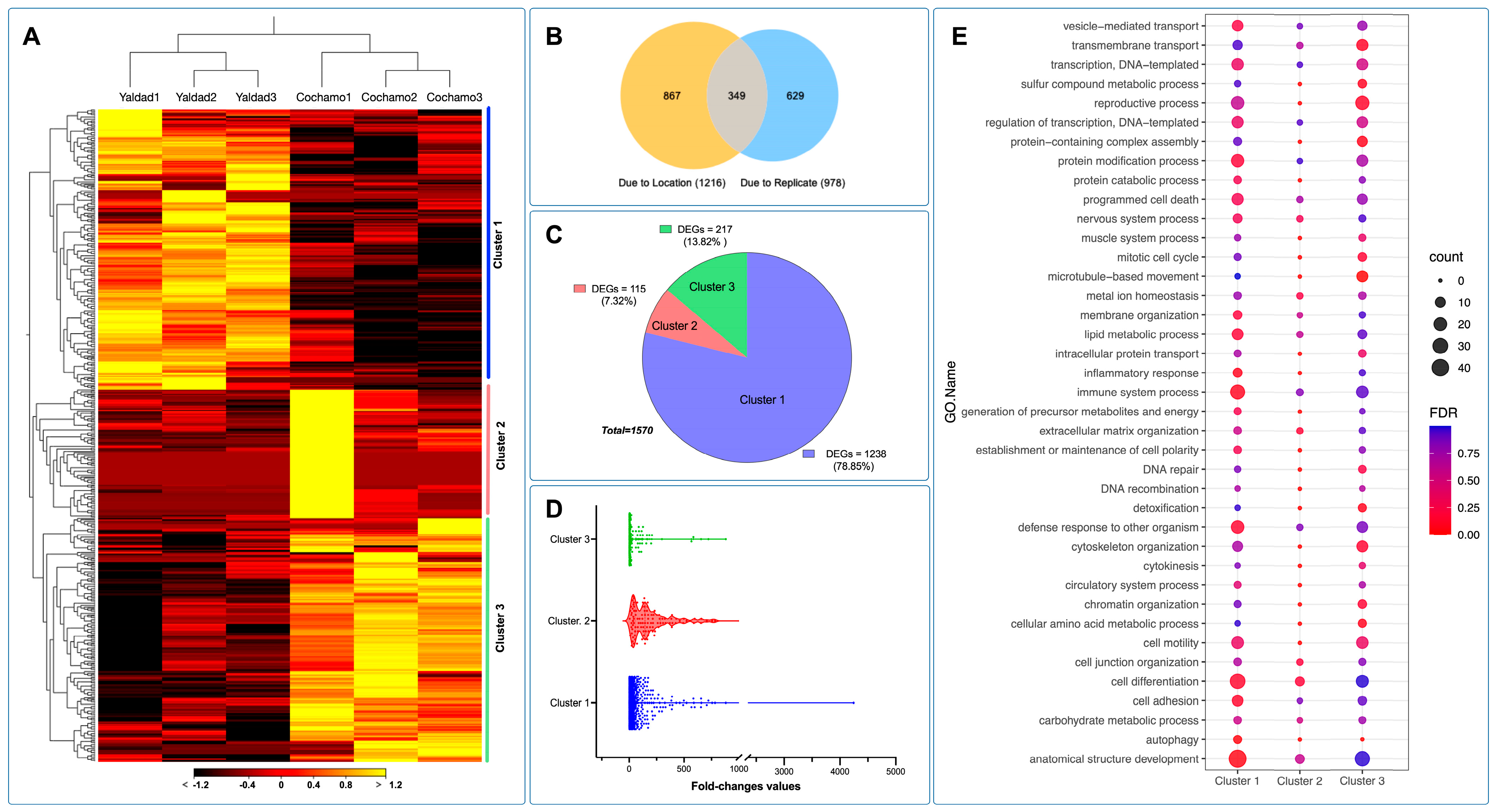
The transcriptome profiling among mussels collected during the austral summer in 2019 from Yaldad and Cochamó evidenced three primary transcriptional clusters. Herein, gene cluster 1 was highly expressed in the gills of mussels exposed to the Yaldad marine conditions; meanwhile, gene clusters 2 and 3 were highly expressed in individuals collected in Cochamó or mussels exposed to estuarine conditions (
Figure 6A). Notably, the RNA-seq from individuals collected as Cochamo1 (replicate) showed a highly expressed gene cluster, indicating a wide transcriptome variation among mussels from this population. The RNA-seq analysis was performed with the mRNA sequences annotated on the
M. chilensis genome. Herein, it is essential to note that in mussel species, specifically in
M. galloprovincialis, the phenomenon of presence–absence variation (PAV) has been described. This fact means that PAVs can bias the analyses of transcriptome profiles in the studied mussel populations. We previously conducted a de novo assembling for the RNA-data sets sequenced from Yaldad and Cochamó populations. The results showed that the number of genes with expression values >1 (total gene reads) did not show statistical differences between both mussel populations. For instance, Yaldad and Cochamó mussels showed 25,086 ± 215 and 25,344 ± 212 (three replicates per population), respectively, of expressed genes in gill tissue (
p-value = 0.98). Collectively, between 72.6% and 73.3% of the annotated genes in the
M. chilensis genome were transcriptionally active in gills independently of the population analyzed.
The evaluation of differentially expressed genes (DEGs) showed that the main factor of differences in the number of DEGs was the population rather than the replicates assessed (
Figure 6B). The proportion of DEGs evaluated among the gene clusters revealed that cluster 1, highly expressed in Yaldad, accounted the 78.85% of the total DEGs analyzed. Clusters 2 and 3 are primarily characterized by high transcription values in the Cochamó population, evidenced by 7.32% and 13.82% of DEGs, respectively. The total number of DEGs analyzed was 1570 (
Figure 6C). Notably, the fold-change values estimated among the replicates and populations revealed high values in gene transcriptional cluster 1, compared with clusters 2 and 3 where the fold-change values were significantly lower (
Figure 6D). The functional analysis showed that cluster 1 was enriched by GO terms related to protein modification processes, programmed cell death, immune system processes, defense response, cell differentiation, and anatomical structure development (
Figure 6E). Clusters 2 and 3 were less enriched, revealing significant GO terms for transmembrane transport, reproductive processes, protein-containing complex assembly, microtube-based movement, cytoskeleton organization, chromatin organization, and metabolic process (
Figure 6E).
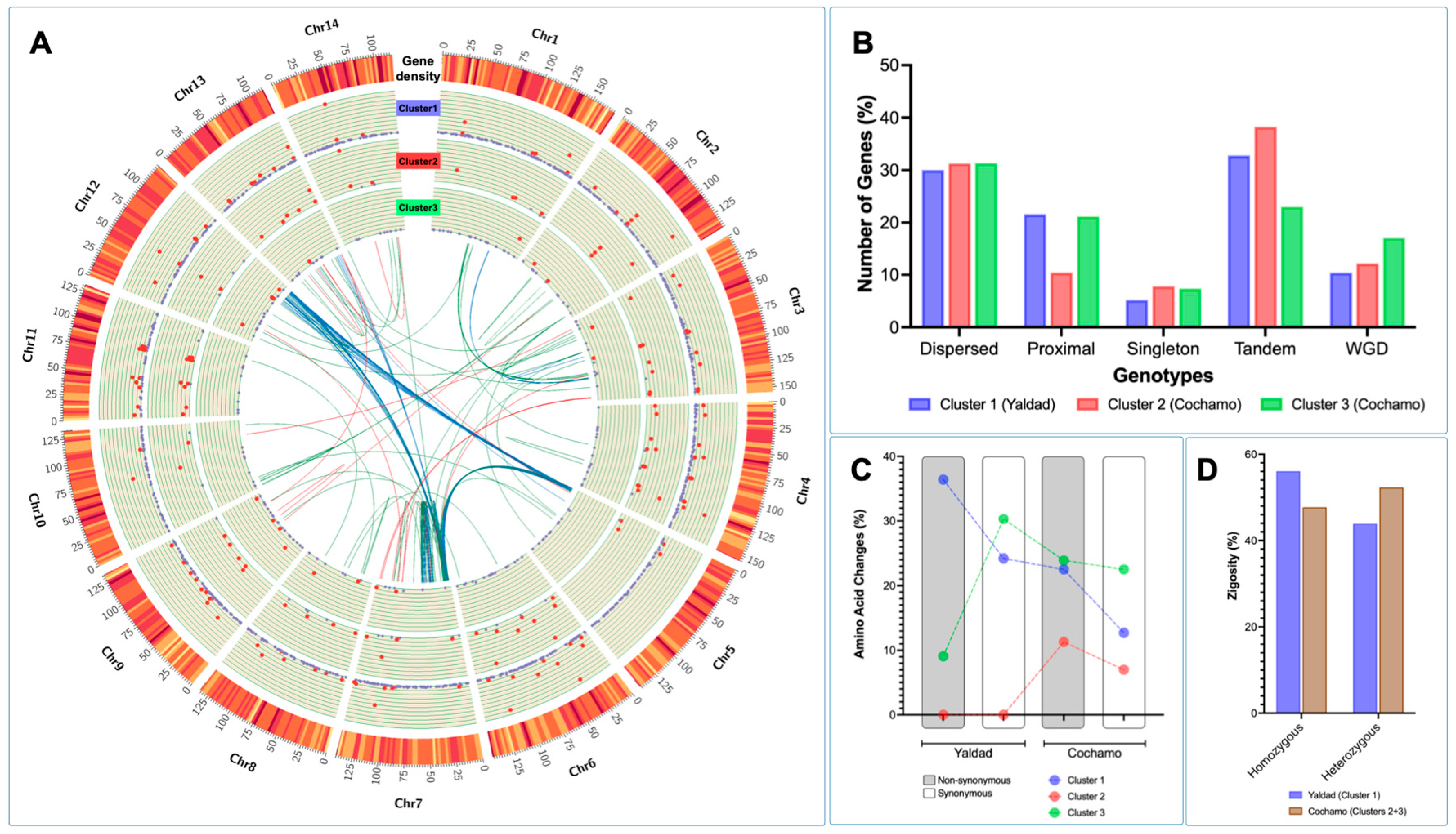
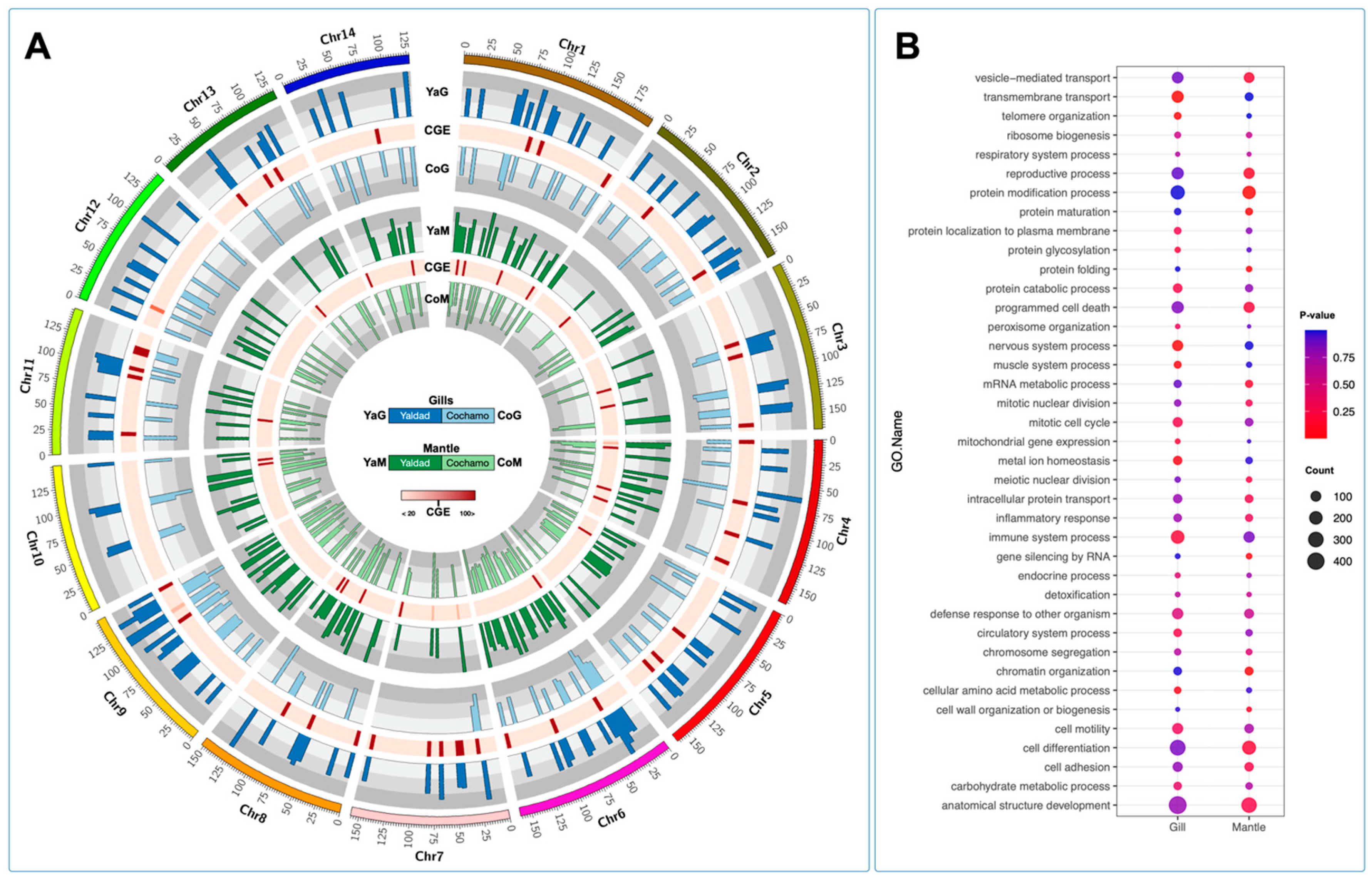
The cluster gene expression analysis was used to identify genetic polymorphisms annotated in differentially expressed genes (DEGs) between the Yaldad and Cochamó mussel populations. The DEGs were evaluated by cluster transcriptome analysis displayed using a Circos plot to visualize specific loci where DEGs were highly transcribed. The fold-change values calculated showed high transcription levels in clusters 1 and 2 through all chromosomes scanned (see red dots in
Figure 7A). Congruently with the previous RNA-seq results in this study, the highest fold-change values were observed in DEGs annotated in cluster 1 (Yaldad population). By contrast, cluster 3 showed a small number of DEGs with high fold-change values. Notably, the physical mapping of DEGs on chromosomes evidenced specific transcriptome patterns, revealing genes differentially expressed through the mussel genome exposed to the marine environment. The synteny analysis for DEGs showed a marked pattern among chromosomes 5, 7, and 12 for cluster 1; meanwhile, the synteny observed for the DEGs annotated in clusters 2 and 3 revealed a wide distribution along the
M. chilensis genome (
Figure 7A). Interestingly, the analysis carried out to detect macro-genome mutation in gene families between the Yaldad and Cochamó populations evidenced a similar number of dispersed genes, suggesting that those might arise from transposition. Tandem or repeatedly duplicated genes were observed with a low proportion in cluster 3 (Cochamó); meanwhile, the proximal genes showed a similar proportion to cluster 1 (Yaldad). These results might suggest small-scale transposition or duplication/insertion events. An interesting finding was observed for whole-genome duplication (WGD). The primary proportion was evidenced in cluster 3 (Cochamó), compared with clusters 1 and 2 from the Yaldad population (
Figure 7B). Furthermore, the bioinformatic analysis conducted for detecting amino acid changes (AAC) in DEGs showed that 38% of non-synonymous AAC were identified in mussels collected from Yaldad. By contrast, the main proportion of synonymous AAC was detected in mussels exposed to Cochamó’s estuarine conditions (
Figure 7C). Notably, the analysis performed for DEGs annotated in cluster 2 did not show non-synonymous and synonymous AAC in mussels collected from Yaldad. Finally, evaluating the zygosity proportion estimated for each mussel population evidenced an inverse pattern between both populations. The Yaldad cluster was higher in the homozygous proportion than Cochamó, where heterozygous AAC were detected in a higher proportion (
Figure 7D).
To explore the transcriptome signatures between the Yaldad and Cochamó mussel populations, we applied the genome chromosome expression (CGE) approach to test differences among tissues and individuals through the
M. chilensis genome. The CGE analysis revealed high differences among chromosome regions, where the gill tissue was more modulated than the mantle tissue (
Figure 8A). Interestingly, there are some levels of congruence among the CGE annotated for both mussel populations. We conducted a gene ontology enrichment analysis using this finding from genes identified by CGE analysis. The results evidenced that gill transcriptomes displayed functional processes associated with transmembrane transport, protein catabolic, nervous system, and metal homeostasis. Notably, immune system process GO terms were highly enriched in gills. Moreover, the chromosome region differentially expressed in mantle tissue revealed that the reproductive process, protein modification process, cell differentiation, anatomical structure development, and gene silencing by RNA were mainly annotated (
Figure 8B). Taken together, the results reported in this study are highly congruent with the previous study conducted by Yévenes et al. [
38], through the transcriptome responses of
M. chilensis collected in ecologically different farm-impacted seedbeds.
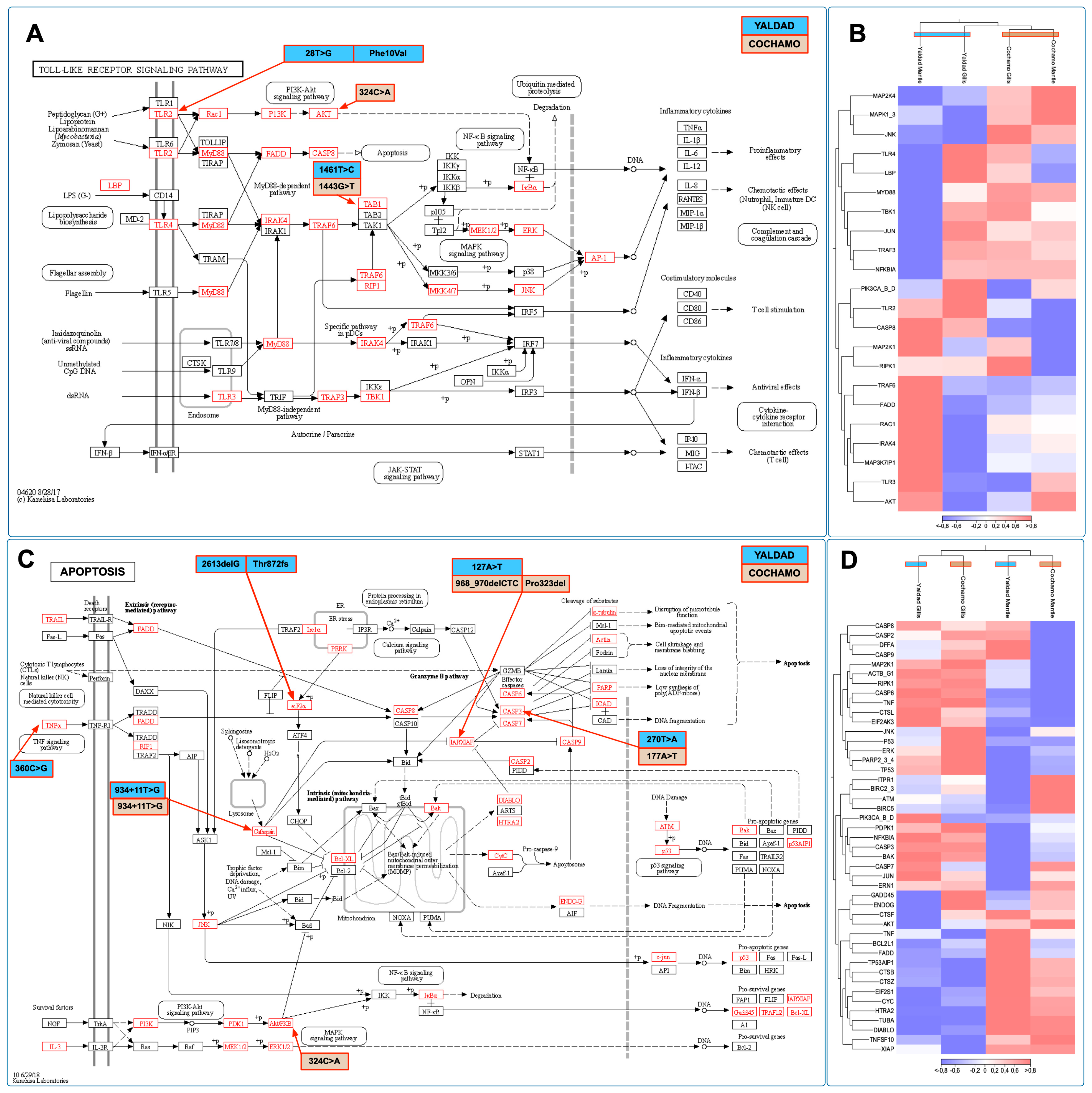
The cumulative findings of this study suggest that the immune system was primarily modulated between mussels exposed to the Yaldad and Cochamó environmental conditions. With the aim of exploring the transcription profiling of immune-related genes, we selected two KEGG pathways annotated in the
M. chilensis genome (
Figure 9). Herein, Toll-like receptor signaling pathway and apoptosis were analyzed in terms of transcription activity and single nucleotide variation (SNV) between mussel populations. Notably, a non-synonymous SNV was detected on the
TLR2 gene (28T>G) in individuals collected from the Yaldad population. The translation evidenced an amino acid change from phenylalanine to valine at position 10 in the ORF (Phe10Val) (
Figure 9A). The analysis also evidenced SNV on genes such as
AKT and
TAB1, where no amino acid changes were detected. The transcriptome profiling for the TLR pathway evidenced a high modulation of genes such as
TLR3,
AKT,
TRAFF6,
FADD,
IRAK4, and
RAC1 in mussels collected from Yaldad. Interestingly, mitogen-activated protein kinases (
MAP2K and
MAPK1) and c-Jun N-terminal kinase (
JNK) were differentially expressed, suggesting putative roles related to stress signaling pathways (
Figure 9B). Furthermore, the apoptosis pathway revealed two SNV localized in eukaryotic initiation factor 2 α (
EIF2α) and inhibitor of apoptosis (
IAP) in mussels sampled from the Yaldad and Cochamó populations, respectively (
Figure 9C). The 2613delG in the
EIF2α gene produces a frameshift at the Thr872; meanwhile, the 968_970delCTC localized in the IAP gene produces a deletion of proline at position 323. The transcriptome profiling of apoptosis-related genes showed a conspicuous differentiation between gills and mantle tissue, where three primary gene expression clusters were identified (
Figure 9D).
Notably, genes such as
P53,
ERK,
TP53,
PARP2, and
JNK were highly expressed in gill tissue. The gene expression analysis in mantle tissue evidenced high transcriptional activity in genes related to the intrinsic (mitochondria-mediated) pathway, such as the B-cell lymphoma (
BCL) gene and the second mitochondria-derived activator of caspase (
DIABLO) gene. Concerning the mitogenome of
M. chilensis, it was previously reported by Gaitán-Espitia et al. [
34] and Śmietanka and Burzyński [
91], evidencing a genome size of 16,748bp and structurally identical to the northern hemisphere
M. edulis and
M. galloprovincialis mitogenomes. Furthermore, the putative adaptive contribution of the mitochondrial genes was recently reported by Yevenes et al. [
37]. The RNA-Seq analysis detected differences in the number of upregulated mitogens between individuals from Cochamó and Yaldad, some being tissue-specific (
ND4L and
COX2). Several monomorphic location-specific mitochondrial genetic variants were detected in samples from Cochamó and Yaldad, representing standing genetic variability to optimize mitochondrial functioning under local habitats. Overall, these mitochondrial transcriptomic differences reflect the impact of environmental conditions on the mitochondrial genome functioning and offer new markers to assess the effects of habitat translocations on mussel fitness, a routine industry practice. Likewise, these mitochondrial markers should help monitor and maintain population differences in this keystone and heavily exploited native species.
Further functional studies will be conducted to validate the association between single nucleotide polymorphisms and the fitness traits observed or how the translocation process associated with aquaculture activity can evolve with the loss of locally adapted alleles. Interestingly, a recent transplant experiment reported by Jahnsen-Guzmán et al. [
92] demonstrated that
M. chilensis individuals are adapted to the subtidal environment (4 m depth), as they exhibit significantly higher fitness (growth and calcification rates) than those transferred to the intertidal environment (1 m depth), which showed increased metabolic stress. Herein, the mussel lives in extreme environmental variability, where their ability to cope with perturbations, and build plasticity and adaptive responses, seems based on the genome architecture.


 ,
, 













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}