
Assessing the Expression of Long INterspersed Elements (LINEs) via Long-Read Sequencing in Diverse Human Tissues and Cell Lines


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
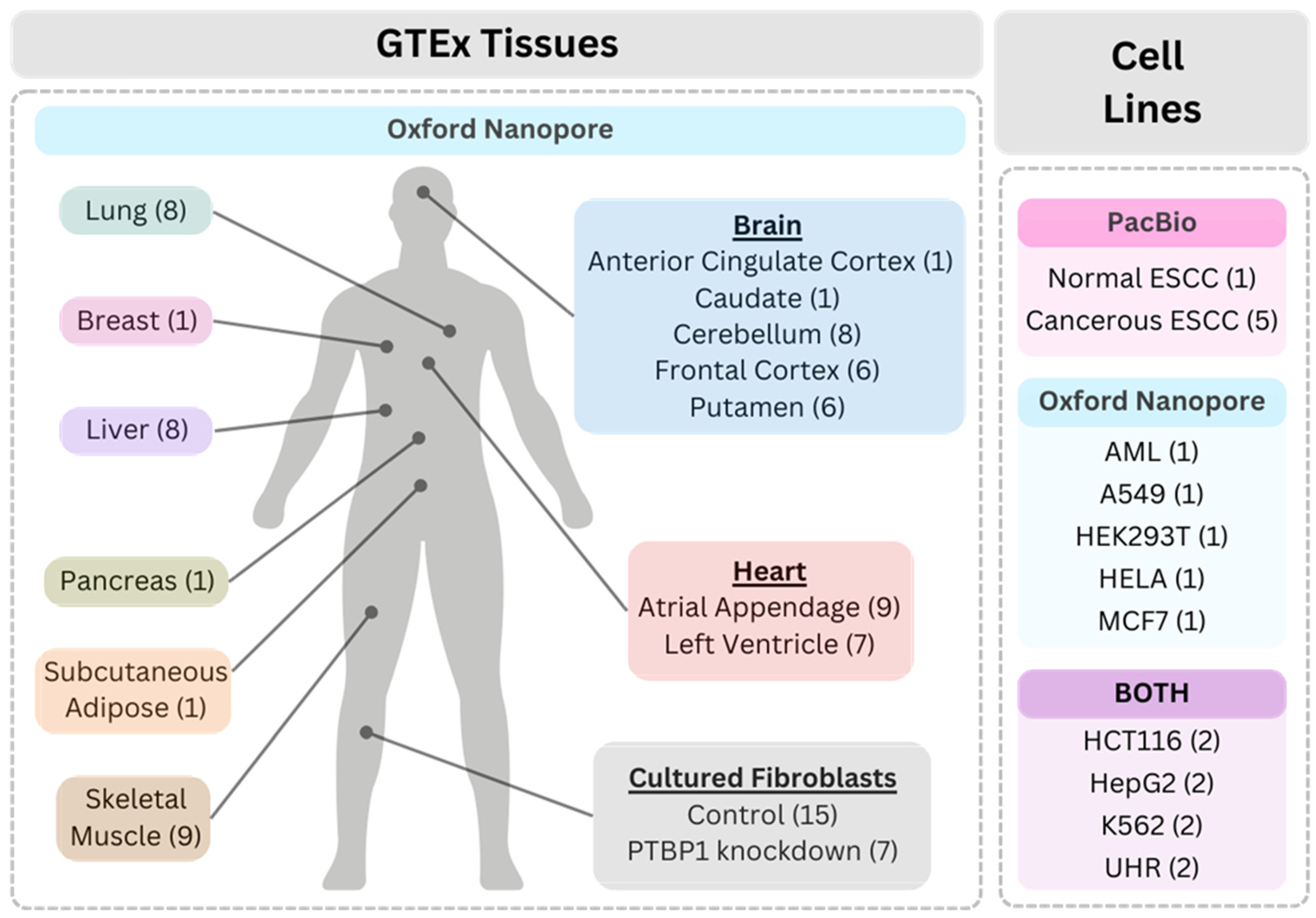
2.1. Datasets
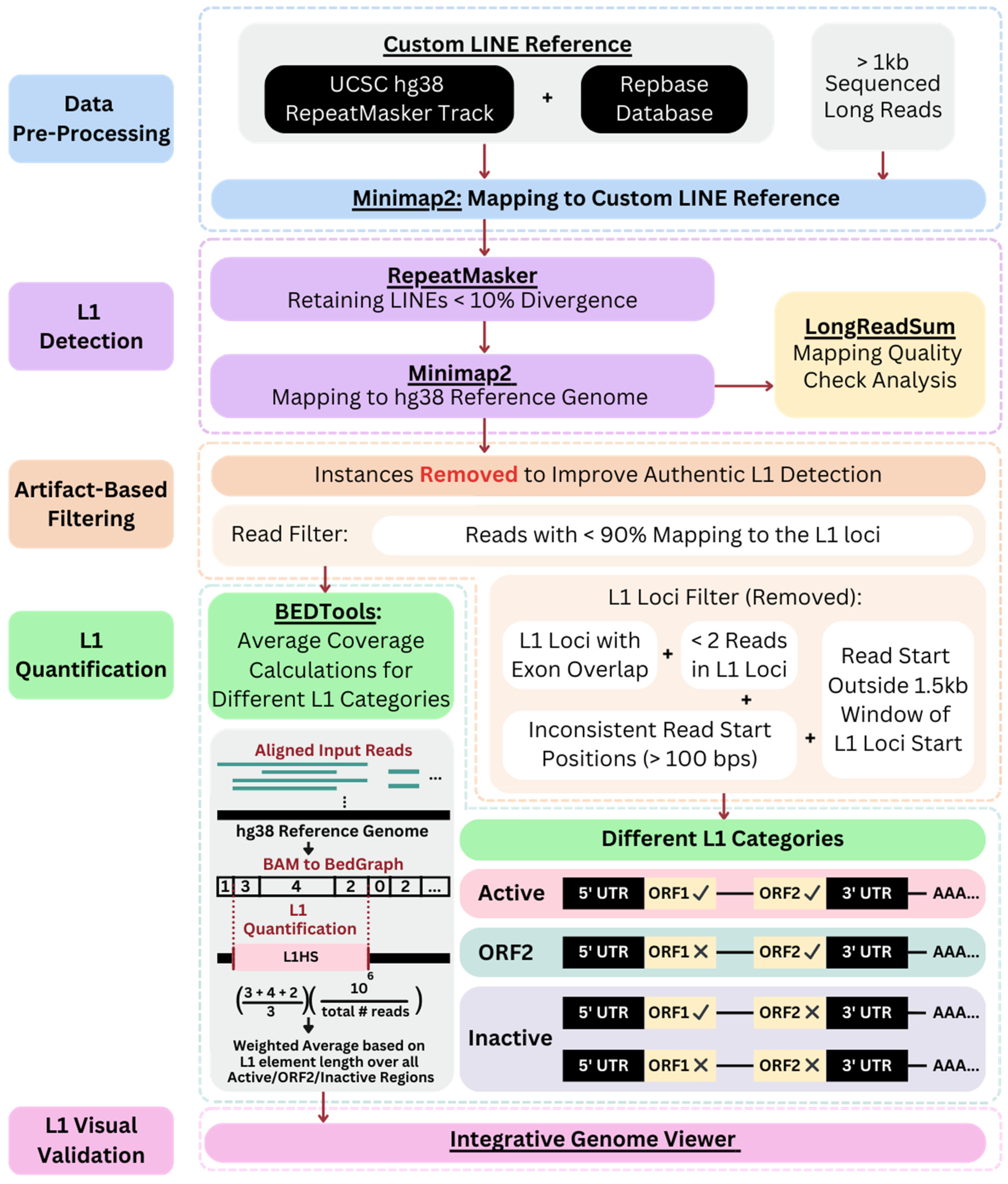
2.2. Data Preprocessing and Analysis Methods
3. Results and Discussion
3.1. General Description of the Datasets
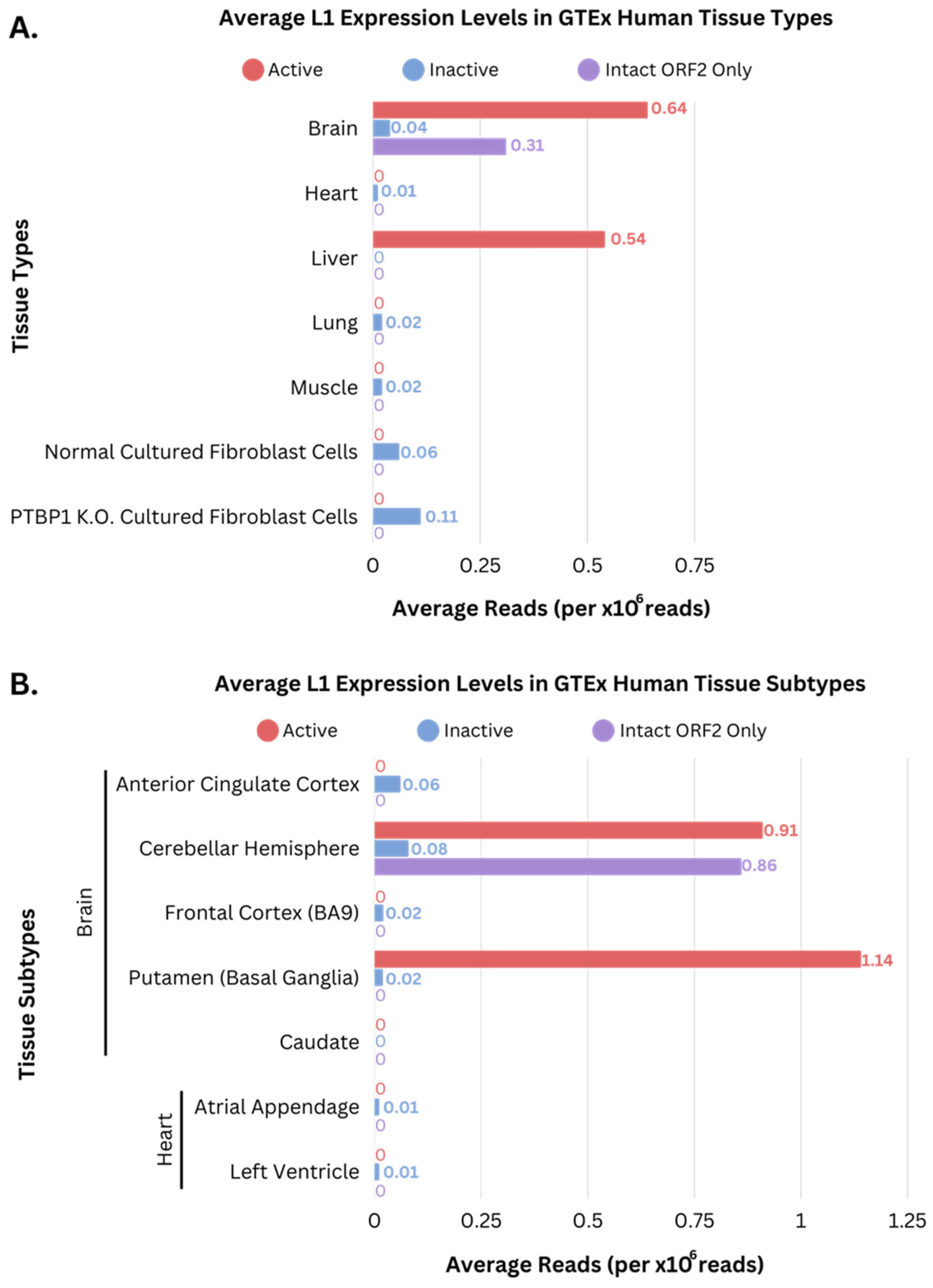
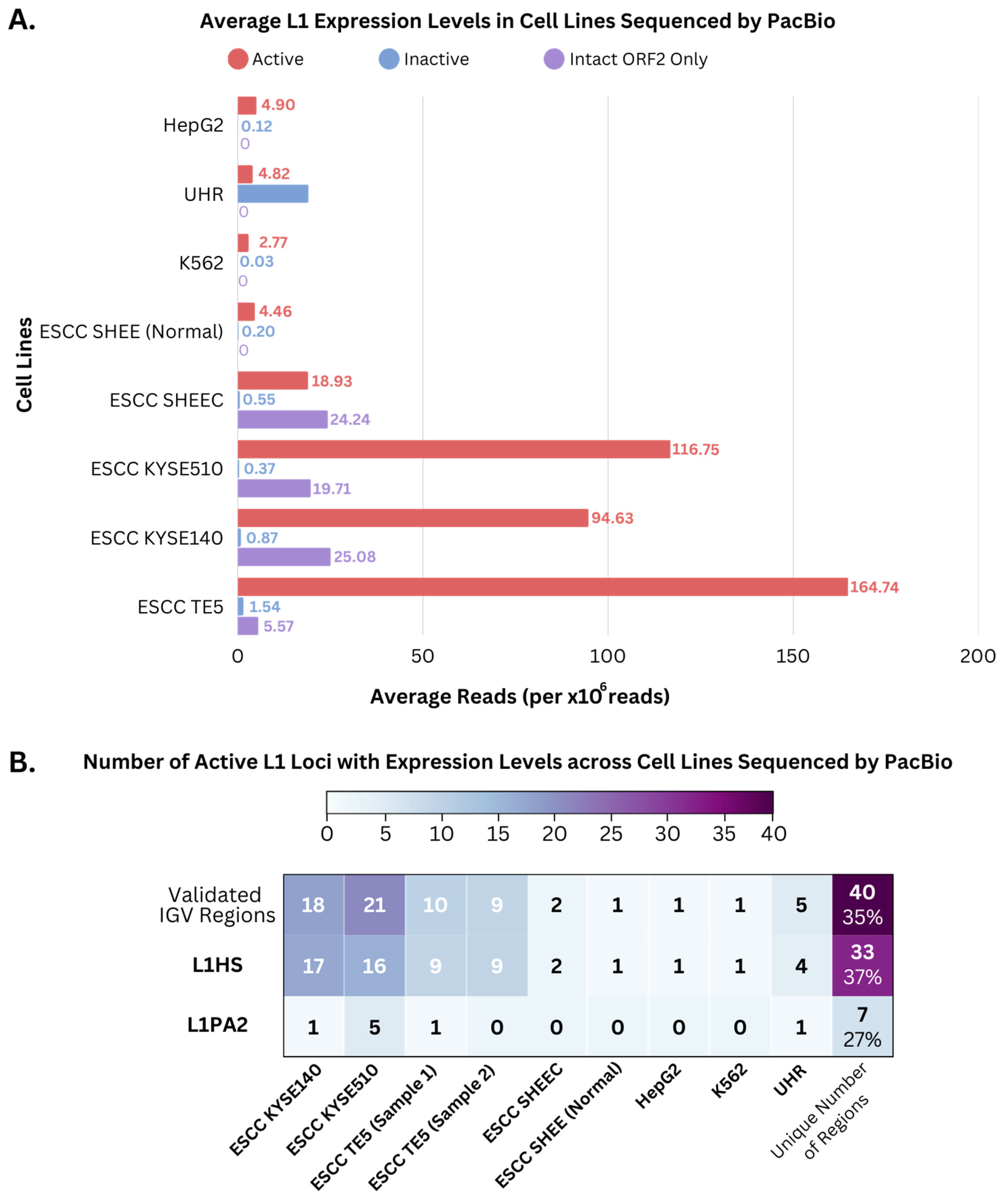
3.2. Overall Expression Levels of L1s
3.3. Expression Levels within L1 Regions with High Coverage and Manual Evaluation in IGV
3.4. Diverse Sequencing Statistics and Their Differential Impact on L1 Profiling
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- McClintock, B. Controlling elements and the gene. Cold Spring Harb. Symp. Quant. Biol. 1956, 21, 197–216. [Google Scholar] [CrossRef] [PubMed]
- Hayward, A. Origin of the retroviruses: When, where, and how? Curr. Opin. Virol. 2017, 25, 23–27. [Google Scholar] [CrossRef] [PubMed]
- Bourque, G.; Burns, K.H.; Gehring, M.; Gorbunova, V.; Seluanov, A.; Hammell, M.; Imbeault, M.; Izsvák, Z.; Levin, H.L.; Macfarlan, T.S. Ten things you should know about transposable elements. Genome Biol. 2018, 19, 199. [Google Scholar] [CrossRef]
- Sotero-Caio, C.G.; Platt, R.N., 2nd; Suh, A.; Ray, D.A. Evolution and Diversity of Transposable Elements in Vertebrate Genomes. Genome Biol. Evol. 2017, 9, 161–177. [Google Scholar] [CrossRef] [PubMed]
- Lander, E.S.; Linton, L.M.; Birren, B.; Nusbaum, C.; Zody, M.C.; Baldwin, J.; Devon, K.; Dewar, K.; Doyle, M.; FitzHugh, W.; et al. Initial sequencing and analysis of the human genome. Nature 2001, 409, 860–921. [Google Scholar] [CrossRef] [PubMed]
- Cordaux, R.; Batzer, M.A. The impact of retrotransposons on human genome evolution. Nat. Rev. Genet. 2009, 10, 691–703. [Google Scholar] [CrossRef] [PubMed]
- Kazazian, H.H., Jr.; Wong, C.; Youssoufian, H.; Scott, A.F.; Phillips, D.G.; Antonarakis, S.E. Haemophilia A resulting from de novo insertion of L1 sequences represents a novel mechanism for mutation in man. Nature 1988, 332, 164–166. [Google Scholar] [CrossRef]
- Nekrutenko, A.; Li, W.-H. Transposable elements are found in a large number of human protein-coding genes. Trends Genet. 2001, 17, 619–621. [Google Scholar] [CrossRef]
- Xing, J.; Wang, H.; Belancio, V.P.; Cordaux, R.; Deininger, P.L.; Batzer, M.A. Emergence of primate genes by retrotransposon-mediated sequence transduction. Proc. Natl. Acad. Sci. USA 2006, 103, 17608–17613. [Google Scholar] [CrossRef]
- Garcia-Perez, J.L.; Widmann, T.J.; Adams, I.R. The impact of transposable elements on mammalian development. Development 2016, 143, 4101–4114. [Google Scholar] [CrossRef]
- Garza, R.; Atacho, D.; Adami, A.; Gerdes, P.; Vinod, M.; Hsieh, P.; Karlsson, O.; Horvath, V.; Johansson, P.A.; Pandiloski, N. L1 retrotransposons drive human neuronal transcriptome complexity and functional diversification. bioRxiv 2023. bioRxiv:2004.531072. [Google Scholar]
- Ravel-Godreuil, C.; Znaidi, R.; Bonnifet, T.; Joshi, R.L.; Fuchs, J. Transposable elements as new players in neurodegenerative diseases. FEBS Lett. 2021, 595, 2733–2755. [Google Scholar] [CrossRef]
- Kazazian, H.H., Jr.; Moran, J.V. Mobile DNA in Health and Disease. N. Engl. J. Med. 2017, 377, 361–370. [Google Scholar] [CrossRef]
- Lanciano, S.; Cristofari, G. Measuring and interpreting transposable element expression. Nat. Rev. Genet. 2020, 21, 721–736. [Google Scholar] [CrossRef]
- Xing, J.; Zhang, Y.; Han, K.; Salem, A.H.; Sen, S.K.; Huff, C.D.; Zhou, Q.; Kirkness, E.F.; Levy, S.; Batzer, M.A.; et al. Mobile elements create structural variation: Analysis of a complete human genome. Genome Res. 2009, 19, 1516–1526. [Google Scholar] [CrossRef] [PubMed]
- Jang, H.S.; Shah, N.M.; Du, A.Y.; Dailey, Z.Z.; Pehrsson, E.C.; Godoy, P.M.; Zhang, D.; Li, D.; Xing, X.; Kim, S.; et al. Transposable elements drive widespread expression of oncogenes in human cancers. Nat. Genet. 2019, 51, 611–617. [Google Scholar] [CrossRef] [PubMed]
- Saito, K.; Kawakami, K.; Matsumoto, I.; Oda, M.; Watanabe, G.; Minamoto, T. Long Interspersed Nuclear Element 1 Hypomethylation Is a Marker of Poor Prognosis in Stage IA Non–Small Cell Lung CancerLINE-1 Methylation and Prognosis in NSCLC. Clin. Cancer Res. 2010, 16, 2418–2426. [Google Scholar] [CrossRef]
- van Hoesel, A.Q.; van de Velde, C.J.; Kuppen, P.J.; Liefers, G.J.; Putter, H.; Sato, Y.; Elashoff, D.A.; Turner, R.R.; Shamonki, J.M.; de Kruijf, E.M. Hypomethylation of LINE-1 in primary tumor has poor prognosis in young breast cancer patients: A retrospective cohort study. Breast Cancer Res. Treat. 2012, 134, 1103–1114. [Google Scholar] [CrossRef]
- Burns, K.H. Transposable elements in cancer. Nat. Rev. Cancer 2017, 17, 415–424. [Google Scholar] [CrossRef]
- Miki, Y.; Nishisho, I.; Horii, A.; Miyoshi, Y.; Utsunomiya, J.; Kinzler, K.W.; Vogelstein, B.; Nakamura, Y. Disruption of the APC gene by a retrotransposal insertion of L1 sequence in a colon cancer. Cancer Res. 1992, 52, 643–645. [Google Scholar]
- Beck, C.R.; Collier, P.; Macfarlane, C.; Malig, M.; Kidd, J.M.; Eichler, E.E.; Badge, R.M.; Moran, J.V. LINE-1 retrotransposition activity in human genomes. Cell 2010, 141, 1159–1170. [Google Scholar] [CrossRef] [PubMed]
- Brouha, B.; Schustak, J.; Badge, R.M.; Lutz-Prigge, S.; Farley, A.H.; Moran, J.V.; Kazazian, H.H., Jr. Hot L1s account for the bulk of retrotransposition in the human population. Proc. Natl. Acad. Sci. USA 2003, 100, 5280–5285. [Google Scholar] [CrossRef]
- Mills, R.E.; Bennett, E.A.; Iskow, R.C.; Devine, S.E. Which transposable elements are active in the human genome? Trends Genet. 2007, 23, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Makałowski, W.; Gotea, V.; Pande, A.; Makałowska, I. Transposable Elements: Classification, Identification, and Their Use as a Tool for Comparative Genomics; Springer: New York, NY, USA, 2019. [Google Scholar]
- Perepelitsa-Belancio, V.; Deininger, P. RNA truncation by premature polyadenylation attenuates human mobile element activity. Nat. Genet. 2003, 35, 363–366. [Google Scholar] [CrossRef] [PubMed]
- Deininger, P.; Morales, M.E.; White, T.B.; Baddoo, M.; Hedges, D.J.; Servant, G.; Srivastav, S.; Smither, M.E.; Concha, M.; DeHaro, D.L.; et al. A comprehensive approach to expression of L1 loci. Nucleic Acids Res. 2017, 45, e31. [Google Scholar] [CrossRef]
- Sultana, T.; van Essen, D.; Siol, O.; Bailly-Bechet, M.; Philippe, C.; El Aabidine, A.Z.; Pioger, L.; Nigumann, P.; Saccani, S.; Andrau, J.-C. The landscape of L1 retrotransposons in the human genome is shaped by pre-insertion sequence biases and post-insertion selection. Mol. Cell 2019, 74, 555–570.e557. [Google Scholar] [CrossRef]
- Ostertag, E.M.; DeBerardinis, R.J.; Goodier, J.L.; Zhang, Y.; Yang, N.; Gerton, G.L.; Kazazian, H.H. A mouse model of human L1 retrotransposition. Nat. Genet. 2002, 32, 655–660. [Google Scholar] [CrossRef]
- Faulkner, G.J.; Kimura, Y.; Daub, C.O.; Wani, S.; Plessy, C.; Irvine, K.M.; Schroder, K.; Cloonan, N.; Steptoe, A.L.; Lassmann, T. The regulated retrotransposon transcriptome of mammalian cells. Nat. Genet. 2009, 41, 563–571. [Google Scholar] [CrossRef]
- Nam, C.H.; Youk, J.; Kim, J.Y.; Lim, J.; Park, J.W.; Oh, S.A.; Lee, H.J.; Park, J.W.; Won, H.; Lee, Y. Widespread somatic L1 retrotransposition in normal colorectal epithelium. Nature 2023, 617, 540–547. [Google Scholar] [CrossRef]
- Lee, E.; Iskow, R.; Yang, L.; Gokcumen, O.; Haseley, P.; Luquette, L.J., III; Lohr, J.G.; Harris, C.C.; Ding, L.; Wilson, R.K. Landscape of somatic retrotransposition in human cancers. Science 2012, 337, 967–971. [Google Scholar] [CrossRef]
- Smit, A.; Hubley, R.; Green, P. RepeatMasker Open-4.0. Available online: http://www.repeatmasker.org (accessed on 1 March 2023).
- Penzkofer, T.; Jäger, M.; Figlerowicz, M.; Badge, R.; Mundlos, S.; Robinson, P.N.; Zemojtel, T. L1Base 2: More retrotransposition-active LINE-1s, more mammalian genomes. Nucleic Acids Res. 2016, 45, D68–D73. [Google Scholar] [CrossRef] [PubMed]
- Glinos, D.A.; Garborcauskas, G.; Hoffman, P.; Ehsan, N.; Jiang, L.; Gokden, A.; Dai, X.; Aguet, F.; Brown, K.L.; Garimella, K. Transcriptome variation in human tissues revealed by long-read sequencing. Nature 2022, 608, 353–359. [Google Scholar] [CrossRef] [PubMed]
- Novoradovskaya, N.; Payette, T.; Novoradovsky, A.; Braman, J.; Chin, N.; Pergamenschikov, A.; Fero, M.; Botstein, D. Pooled, high-quality reference RNA for human microarrays. Strategies 2000, 13, 121–122. [Google Scholar]
- Chen, Y.; Davidson, N.M.; Wan, Y.K.; Patel, H.; Yao, F.; Low, H.M.; Hendra, C.; Watten, L.; Sim, A.; Sawyer, C. A systematic benchmark of Nanopore long read RNA sequencing for transcript level analysis in human cell lines. bioRxiv 2021. bioRxiv:2021.440736. [Google Scholar]
- Graham, F.L.; Smiley, J.; Russell, W.; Nairn, R. Characteristics of a human cell line transformed by DNA from human adenovirus type 5. J. Gen. Virol. 1977, 36, 59–72. [Google Scholar] [CrossRef] [PubMed]
- Tavakoli, S.; Nabizadeh, M.; Makhamreh, A.; Gamper, H.; McCormick, C.A.; Rezapour, N.K.; Hou, Y.-M.; Wanunu, M.; Rouhanifard, S.H. Semi-quantitative detection of pseudouridine modifications and type I/II hypermodifications in human mRNAs using direct long-read sequencing. Nat. Commun. 2023, 14, 334. [Google Scholar] [CrossRef]
- De Kouchkovsky, I.; Abdul-Hay, M. Acute myeloid leukemia: A comprehensive review and 2016 update. Blood Cancer J. 2016, 6, e441. [Google Scholar] [CrossRef]
- UHR_IsoSeq. Pacbcloud. Available online: https://downloads.pacbcloud.com/public/dataset/UHR_IsoSeq/ (accessed on 2 June 2023).
- Cheng, Y.-W.; Chen, Y.-M.; Zhao, Q.-Q.; Zhao, X.; Wu, Y.-R.; Chen, D.-Z.; Liao, L.-D.; Chen, Y.; Yang, Q.; Xu, L.-Y. Long read single-molecule real-time sequencing elucidates transcriptome-wide heterogeneity and complexity in esophageal squamous cells. Front. Genet. 2019, 10, 915. [Google Scholar] [CrossRef]
- Oxford Nanopore Technologies. Oxford Nanopore Technologies Guppy6 Basecaller. Available online: https://community.nanoporetech.com (accessed on 1 March 2023).
- Karolchik, D.; Hinrichs, A.S.; Furey, T.S.; Roskin, K.M.; Sugnet, C.W.; Haussler, D.; Kent, W.J. The UCSC Table Browser data retrieval tool. Nucleic Acids Res. 2004, 32, D493–D496. [Google Scholar] [CrossRef]
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef]
- Frankish, A.; Diekhans, M.; Jungreis, I.; Lagarde, J.; Loveland, J.E.; Mudge, J.M.; Sisu, C.; Wright, J.C.; Armstrong, J.; Barnes, I. GENCODE 2021. Nucleic Acids Res. 2021, 49, D916–D923. [Google Scholar] [CrossRef] [PubMed]
- Perdomo, J.E.; Ahsan, M.U.; Liu, Q.; Fang, L.; Wang, K. LongReadSum: A fast and flexible quality control tool for long-read sequencing data. In Proceedings of the Poster Presented at: American Society of Human Genetics (ASHG) Annual Meeting, Los Angeles, CA, USA, 25–29 October 2022. [Google Scholar]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.T.; Thorvaldsdóttir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genomics viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef]
- Sudmant, P.H.; Rausch, T.; Gardner, E.J.; Handsaker, R.E.; Abyzov, A.; Huddleston, J.; Zhang, Y.; Ye, K.; Jun, G.; Hsi-Yang Fritz, M. An integrated map of structural variation in 2,504 human genomes. Nature 2015, 526, 75–81. [Google Scholar] [CrossRef]
- Navarro, F.C.; Hoops, J.; Bellfy, L.; Cerveira, E.; Zhu, Q.; Zhang, C.; Lee, C.; Gerstein, M.B. TeXP: Deconvolving the effects of pervasive and autonomous transcription of transposable elements. PLoS Comput. Biol. 2019, 15, e1007293. [Google Scholar] [CrossRef]
- Ponomaryova, A.A.; Rykova, E.Y.; Gervas, P.A.; Cherdyntseva, N.V.; Mamedov, I.Z.; Azhikina, T.L. Aberrant methylation of LINE-1 transposable elements: A search for cancer biomarkers. Cells 2020, 9, 2017. [Google Scholar] [CrossRef] [PubMed]
- Iwagami, S.; Baba, Y.; Watanabe, M.; Shigaki, H.; Miyake, K.; Ishimoto, T.; Iwatsuki, M.; Sakamaki, K.; Ohashi, Y.; Baba, H. LINE-1 hypomethylation is associated with a poor prognosis among patients with curatively resected esophageal squamous cell carcinoma. Ann. Surg. 2013, 257, 449–455. [Google Scholar] [CrossRef]
- Erwin, J.A.; Paquola, A.C.; Singer, T.; Gallina, I.; Novotny, M.; Quayle, C.; Bedrosian, T.A.; Alves, F.I.; Butcher, C.R.; Herdy, J.R. L1-associated genomic regions are deleted in somatic cells of the healthy human brain. Nat. Neurosci. 2016, 19, 1583–1591. [Google Scholar] [CrossRef]
- Suarez, N.A.; Macia, A.; Muotri, A.R. LINE-1 retrotransposons in healthy and diseased human brain. Dev. Neurobiol. 2018, 78, 434–455. [Google Scholar] [CrossRef]
- Coufal, N.G.; Garcia-Perez, J.L.; Peng, G.E.; Yeo, G.W.; Mu, Y.; Lovci, M.T.; Morell, M.; O’Shea, K.S.; Moran, J.V.; Gage, F.H. L1 retrotransposition in human neural progenitor cells. Nature 2009, 460, 1127–1131. [Google Scholar] [CrossRef]
- Capy, P.; Gasperi, G.; Biémont, C.; Bazin, C. Stress and transposable elements: Co-evolution or useful parasites? Heredity 2000, 85, 101–106. [Google Scholar] [CrossRef] [PubMed]
- Horváth, V.; Merenciano, M.; González, J. Revisiting the relationship between transposable elements and the eukaryotic stress response. Trends Genet. 2017, 33, 832–841. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.D.; Heller, E.A. Convergent actions of stress and stimulants via epigenetic regulation of neural circuitry. Trends Neurosci. 2022, 45, 955–967. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhao, Y.; Bollas, A.; Wang, Y.; Au, K.F. Nanopore sequencing technology, bioinformatics and applications. Nat. Biotechnol. 2021, 39, 1348–1365. [Google Scholar] [CrossRef] [PubMed]
- Pradhan, R.K.; Ramakrishna, W. Transposons: Unexpected players in cancer. Gene 2022, 808, 145975. [Google Scholar] [CrossRef]
- Philippe, C.; Vargas-Landin, D.B.; Doucet, A.J.; van Essen, D.; Vera-Otarola, J.; Kuciak, M.; Corbin, A.; Nigumann, P.; Cristofari, G. Activation of individual L1 retrotransposon instances is restricted to cell-type dependent permissive loci. Elife 2016, 5, e13926. [Google Scholar] [CrossRef]
- Miret, N.; Zappia, C.D.; Altamirano, G.; Pontillo, C.; Zárate, L.; Gómez, A.; Lasagna, M.; Cocca, C.; Kass, L.; Monczor, F. AhR ligands reactivate LINE-1 retrotransposon in triple-negative breast cancer cells MDA-MB-231 and non-tumorigenic mammary epithelial cells NMuMG. Biochem. Pharmacol. 2020, 175, 113904. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rybacki, K.; Xia, M.; Ahsan, M.U.; Xing, J.; Wang, K. Assessing the Expression of Long INterspersed Elements (LINEs) via Long-Read Sequencing in Diverse Human Tissues and Cell Lines. Genes 2023, 14, 1893. https://doi.org/10.3390/genes14101893
Rybacki K, Xia M, Ahsan MU, Xing J, Wang K. Assessing the Expression of Long INterspersed Elements (LINEs) via Long-Read Sequencing in Diverse Human Tissues and Cell Lines. Genes. 2023; 14(10):1893. https://doi.org/10.3390/genes14101893
Chicago/Turabian StyleRybacki, Karleena, Mingyi Xia, Mian Umair Ahsan, Jinchuan Xing, and Kai Wang. 2023. "Assessing the Expression of Long INterspersed Elements (LINEs) via Long-Read Sequencing in Diverse Human Tissues and Cell Lines" Genes 14, no. 10: 1893. https://doi.org/10.3390/genes14101893
APA StyleRybacki, K., Xia, M., Ahsan, M. U., Xing, J., & Wang, K. (2023). Assessing the Expression of Long INterspersed Elements (LINEs) via Long-Read Sequencing in Diverse Human Tissues and Cell Lines. Genes, 14(10), 1893. https://doi.org/10.3390/genes14101893