
The Genetics of Primary Biliary Cholangitis: A GWAS and Post-GWAS Update

Abstract
1. Introduction
2. Genetic Factors Associated with PBC
2.1. Human Leukocyte Antigen (HLA)
2.2. PBC Susceptibility Loci Outside HLA
2.3. Specific and Non-Specific PBC Susceptibility Genes
3. Post-GWAS Studies
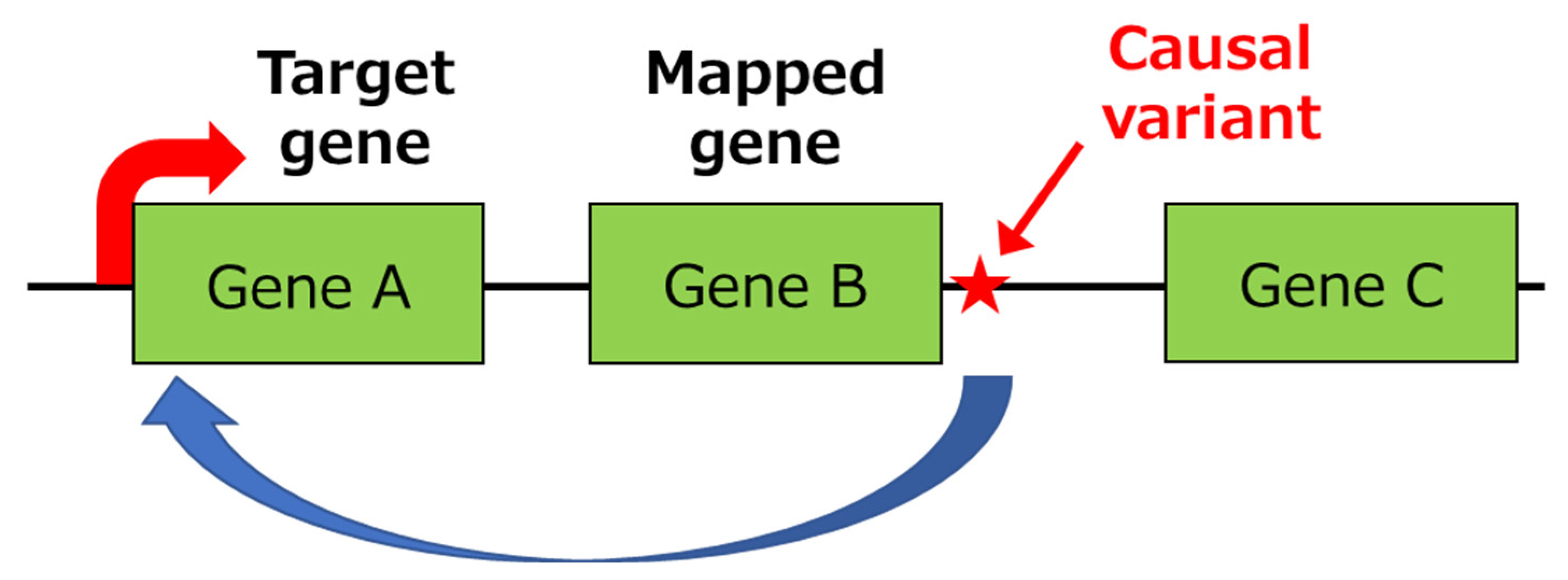
3.1. Identification of Primary Functional (Causal) Variants and Effector Genes
3.2. In Silico Gene Set Analysis
3.2.1. IL-12-Mediated Signaling Pathway
3.2.2. Cellular Response to TNF
3.2.3. Activation, Maturation, and Differentiation of B Cells
3.3. Integration of GWAS and Transcriptome Data
3.4. In Silico Drug Efficacy Screening
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shimoda, S.; Nakamura, M.; Ishibashi, H.; Hayashida, K.; Niho, Y. HLA DRB4 0101-restricted immunodominant T cell autoepitope of pyruvate dehydrogenase complex in primary biliary cirrhosis: Evidence of molecular mimicry in human autoimmune diseases. J. Exp. Med. 1995, 181, 1835–1845. [Google Scholar] [CrossRef]
- Shimoda, S.; Van De Water, J.; Ansari, A.; Nakamura, M.; Ishibashi, H.; Coppel, R.L.; Lake, J.; Keeffe, E.B.; Roche, T.E.; Gershwin, M.E. Identification and precursor frequency analysis of a common T cell epitope motif in mitochondrial autoantigens in primary biliary cirrhosis. J. Clin. Investig. 1998, 102, 1831–1840. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, M.M.; Gershwin, M.E. Primary biliary cirrhosis. N. Engl. J. Med. 2005, 353, 1261–1273. [Google Scholar] [CrossRef]
- Selmi, C.; Bowlus, C.L.; Gershwin, M.E.; Coppel, R.L. Primary biliary cirrhosis. Lancet 2011, 377, 1600–1609. [Google Scholar] [CrossRef] [PubMed]
- Honda, A.; Tanaka, A.; Kaneko, T.; Komori, A.; Abe, M.; Inao, M.; Namisaki, T.; Hashimoto, N.; Kawata, K.; Takahashi, A.; et al. Bezafibrate Improves GLOBE and UK-PBC Scores and Long-Term Outcomes in Patients With Primary Biliary Cholangitis. Hepatology 2019, 70, 2035–2046. [Google Scholar] [CrossRef] [PubMed]
- Gershwin, M.E.; Mackay, I.R. The causes of primary biliary cirrhosis: Convenient and inconvenient truths. Hepatology 2008, 47, 737–745. [Google Scholar] [CrossRef] [PubMed]
- Shimoda, S.; Hisamoto, S.; Harada, K.; Iwasaka, S.; Chong, Y.; Nakamura, M.; Bekki, Y.; Yoshizumi, T.; Shirabe, K.; Ikegami, T.; et al. Natural killer cells regulate T cell immune responses in primary biliary cirrhosis. Hepatology 2015, 62, 1817–1827. [Google Scholar] [CrossRef]
- Gerussi, A.; Cristoferi, L.; Carbone, M.; Asselta, R.; Invernizzi, P. The immunobiology of female predominance in primary biliary cholangitis. J. Autoimmun. 2018, 95, 124–132. [Google Scholar] [CrossRef]
- Nakamura, M. Clinical Significance of Autoantibodies in Primary Biliary Cirrhosis. Semin. Liver Dis. 2014, 34, 334–340. [Google Scholar] [CrossRef]
- Leung, P.S.C.; Coppel, R.L.; Ansari, A.; Munoz, S.; Gershwin, M.E. Antimitochondrial Antibodies in Primary Biliary Cirrhosis. Semin. Liver Dis. 1997, 17, 61–69. [Google Scholar] [CrossRef]
- Kakuda, Y.; Harada, K.; Sawada-Kitamura, S.; Ikeda, H.; Sato, Y.; Sasaki, M.; Okafuji, H.; Mizukoshi, E.; Terasaki, S.; Ohta, H.; et al. Evaluation of a new histologic staging and grading system for primary biliary cirrhosis in comparison with classical systems. Hum. Pathol. 2013, 44, 1107–1117. [Google Scholar] [CrossRef]
- Selmi, C.; Meroni, P.L.; Gershwin, M.E. Primary biliary cirrhosis and Sjögren’s syndrome: Autoimmune epithelitis. J. Autoimmun. 2012, 39, 34–42. [Google Scholar] [CrossRef]
- Corpechot, C.; Chr´etien, Y.; Chazouill`eres, O.; Poupon, R. Demographic, lifestyle, medical and familial factors associated with primary biliary cirrhosis. J. Hepatol. 2010, 53, 162–169. [Google Scholar] [CrossRef]
- Jones, D.E.; Watt, F.E.; Metcalf, J.V.; Bassendine, M.F.; James, O.F. Familial primary biliary cirrhosis reassessed: A geographically-based population study. J. Hepatol. 1999, 30, 402–407. [Google Scholar] [CrossRef] [PubMed]
- Selmi, C.; Mayo, M.J.; Bach, N.; Ishibashi, H.; Invernizzi, P.; Gish, R.; Gordon, S.C.; Wright, H.I.; Zweiban, B.; Podda, M.; et al. Primary biliary cirrhosis in monozygotic and dizygotic twins: Genetics, epigenetics, and environment. Gastroenterology 2004, 127, 485–492. [Google Scholar] [CrossRef]
- Welter, D.; MacArthur, J.; Morales, J.; Burdett, T.; Hall, P.; Junkins, H.; Klemm, A.; Flicek, P.; Manolio, T.; Hindorff, L.; et al. The NHGRI GWAS Catalog, a curated resource of SNP-trait associations. Nucleic Acids Res. 2014, 42, D1001-6. [Google Scholar] [CrossRef]
- Tsuchiya, N.; Ohashi, J. Human immune system diversity and its implications in diseases. J. Hum. Genet. 2015, 60, 655–656. [Google Scholar] [CrossRef]
- Barreiro, L.B.; Quintana-Murci, L. From evolutionary genetics to human immunology: How selection shapes host defence genes. Nat. Rev. Genet. 2010, 11, 17–30. [Google Scholar] [CrossRef] [PubMed]
- Donaldson, P.; Agarwal, K.; Craggs, A.; Craig, W.; James, O.; Jones, D. HLA and interleukin 1 gene polymorphisms in primary biliary cirrhosis: Associations with disease progression and disease susceptibility. Gut 2001, 48, 397–402. [Google Scholar] [CrossRef] [PubMed]
- Wassmuth, R.; Depner, F.; Danielsson, A.; Hultcrantz, R.; Lööf, L.; Olson, R.; Prytz, H.; Sandberg-Gertzen, H.; Wallerstedt, S.; Lindgren, S. HLA class II markers and clinical heterogeneity in Swedish patients with primary biliary cirrhosis. Tissue Antigens 2002, 59, 381–387. [Google Scholar] [CrossRef] [PubMed]
- Mullarkey, M.; Stevens, A.; McDonnell, W.; Loubière, L.; Brackensick, J.; Pang, J.; Porter, A.; Galloway, D.; Nelson, J. Human leukocyte antigen class II alleles in Caucasian women with primary biliary cirrhosis. Tissue Antigens 2005, 65, 199–205. [Google Scholar] [CrossRef] [PubMed]
- Donaldson, P.T.; Baragiotta, A.; Heneghan, M.A.; Floreani, A.; Venturi, C.; Underhill, J.A.; Jones, D.E.; James, O.F.; Bassendine, M.F. HLA class II alleles, genotypes, haplotypes, and amino acids in primary biliary cirrhosis: A large-scale study. Hepatology 2006, 44, 667–674. [Google Scholar] [CrossRef] [PubMed]
- Invernizzi, P.; Selmi, C.; Poli, F.; Frison, S.; Floreani, A.; Alvaro, D.; Almasio, P.; Rosina, F.; Marzioni, M.; Fabris, L.; et al. Human leukocyte antigen polymorphisms in italian primary biliary cirrhosis: A multicenter study of 664 patients and 1992 healthy controls. Hepatology 2008, 48, 1906–1912. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, M.; Yasunami, M.; Kondo, H.; Horie, H.; Aiba, Y.; Komori, A.; Migita, K.; Yatsuhashi, H.; Ito, M.; Shimoda, S.; et al. Analysis of HLA-DRB1 polymorphisms in Japanese patients with primary biliary cirrhosis (PBC): The HLA-DRB1polymorphism determines the relative risk of antinuclear antibodies for disease progression in PBC. Hepatol. Res. 2010, 40, 494–504. [Google Scholar] [CrossRef] [PubMed]
- Umemura, T.; Joshita, S.; Ichijo, T.; Yoshizawa, K.; Katsuyama, Y.; Tanaka, E.; Ota, M. The Shinshu PBC Study Group Human leukocyte antigen class II molecules confer both susceptibility and progression in Japanese patients with primary biliary cirrhosis. Hepatology 2012, 55, 506–511. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.-T.; Liao, H.-Y.; Zhang, X.; Liu, Y.-M.; Zhao, Y.; Zhang, H.-P.; Sun, L.-M.; Ma, Y.-X.; Yan, H.-P. Human leucocyte antigen alleles and haplotypes and their associations with antinuclear antibodies features in Chinese patients with primary biliary cirrhosis. Liver Int. 2014, 34, 220–226. [Google Scholar] [CrossRef]
- Yasunami, M.; Nakamura, H.; Tokunaga, K.; Kawashima, M.; Nishida, N.; Hitomi, Y.; Nakamura, M. Principal contribution of HLA-DQ alleles, DQB1*06:04 and DQB1*03:01, to disease resistance against primary biliary cholangitis in a Japanese population. Sci. Rep. 2017, 7, 11093. [Google Scholar] [CrossRef]
- Wang, C.; Zheng, X.; Tang, R.; Han, C.; Jiang, Y.; Wu, J.; Shao, Y.; Gao, Y.; Yu, J.; Hu, Z.; et al. Fine mapping of the MHC region identifies major independent variants associated with Han Chinese primary biliary cholangitis. J. Autoimmun. 2020, 107, 102372. [Google Scholar] [CrossRef]
- Mulinacci, G.; Palermo, A.; Gerussi, A.; Asselta, R.; Gershwin, M.E.; Invernizzi, P. New insights on the role of human leukocyte antigen complex in primary biliary cholangitis. Front. Immunol. 2022, 13, 975115. [Google Scholar] [CrossRef]
- Barone, J.C.; Saito, K.; Beutner, K.; Campo, M.; Dong, W.; Goswami, C.P.; Johnson, E.S.; Wang, Z.-X.; Hsu, S. HLA-genotyping of clinical specimens using Ion Torrent-based NGS. Hum. Immunol. 2015, 76, 903–909. [Google Scholar] [CrossRef]
- Khor, S.-S.; Yang, W.; Kawashima, M.; Kamitsuji, S.; Zheng, X.; Nishida, N.; Sawai, H.; Toyoda, H.; Miyagawa, T.; Honda, M.; et al. High-accuracy imputation for HLA class I and II genes based on high-resolution SNP data of population-specific references. Pharm. J. 2016, 15, 530–537. [Google Scholar] [CrossRef] [PubMed]
- Hirschfield, G.M.; Liu, X.; Xu, C.; Lu, Y.; Xie, G.; Lu, Y.; Gu, X.; Walker, E.J.; Jing, K.; Juran, B.D.; et al. Primary biliary cholangitis associated with HLA, IL12A, and IL12RB2 variants. N. Engl. J. Med. 2009, 360, 2544–2555. [Google Scholar] [CrossRef] [PubMed]
- Hirschfield, G.M.; Liu, X.; Han, Y.; Gorlov, I.P.; Lu, Y.; Xu, C.; Lu, Y.; Chen, W.; Juran, B.D.; Coltescu, C.; et al. Variants at IRF5-TNPO3, 17q12-21 and MMEL1 are associated with primary biliary cholangitis. Nat. Genet. 2010, 42, 655–657. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Invernizzi, P.; Lu, Y.; Kosoy, R.; Lu, Y.; Bianchi, I.; Podda, M.; Xu, C.; Xie, G.; Macciardi, F.; et al. Genome-wide meta-analyses identify three loci associated with primary biliary cholangitis. Nat. Genet. 2010, 42, 658–660. [Google Scholar] [CrossRef] [PubMed]
- Mells, G.F.; Floyd, J.A.; Morley, K.I.; Cordell, H.J.; Franklin, C.S.; Shin, S.Y.; Heneghan, M.A.; Neuberger, J.M.; Donaldson, P.T.; Day, D.B.; et al. Genome-wide association study identifies 12 new susceptibility loci for primary biliary cholangitis. Nat. Genet. 2011, 43, 329–332. [Google Scholar] [CrossRef]
- Hirschfield, G.M.; Xie, G.; Lu, E.; Sun, Y.; Juran, B.D.; Chellappa, V.; Coltescu, C.; Mason, A.L.; Milkiewicz, P.; Myers, P.R.; et al. Association of primary biliary cholangitis with variants in the CLEC16A, SOCS1, SPIB and SIAE immunomodulatory genes. Genes Immun. 2012, 13, 328–335. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.Z.; Almarri, M.A.; Gaffney, D.J.; Mells, G.F.; Jostins, L.; Cordell, H.J.; Ducker, S.J.; Day, D.B.; Heneghan, M.A.; Neuberger, J.M.; et al. Dense fine-mapping study identifies new susceptibility loci for primary biliary cholangitis. Nat. Genet. 2012, 44, 1137–1141. [Google Scholar] [CrossRef] [PubMed]
- Juran, B.D.; Hirschfield, G.M.; Invernizzi, P.; Atkinson, E.J.; Li, Y.; Xie, G.; Kosoy, R.; Ransom, M.; Sun, Y.; Bianchi, I.; et al. Immunochip analyses identify a novel risk locus for primary biliary cirrhosis at 13q14, multiple independent associations at four established risk loci and epistasis between 1p31 and 7q32 risk variants. Hum. Mol. Genet. 2012, 21, 5209–5221. [Google Scholar] [CrossRef] [PubMed]
- Cordell, H.J.; Han, Y.; Mells, G.F.; Li, Y.; Hirschfield, G.M.; Greene, C.S.; Xie, G.; Juran, B.D.; Zhu, D.; Qian, D.C.; et al. International genome-wide meta-analysis identifies new primary biliary cholangitis risk loci and targetable pathogenic pathways. Nat. Commun. 2015, 6, 8019. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, M.; Nishida, N.; Kawashima, M.; Aiba, Y.; Tanaka, A.; Yasunami, M.; Nakamura, H.; Komori, A.; Nakamuta, M.; Zeniya, M.; et al. Genome-wide association study identified TNFSF15 and POU2AF1 as susceptibility locus for primary biliary cholangitis in the Japanese population. Am. J. Hum. Genet. 2012, 91, 721–728. [Google Scholar] [CrossRef]
- Kawashima, M.; Hitomi, Y.; Aiba, Y.; Nishida, N.; Kojima, K.; Kawai, Y.; Nakamura, H.; Tanaka, A.; Zeniya, M.; Hashimoto, E.; et al. Genome-wide association study identified PRKCB as a genetic susceptibility locus for primary biliary cholangitis in a Japanese population. Hum. Mol. Genet. 2017, 26, 650–659. [Google Scholar] [PubMed]
- Hitomi, Y.; Ueno, K.; Kawai, Y.; Nishida, N.; Kojima, K.; Kawashima, M.; Aiba, Y.; Nakamura, H.; Kouno, H.; Kouno, H.; et al. POGLUT1, the putative effector gene driven by rs2293370 in primary biliary cholangitis susceptibility locus chromosome 3q13.33. Sci. Rep. 2019, 9, 102. [Google Scholar] [CrossRef] [PubMed]
- Qiu, F.; Tang, R.; Zuo, X.; Shi, X.; Wei, Y.; Zheng, X.; Dai, Y.; Gong, Y.; Wang, L.; Xu, P.; et al. A genome-wide association study identifies six novel risk loci for primary biliary cholangitis. Nat. Commun. 2017, 8, 14828. [Google Scholar] [CrossRef] [PubMed]
- Cordell, H.J.; Fryett, J.J.; Ueno, K.; Darlay, R.; Aiba, Y.; Hitomi, Y.; Kawashima, M.; Nishida, N.; Khor, S.S.; Gervais, O.; et al. An international genome-wide meta-analysis of primary biliary cholangitis: Novel risk loci and a hierarchy of candidate drugs. J. Hepatol. 2021, 75, 572–581. [Google Scholar] [CrossRef]
- Asselta, R.; Paraboschi, E.M.; Gerussi, A.; Cordell, H.J.; Mells, G.F.; Sandford, R.N.; Jones, D.E.; Nakamura, M.; Ueno, K.; Hitomi, Y.; et al. X Chromosome Contribution to the Genetic Architecture of Primary Biliary Cholangitis. Gastroenterology 2021, 160, 2483–2495.e26. [Google Scholar] [CrossRef]
- Ge, D.; Fellay, J.; Thompson, A.J.; Simon, J.S.; Shianna, K.V.; Urban, T.J.; Heinzen, E.L.; Qiu, P.; Bertelsen, A.H.; Muir, A.J.; et al. Genetic variation in IL28B predicts hepatitis C treatment-induced viral clearance. Nature 2009, 461, 399–401. [Google Scholar] [CrossRef]
- Tanaka, Y.; Nishida, N.; Sugiyama, M.; Kurosaki, M.; Matsuura, K.; Sakamoto, N.; Nakagawa, M.; Korenaga, M.; Hino, K.; Hige, S.; et al. Genome-wide association of IL28B with response to pegylated interferon-α and ribavirin therapy for chronic hepatitis C. Nat. Genet. 2009, 41, 1105–1109. [Google Scholar] [CrossRef]
- Suppiah, V.; Moldovan, M.; Ahlenstiel, G.; Berg, T.; Weltman, M.; Abate, M.L.; Bassendine, M.; Spengler, U.; Dore, G.J.; Powell, E.; et al. IL28B is associated with response to chronic hepatitis C interferon-α and ribavirin therapy. Nat. Genet. 2009, 41, 1100–1104. [Google Scholar] [CrossRef]
- Fellay, J.; Thompson, A.J.; Ge, D.; Gumbs, C.E.; Urban, T.J.; Shianna, K.V.; Little, L.D.; Qiu, P.; Bertelsen, A.H.; Watson, M.; et al. ITPA gene variants protect against anaemia in patients treated for chronic hepatitis C. Nature 2010, 464, 405–408. [Google Scholar] [CrossRef]
- COVID-19 Host Genetics Initiative. Mapping the human genetic architecture of COVID-19. Nature 2021, 600, 472–477. [Google Scholar]
- Nishida, N.; Aiba, Y.; Hitomi, Y.; Kawashima, M.; Kojima, K.; Kawai, Y.; Ueno, K.; Nakamura, H.; Yamashiki, N.; Tanaka, T.; et al. NELFCD and CTSZ loci are associated with jaundice-stage progression in primary biliary cholangitis in the Japanese population. Sci. Rep. 2018, 8, 8071. [Google Scholar] [CrossRef]
- Gervais, O.; Ueno, K.; Kawai, Y.; Hitomi, Y.; Aiba, Y.; Ueta, M.; Nakamura, M.; Tokunaga, K.; Nagasaki, M. Regional heritability mapping identifies several novel loci (STAT4, ULK4, and KCNH5) for primary biliary cholangitis in the Japanese population. Eur. J. Hum. Genet. 2021, 29, 1282–1291. [Google Scholar] [CrossRef] [PubMed]
- Gerussi, A.; Carbone, M.; Corpechot, C.; Schramm, C.; Asselta, R.; Invernizzi, P. The genetic architecture of primary biliary cholangitis. Eur. J. Med. Genet. 2021, 64, 104292. [Google Scholar] [CrossRef] [PubMed]
- GTEx Consortium. The Genotype-Tissue Expression (GTEx) project. Nat Genet 2013, 45, 580–585. [Google Scholar] [CrossRef]
- Ota, M.; Nagafuchi, Y.; Hatano, H.; Ishigaki, K.; Terao, C.; Takeshima, Y.; Yanaoka, H.; Kobayashi, S.; Okubo, M.; Shirai, H.; et al. Dynamic landscape of immune cell-specific gene regulation in immune-mediated diseases. Cell 2021, 184, 3006–3021.e17. [Google Scholar] [CrossRef]
- Hitomi, Y.; Kojima, K.; Kawashima, M.; Kawai, Y.; Nishida, N.; Aiba, Y.; Yasunami, M.; Nagasaki, M.; Nakamura, M.; Tokunaga, K. Identification of the functional variant driving ORMDL3 and GSDMB expression in human chromosome 17q12-21 in primary biliary cholangitis. Sci. Rep. 2017, 7, 2904. [Google Scholar] [CrossRef] [PubMed]
- Hitomi, Y.; Kawashima, M.; Aiba, Y.; Nishida, N.; Matsuhashi, M.; Okazaki, H.; Nakamura, M.; Tokunaga, K. Human primary biliary cirrhosis-susceptible allele of rs4979462 enhances TNFSF15 expression by binding NF-1. Hum. Genet. 2015, 134, 737–747. [Google Scholar] [CrossRef] [PubMed]
- Hitomi, Y.; Nakatani, K.; Kojima, K.; Nishida, N.; Kawai, Y.; Kawashima, M.; Aiba, Y.; Nagasaki, M.; Nakamura, M.; Tokunaga, K. NFKB1 and MANBA Confer Disease Susceptibility to Primary Biliary Cholangitis via Independent Putative Primary Functional Variants. Cell. Mol. Gastroenterol. Hepatol. 2019, 7, 515–532. [Google Scholar] [CrossRef] [PubMed]
- Hitomi, Y.; Aiba, Y.; Ueno, K.; Nishida, N.; Kawai, Y.; Kawashima, M.; Yasunami, M.; Gervais, O.; Ito, M.; Cordell, H.J.; et al. rs9459874 and rs1012656 in CCR6/FGFR1OP confer susceptibility to primary biliary cholangitis. J. Autoimmun. 2022, 126, 102775. [Google Scholar] [CrossRef] [PubMed]
- Hitomi, Y.; Aiba, Y.; Kawai, Y.; Kojima, K.; Ueno, K.; Nishida, N.; Kawashima, M.; Gervais, O.; Khor, S.S.; Nagasak, M.; et al. rs1944919 on chromosome 11q23.1 and its effector genes COLCA1/COLCA2 confer susceptibility to primary biliary cholangitis. Sci. Rep. 2021, 11, 4557. [Google Scholar] [CrossRef]
- Hitomi, Y.; Aiba, Y.; Ueno, K.; Nishida, N.; Kawai, Y.; Kawashima, M.; Tsuiji, M.; Iwabuchi, C.; Takada, S.; Miyake, N.; et al. rs2013278 in the multiple immunological-trait susceptibility locus CD28 regulates the production of non-functional splicing isoforms. Hum. Genom. 2022, 16, 46. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Nastou, K.C.; Lyon, D.; Kirsch, R.; Pyysalo, S.; Doncheva, N.T.; Legeay, M.; Fang, T.; Bork, P.; et al. The STRING database in 2021: Customizable protein–protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic. Acids Res. 2021, 49, D605-12. [Google Scholar] [CrossRef] [PubMed]
- Aiba, Y.; Harada, K.; Komori, A.; Ito, M.; Shimoda, S.; Nakamura, H.; Nagaoka, S.; Abiru, S.; Migita, K.; Ishibashi, H.; et al. Systemic and local expression levels of TNF-like ligand 1A and its decoy receptor 3 are increased in primary biliary cirrhosis. Liver Int. 2013, 34, 679–688. [Google Scholar] [CrossRef] [PubMed]
- Ueno, K.; Aiba, Y.; Hitomi, Y.; Shimoda, S.; Nakamura, H.; Gervais, O.; Kawai, Y.; Kawashima, M.; Nishida, N.; Kohn, S.; et al. Integrated GWAS and mRNA Microarray Analysis Identified IFNG and CD40L as the Central Upstream Regulators in Primary Biliary Cholangitis. Hepatol. Commun. 2020, 4, 724–738. [Google Scholar] [CrossRef] [PubMed]
- Takii, Y.; Nakamura, M.; Ito, M.; Yokoyama, T.; Komori, A.; Shimizu-Yoshida, Y.; Nakao, R.; Kusumoto, K.; Nagaoka, S.; Yano, K.; et al. Enhanced expression of type I interferon and toll-like receptor-3 in primary biliary cirrhosis. Lab. Investig. 2005, 85, 908–920. [Google Scholar] [CrossRef]
- Bae, H.R.; Leung, P.S.; Tsuneyama, K.; Valencia, J.C.; Hodge, D.L.; Kim, S.; Back, T.; Karwan, M.; Merchant, A.S.; Baba, N.; et al. Chronic expression of interferon-gamma leads to murine autoimmune cholangitis with a female predominance. Hepatology 2016, 64, 1189–1201. [Google Scholar] [CrossRef]
- Hanada, S.; Harada, M.; Koga, H.; Kawaguchi, T.; Taniguchi, E.; Kumashiro, R.; Takato, U.; Yoshiyuki, U.; Motoyasu, I.; Shotaro, S.; et al. Tumor necrosis factor-alpha and interferon-gamma directly impair epithelial barrier function in cultured mouse cholangiocytes. Liver Int. 2003, 23, 3–11. [Google Scholar]
- Tummers, B.; Green, D.R. Caspase-8: Regulating life and death. Immunol. Rev. 2017, 277, 76–89. [Google Scholar] [CrossRef] [PubMed]
- Cerutti, A.; Schaffer, A.; Goodwin, R.G.; Shah, S.; Zan, H.; Ely, S.; Casali, P. Engagement of CD153 (CD30 Ligand) by CD30+ T Cells Inhibits Class Switch DNA Recombination and Antibody Production in Human IgD+ IgM+ B Cells. J. Immunol. 2000, 165, 786–794. [Google Scholar] [CrossRef] [PubMed]
- Guerrini, M.M.; Takayanagi, H. The immune system, bone and RANKL. Arch. Biochem. Biophys. 2014, 561, 118–123. [Google Scholar] [CrossRef]
- Vondenhoff, M.F.; Greuter, M.; Goverse, G.; Elewaut, D.; Dewint, P.; Ware, C.F.; Hoorweg, K.; Kraal, G.; Mebius, R.E. LTbetaR signaling induces cytokine expression and up-regulates lymphangiogenic factors in lymph node anlagen. J. Immunol. 2009, 182, 5439–5445. [Google Scholar] [CrossRef]
- Borelli, A.; Irla, M. Lymphotoxin: From the physiology to the regeneration of the thymic function. Cell Death Differ. 2021, 28, 2305–2314. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Liu, X.; Xu, L.; Hunter, Z.R.; Cao, Y.; Yang, G.; Carrasco, R.; Treon, S.P. Transcriptional repression of plasma cell differentiation is orchestrated by aberrant over-expression of the ETS factor SPIB in Waldenström macroglobulinaemia. Br. J. Haematol. 2014, 166, 677–689. [Google Scholar] [CrossRef]
- Cortés, M.; Georgopoulos, K. Aiolos Is Required for the Generation of High Affinity Bone Marrow Plasma Cells Responsible for Long-Term Immunity. J. Exp. Med. 2004, 199, 209–219. [Google Scholar] [CrossRef] [PubMed]
- Moser, B.; Schaerli, P.; Loetscher, P. CXCR5+ T cells: Follicular homing takes center stage in T-helper-cell responses. Trends Immunol. 2002, 23, 250–254. [Google Scholar] [CrossRef]
- Long, D.; Chen, Y.; Wu, H.; Zhao, M.; Lu, Q. Clinical significance and immunobiology of IL-21 in autoimmunity. J. Autoimmun. 2019, 99, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Davis, R.S. FCRL regulation in innate-like B cells. Ann. N. Y. Acad. Sci. 2015, 1362, 110–116. [Google Scholar] [CrossRef] [PubMed]
- Chalayer, E.; Gramont, B.; Zekre, F.; Goguyer-Deschaumes, R.; Waeckel, L.; Grange, L.; Paul, S.; Chung, A.W.; Killian, M. Fc receptors gone wrong: A comprehensive review of their roles in autoimmune and inflammatory diseases. Autoimmun. Rev. 2022, 21, 103016. [Google Scholar] [CrossRef] [PubMed]
- Okada, Y.; Wu, D.; Trynka, G.; Raj, T.; Terao, C.; Ikari, K.; Kochi, Y.; Ohmura, K.; Suzuki, A.; Yoshida, S.; et al. Genetics of rheumatoid arthritis contributes to biology and drug discovery. Nature 2014, 506, 376–381. [Google Scholar] [CrossRef]
- Zhang, J.; Jiang, K.; Lv, L.; Wang, H.; Shen, Z.; Gao, Z.; Wang, B.; Yang, Y.; Ye, Y.; Wang, S. Use of Genome-Wide Association Studies for Cancer Research and Drug Repositioning. PLoS ONE 2015, 10, e0116477. [Google Scholar] [CrossRef]
- Imamura, M.; Takahashi, A.; Yamauchi, T.; Hara, K.; Yasuda, K.; Grarup, N.; Zhao, W.; Wang, X.; Huerta-Chagoya, A.; Hu, C.; et al. Genome-wide association studies in the Japanese population identify seven novel loci for type 2 diabetes. Nat. Commun. 2016, 7, 10531. [Google Scholar] [CrossRef] [PubMed]
- Kawai, Y.; Hitomi, Y.; Ueta, M.; Khor, S.-S.; Nakatani, K.; Sotozono, C.; Kinoshita, S.; Nagasaki, M.; Tokunaga, K. Mapping of susceptible variants for cold medicine-related Stevens–Johnson syndrome by whole-genome resequencing. NPJ Genom. Med. 2021, 6, 9. [Google Scholar] [CrossRef] [PubMed]
- Schwanhäusser, B.; Busse, D.; Li, N.; Dittmar, G.; Schuchhardt, J.; Wolf, J.; Chen, W.; Selbach, M. Global quantification of mammalian gene expression control. Nature 2011, 473, 337–342. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chromosome | Location | Nearest Gene | GWAS-Lead SNP | Pa | e-QTL Genes (GTEx) b | e-QTL Genes (ImmuNexUT) c | Population | Ref |
|---|---|---|---|---|---|---|---|---|
| 1 | 1p36.32 | MMEL1 | rs867436 | 2.99 × 10−9 | - | - | European | [33] |
| 1p31.3 | IL12RB2 | rs6679356 | 4.85 × 10−65 | IL12RB2 (Whole Blood) | - | European | [32] | |
| 1p13.1 | CD58 | rs10802191 | 3.68 × 10−17 | - | CD58 (T cell, B cell) | European, Asian | [43] | |
| 1q23.1 | FCRL3 | rs945635 | 1.59 × 10−8 | FCRL1 (Whole blood, Spleen) FCRL3 (Whole blood, Spleen) | FCRL1 (T cell, B cell, Monocyte) FCRL2 (T cell, B cell) FCRL3 (T cell, B cell, NK cell, Neutrophil) FCRL5 (B cell, DC) | European | [44] | |
| 1p31.3 | DENND1B | rs12123169 | 9.75 × 10−18 | - | - | European | [35] | |
| 1q32.1 | CACNA1S | rs55734382 | 2.06 × 10−9 | GPR25 (Whole blood) | - | European | [44] | |
| 2 | 2p25.1 | LINC00299 | rs13416555 | 2.95 × 10−8 | LINC00299 (Liver) | LINC00299 (NK cell) ID2 (Monocyte) | International Meta | [44] |
| 2p23.3 | DNMT3A | rs34655300 | 5.23 × 10−10 | - | - | European | [44] | |
| 2q21.3 | TMEM163 | rs859767 | 8.94 × 10−16 | TMEM163 (Whole Blood) | TMEM163 (NK cell) | European, Asian | [44] | |
| 2q32.2 | STAT4 | rs11889341 | 3.41 × 10−31 | - | - | European, Asian | [35] | |
| RHM d | - | - | - | Asian | [52] | |||
| 2q33.2 | CD28 | rs10581773 | 3.82 × 10−13 | - | CD28 (T cell) | Asian | [43] | |
| 3 | 3p24.3 | PLCL2 | rs1156336 | 7.13 × 10−12 | PLCL2 (Whole Blood) | PLCL2 (T cell, Neutrophil) | European | [35] |
| 3p24.2 | RARB | rs6550965 | 1.50 × 10−14 | - | - | European | [44] | |
| 3p22.1 | ULK4 | RHM d | - | - | - | Asian | [52] | |
| 3q13.33 | TIMMDC1 | rs2293370 | 2.05 × 10−31 | POGLUT1 (Whole Blood) | POGLUT1 (T cell, Monocyte, DC) | European, Asian | [35] | |
| 3q25.33 | IL12A | rs589446 | 1.96 × 10−55 | - | - | European | [32] | |
| 4 | 4q24 | NFKB1 | rs6533022 | 2.21 × 10−32 | MANBA (Spleen, Liver) | NFKB1 (T cell, Neutrophil, DC) MANBA (T cell, NK cell, Monocyte, Neutrophil, DC) UBE2D3 (T cell, Monocyte) CISD2 (DC) | European, Asian | [35] |
| 4q24 | TET2 | rs2007403 | 6.19 × 10−10 | - | - | European | [44] | |
| 5 | 5p13.2 | IL7R | rs11742270 | 4.44 × 10−24 | - | - | European, Asian | [35] |
| 5q21.1 | ST8SIA4 | rs60643069 | 2.48 × 10−9 | - | - | International Meta | [44] | |
| 5q31.3 | NDFIP1 | rs6874308 | 4.67 × 10−8 | NDFIP1 (Whole Blood, Liver) | NDFIP1 (T cell, B cell, Monocyte, Neutrophil, DC) GNPDA1 (T cell) | International Meta | [44] | |
| 5q33.3 | IL12B | rs2546890 | 4.61 × 10−12 | - | RNF145 (T cell, Monocyte) | European | [39] | |
| 6 | 6p21.32 | HLA genes | rs7774434 | 2.72 × 10−116 | HLA-DRB6 (Whole Blood) HLA-DQA2 (Whole Blood) HLA-DRB1 (Whole Blood) | - | European, Asian | [44] |
| 6q21 | ATG5 | rs742108 | 3.16 × 10−8 | - | PRDM1 (T cell) | International Meta | [44] | |
| 6q23.3 | LINC03004 | rs2327832 | 2.31 × 10−10 | - | - | European | [39] | |
| 6q27 | CCR6 | rs4709148 | 2.18 × 10−10 | CCR6 (Whole Blood) FGFR1OP (Whole Blood) RNASET2 (Whole Blood) | CCR6 (T cell, B cell, DC) FGFR1OP (T cell, DC) RNASET2 (T cell, B cell, NK cell, Monocyte, DC) | Asian | [44] | |
| 7 | 7p21.1 | ITGB8 | rs7805218 | 4.12 × 10−8 | - | - | European | [44] |
| 7p14.1 | ELMO1 | rs60600003 | 4.70 × 10−13 | - | - | European | [35] | |
| 7q32.1 | TNPO3 | rs12531711 | 8.57 × 10−41 | TNPO5 (Whole Blood) IRF5 (Whole Blood) | - | European | [33] | |
| 7q34 | ZC3HAV1 | rs370193557 | 9.37 × 10−10 | - | - | European | [44] | |
| 8 | 8q24.21 | MYC | rs4733851 | 4.98 × 10−8 | - | - | International Meta | [44] |
| 9 | 9q22.33 | ANP32B | rs112500293 | 7.63 × 10−9 | ANP32B (Whole Blood) | TRIM14 (T cell, DC) | European | [44] |
| 9q32 | TNFSF15 | rs1322057 | 5.48 × 10−26 | - | TNFSF15 (B cell, Monocyte, Neutrophil, DC) TNFSF8 (T cell, NK cell, Monocyte) TNC (T cell) | Asian | [40] | |
| 10 | 10q11.23 | WDFY4 | rs7097397 | 2.42 × 10−10 | - | WDFY4 (DC) | European | [44] |
| 11 | 11p15.5 | DEAF1 | rs58523027 | 4.00 × 10−8 | - | - | European | [44] |
| 11q13.1 | CCDC88B | rs11601860 | 1.45 × 10−10 | - | - | European | [35] | |
| 11q23.1 | POU2AF1 | rs12419634 | 1.22 × 10−13 | COLCA1 (Spleen) | POU2AF1 (B cell) COLCA1 (B cell) COLCA2 (B cell) | European, Asian | [40] | |
| 11q23.3 | CXCR5 | rs7119044 | 6.85 × 10−38 | TREH (Liver) DDX6 (Whole Blood) | CXCR5 (B cell) | European, Asian | [35] | |
| 11q24.3 | ETS1 | rs10893872 | 9.77 × 10−9 | - | - | International Meta | [44] | |
| 12 | 12p13.31 | TNFRSF1A | rs1800693 | 1.19 × 10−16 | - | TNFRSF1A (T cell, NK cell, Neutrophil) LTBR (Neutrophil) | European | [35] |
| 12q24.12 | ATXN2 | rs35350651 | 5.50 × 10−19 | - | - | European | [37] | |
| 13 | 13q14.11 | TNFSF11 | rs9533122 | 5.83 × 10−13 | - | TNFSF11 (NK cell) | European | [38] |
| 13q14.2 | DLEU1 | rs9591325 | 2.14 × 10−19 | - | - | European | [39] | |
| 14 | 14q13.2 | SRP54 | rs799469 | 1.73 × 10−9 | SRP54 (Whole Blood) FAM177A1 (Whole Blood) | SPR54 (B cell, Neutrophil) PPP2R3C (Monocyte, Neutrophil, DC) FAM177A1 (T cell, Monocyte, Neutrophil, DC) KIAA0391 (T cell, NK cell, Monocyte, Neutrophil, DC) BAZ1A (Neutrophil) PSMA6 (Monocyte) | International Meta | [44] |
| 14q23.2 | KCNH5 | RHM d | - | - | - | Asian | [52] | |
| 14q24.1 | RAD51B | rs3784099 | 8.31 × 10−17 | - | - | European | [35] | |
| 14q32.12 | RIN3 | rs72699866 | 2.89 × 10−11 | - | - | European | [44] | |
| 14q32.32 | EXOC3L4 | rs59643720 | 2.73 × 10−38 | - | TNFAIP2 (B cell, Monocyte) | European | [35] | |
| 16 | 16p13.13 | CLEC16A | rs12919083 | 3.63 × 10−26 | - | - | European | [35] |
| 16p12.1 | IL21R | rs1119132 | 4.09 × 10−14 | - | IL21R (T cell, B cell, DC) IL4R (DC) | European | [43] | |
| 16q22.1 | DPEP2 | rs79577483 | 1.23 × 10−11 | DPEP3 (Whole Blood) LCAT (Whole Blood) SLC12A4 (Whole Blood) DUS2 (Whole Blood) PRMT7 (Whole Blood) STRL (Whole Blood) ELMO3 (Whole Blood) FAM65A (Whole Blood) PARD6A (Whole Blood) PRMT7 (Spleen) | DPEP2 (T cell, NK cell, Monocyte, DC) DUS2 (B cell, Monocyte, Neutrophil, DC) SLC7A6 (T cell, Monocyte, Neutrophil, DC) NFATC3 (T cell, B cell, NK cell, Neutrophil) PLA2G15 (B cell, Monocyte) DPEP3 (Monocyte, Neutrophil) PRMT7 (T cell, B cell, NK cell, Monocyte) DPRP2 (T cell, NK cell, Monocyte, DC) SLC12A4 (Neutrophil, DC) SMPD3 (B cell) FBXL8 (B cell) DDX28 (T cell) PSKH1 (B cell) | European | [44] | |
| 16q24.1 | IRF8 | rs11117432 | 2.82 × 10−24 | - | - | European | [35] | |
| 17 | 17q12 | IKZF3 | rs71152606 | 8.36 × 10−44 | - | IKZF3 (T cell, NK cell) ORMDL3 (T cell, B cell, DC) GSDMB (T cell, B cell, Monocyte, DC) GSDMA (T cell) ZPBP2 (B cel) PGAP3 (B cell) STARD3 (T cell, Monocyte, DC) | European, Asian | [34] |
| 17q21.31 | HROB | Chr17:44149348 A > C | 8.16 × 10−11 | - | - | European | [37] | |
| 18 | 18q22.2 | CD226 | rs1808094 | 1.66 × 10−10 | DOC6 (Whole Blood, Spleen) | CD226 (T cell, B cell, NK cell, DC) DOC6 (T cell, NK cell) | European | [44] |
| 19 | 19p13.2 | TYK2 | rs2304256 | 4.43 × 10−17 | TYK2 (Whole Blood) AP1M2 (Whole Blood) | TNK2 (T cell, B cell, NK cell, Monocyte, Neutrophil, DC) PDE4A (T cell, B cell, Monocyte) ICAM3 (Monocyte, Neutrophil) CDC37 (T cell) RAVER1 (Neutrophil) | European | [37] |
| 19q13.33 | SPIB | rs3745516 | 2.65 × 10−30 | KAPSB (Whole Blood) | SPIB (B cell, DC) | European | [34] | |
| 22 | 22q13.1 | SYNGR1 | rs137687 | 2.37 × 10−23 | SYNGR1 (Whole Blood, Liver, Spleen) | SYNGR1 (T cell, B cell, NK cell, Monocyte, DC) | European | [35] |
| X | Xp11.23 | GRIPAP1 | rs7059064 | 6.20 × 10−9 | PCSK1N (Whole Blood) KCND1 (Whole Blood) | - | Asian | [45] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hitomi, Y.; Nakamura, M. The Genetics of Primary Biliary Cholangitis: A GWAS and Post-GWAS Update. Genes 2023, 14, 405. https://doi.org/10.3390/genes14020405
Hitomi Y, Nakamura M. The Genetics of Primary Biliary Cholangitis: A GWAS and Post-GWAS Update. Genes. 2023; 14(2):405. https://doi.org/10.3390/genes14020405
Chicago/Turabian StyleHitomi, Yuki, and Minoru Nakamura. 2023. "The Genetics of Primary Biliary Cholangitis: A GWAS and Post-GWAS Update" Genes 14, no. 2: 405. https://doi.org/10.3390/genes14020405
APA StyleHitomi, Y., & Nakamura, M. (2023). The Genetics of Primary Biliary Cholangitis: A GWAS and Post-GWAS Update. Genes, 14(2), 405. https://doi.org/10.3390/genes14020405