
Pacing Dynamics Determines the Arrhythmogenic Mechanism of the CPVT2-Causing CASQ2G112+5X Mutation in a Guinea Pig Ventricular Myocyte Computational Model




Abstract
1. Introduction
2. Materials and Methods
2.1. Model Development
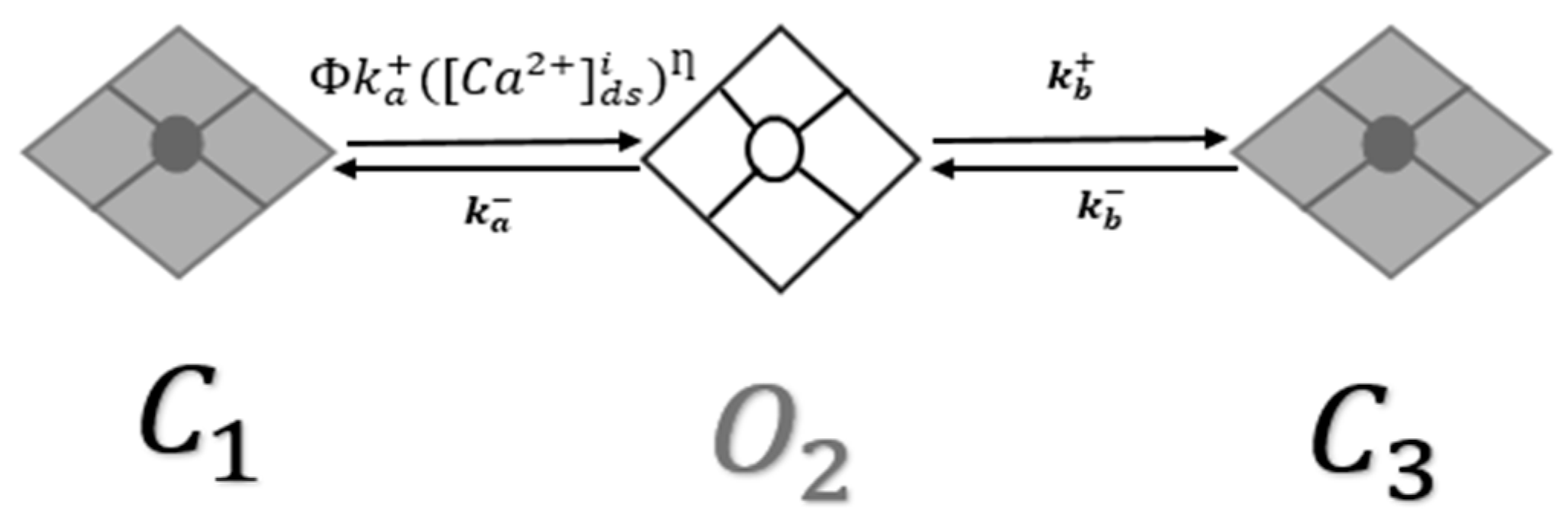
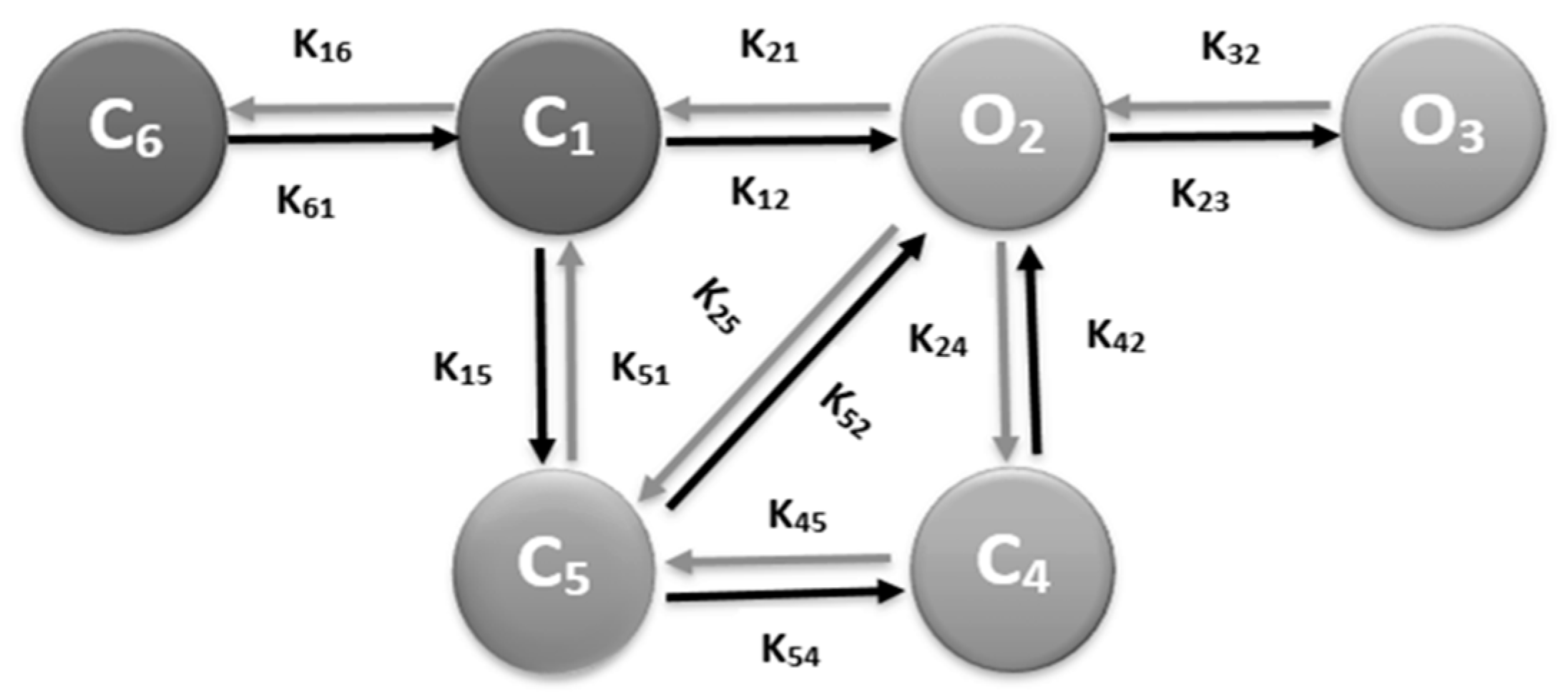
2.2. RyR2 Model
2.3. L-type Ca2+ Channel Model
2.4. Simulation Protocols
β-AR Stimulation Parameters
- (a)
- Increase in L-type channel activity: Experiments observe an amplified L-type current when the level of catecholamine increases by the activation of adrenergic receptors in the sarcolemma. An increase of 40% for L-type current (P_dhpr, the single channel permeability) replicates experimental findings. Ginsburg and Bers [33] found a 53% increase in the L-type Ca2+ channel peak value with the treatment of isoproterenol (ISO).
- (b)
- Increase in luminal dependence: CASQ2 is the major Ca2+ storage protein in the SR and its absence has a major implication in the availability of free Ca2+ in the SR luminal. Luminal sensitivity regulation function (ϕ) depends upon free Ca2+ in the SR as shown in Figure 1. Greater free Ca2+ available in the lumen enhances the opening rate of RyR2. Since there is a total reduction in Ca2+ buffer in CASQ2G112+5X, a 90% increase in luminal dependence (Kjsro) provided a steady-state Ca2+ transient by distressing free SR Ca2+ availability. Gyorke [35] reported the RyR2 open probability increased 0.26 ± 0.04 to 0.49 ± 0.09 with doubling luminal doubling SR free [Ca2+].
- (c)
- Enhanced SERCA-ATPase cycling rate (Vcycle): Increased Ca2+ in cytosol due to an increase in L-type current as well as increased RyR2 channel activity, affects the SERCA2A cycling rates, and they pump Ca2+ back to SR rapidly increasing SR load [16,36,37]. In the simulation, the whole-cell SERCA pump flux is given bywhere is cycling rate per molecule, is the concentration of SERCA molecules per liter cytosol and the unit of Ca2+ flux, and has units of mol s−1.
- (d)
- Increased SR volume: The morphometric analysis of volume fraction of SR membrane of CASQ2 knockout myocyte by Knollmann et al. found that the SR to cytoplasm volume ratio increased by ~51% while the volume related to myofibril was increased by ~45% [32]. They also found a 50% increase in the SR volume which effectively maintains the normal SR Ca2+ storage capacity.
- (e)
- Abolition of SR buffering capacity: CASQ2G115+5x deletion mutation removes the entirety of CASQ2 protein leaving no place to SR Ca2+ to buffer [15]. The amount of SR Ca2+ was reduced to 5% of control to reflect residual Ca2+ buffering in the SR.
2.5. Numerical Methods
2.6. Statistical Analysis
2.7. Model Simulation Protocols
3. Results
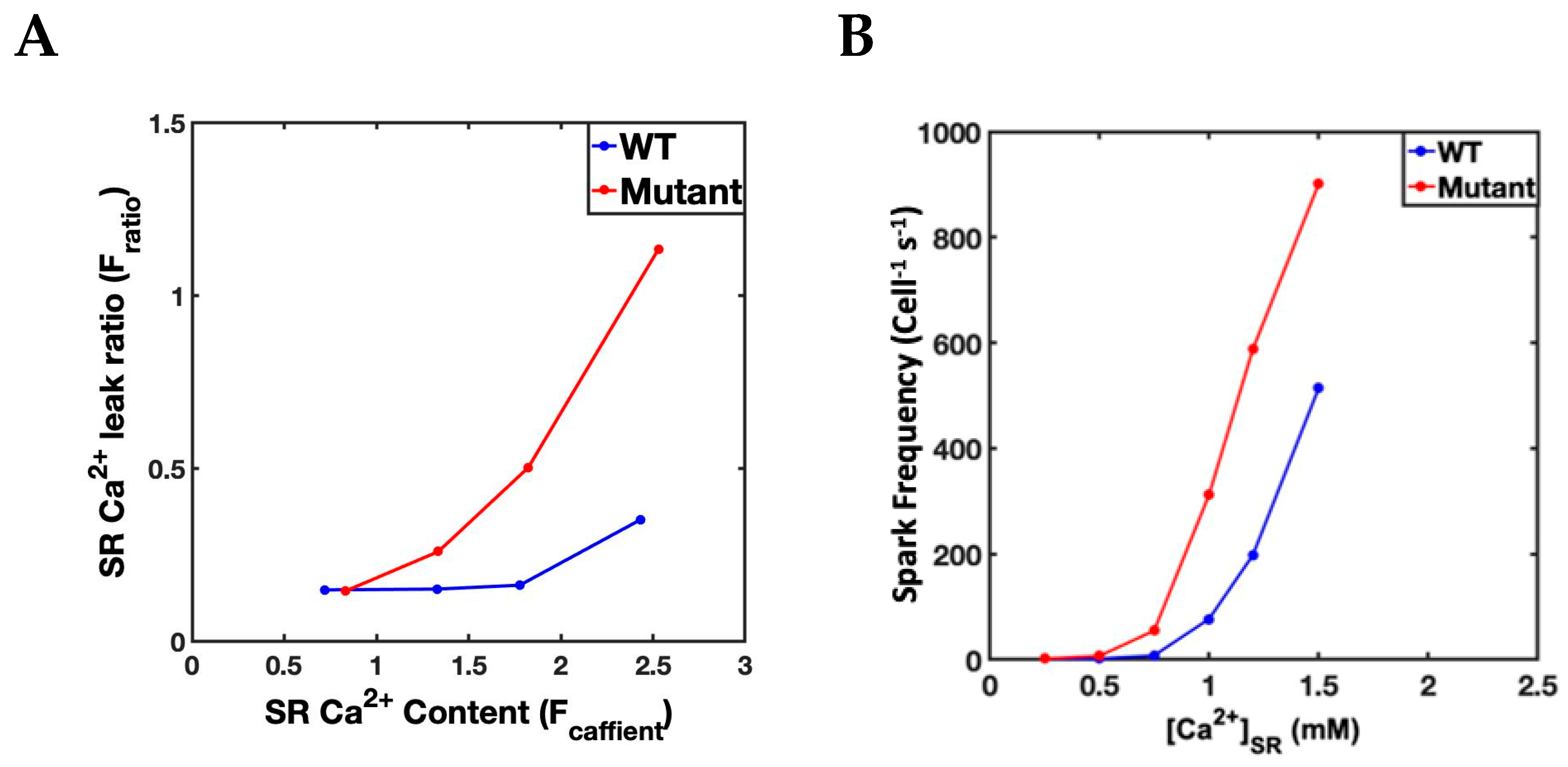
3.1. Simulation at 1 Hz
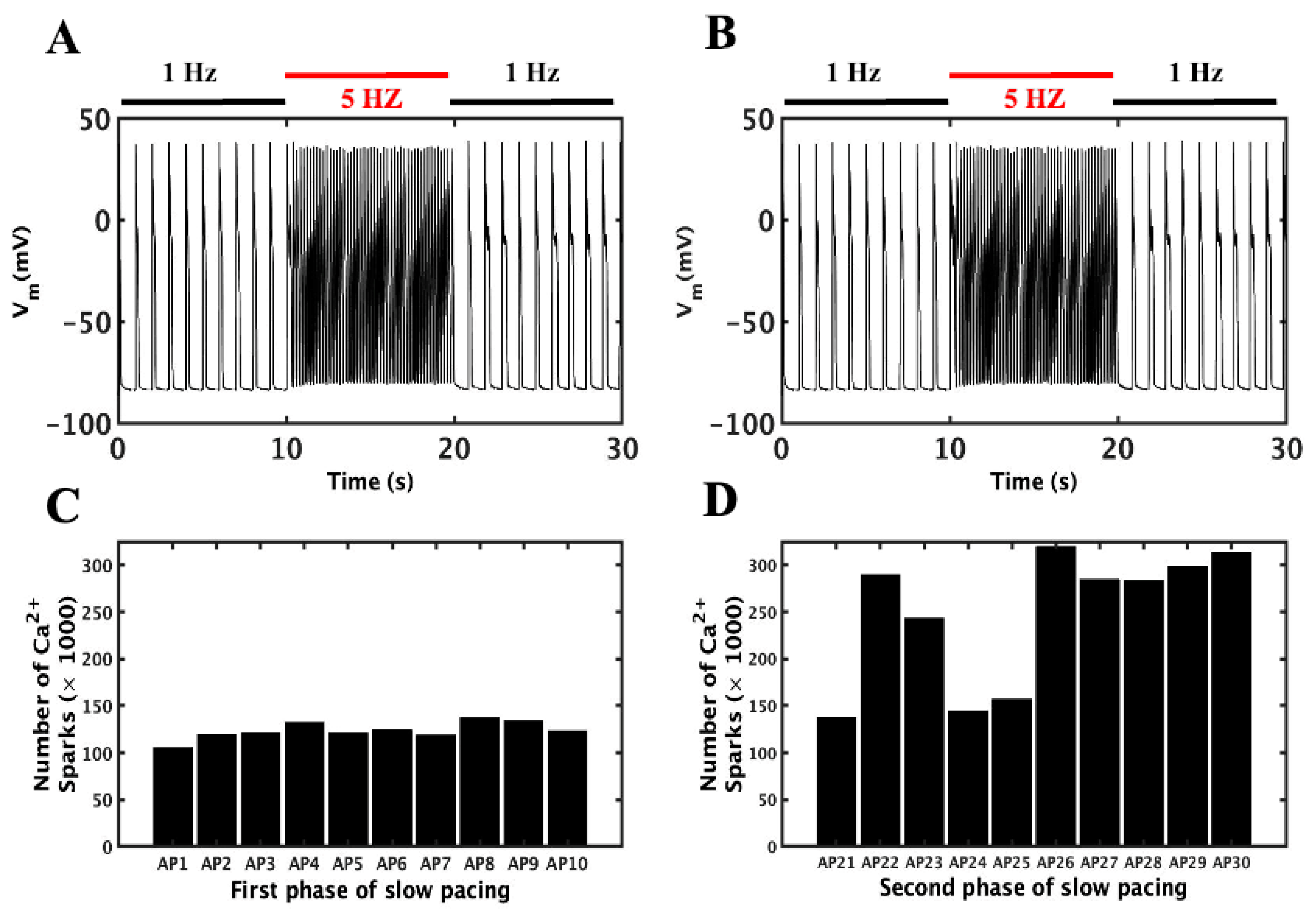
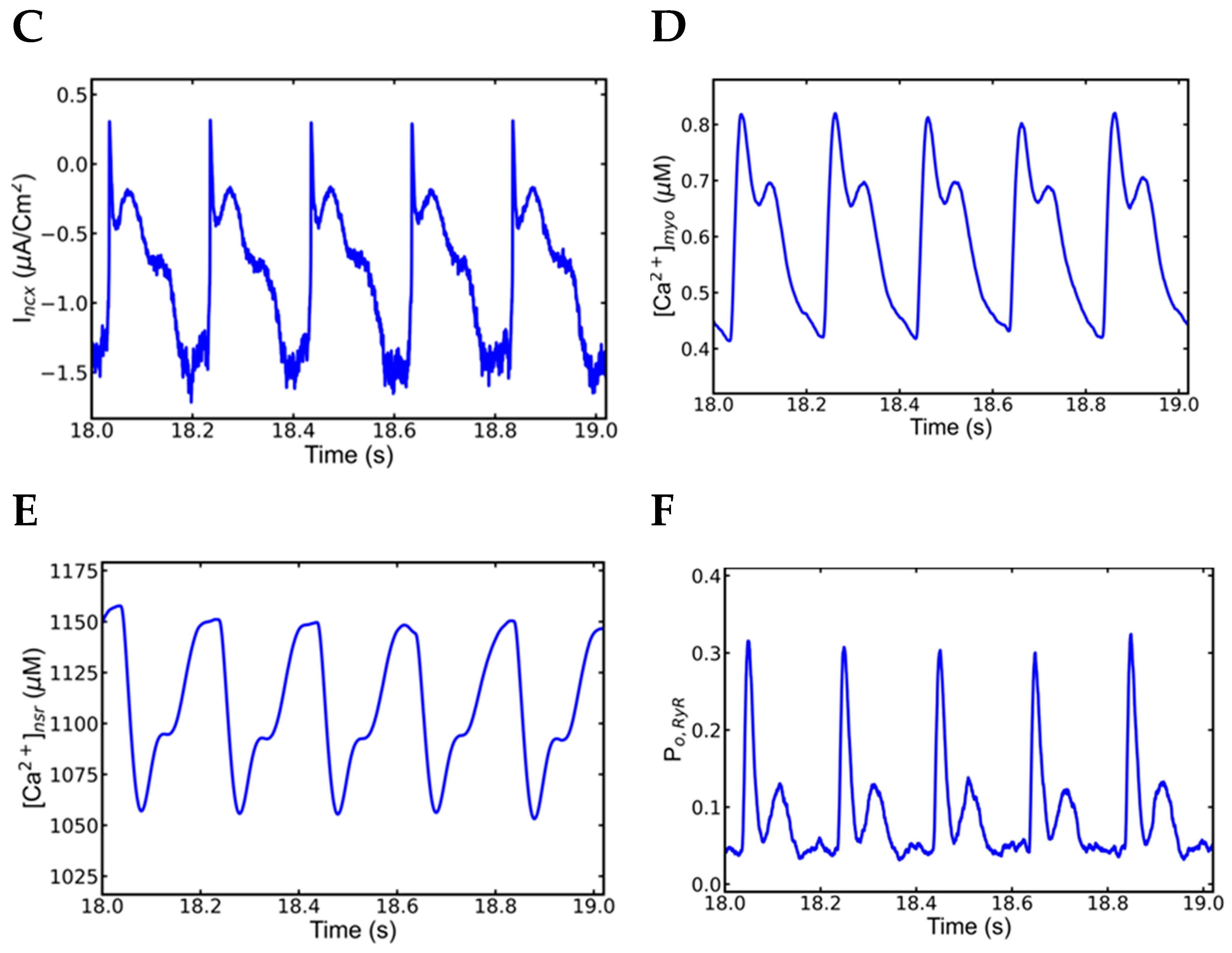
3.2. Slow–Rapid–Slow (S-R-S) Pacing Developed EADs
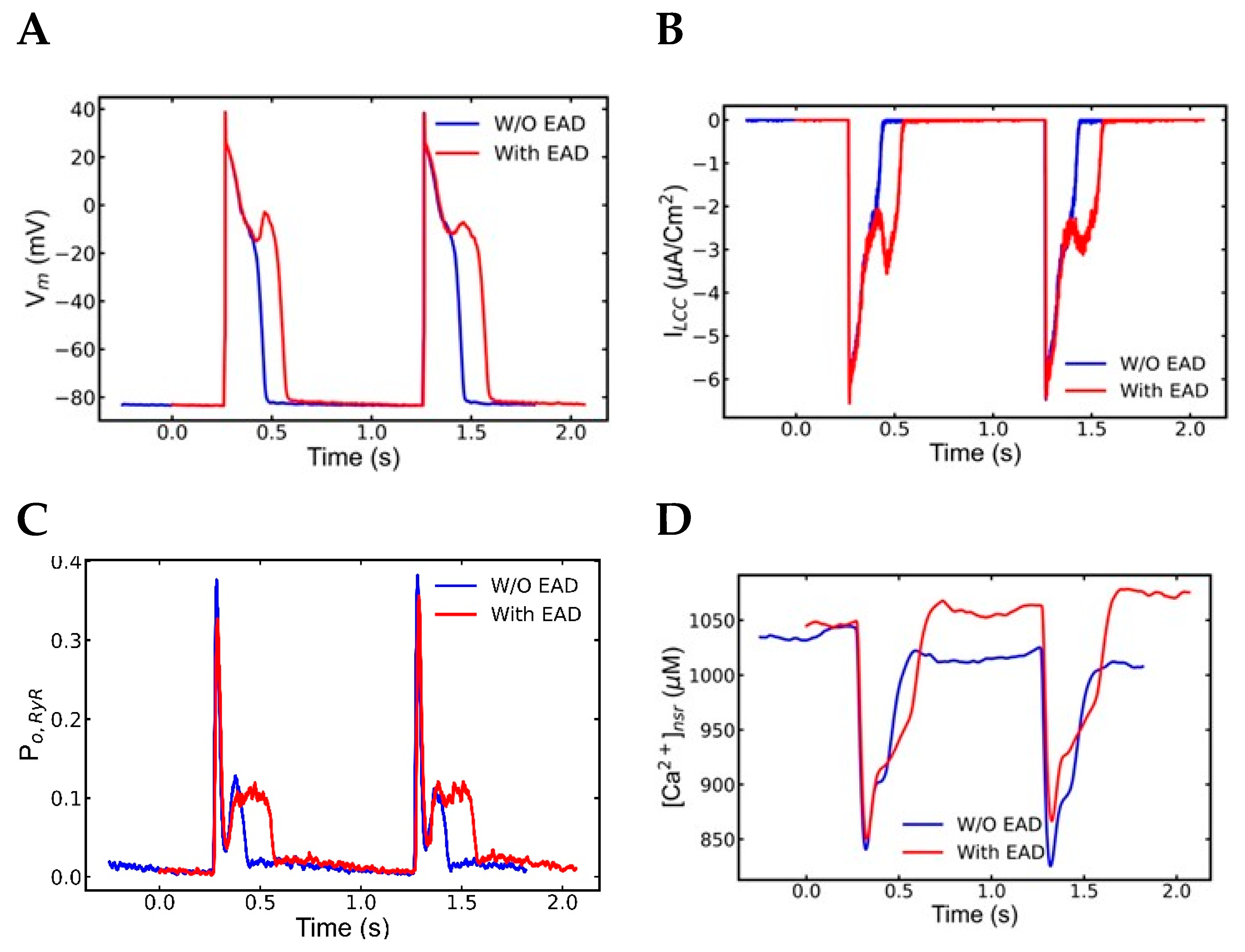
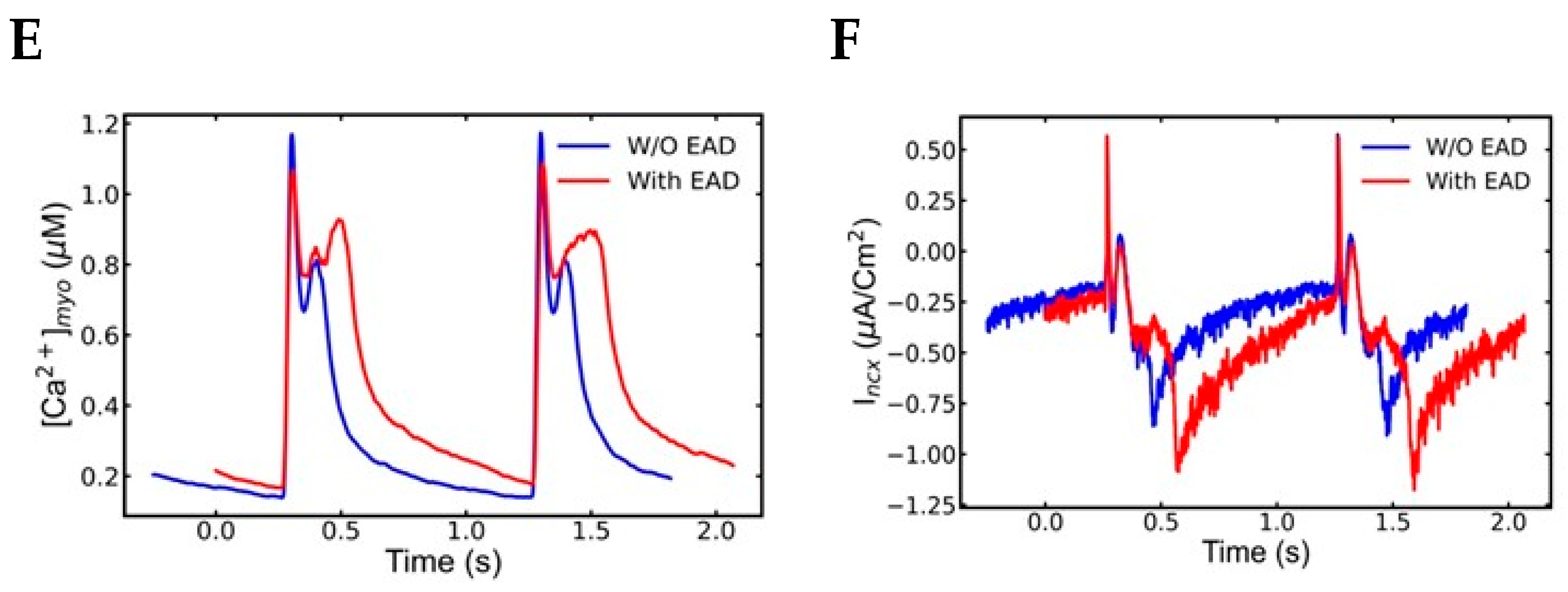
3.3. Mechanism of EADs
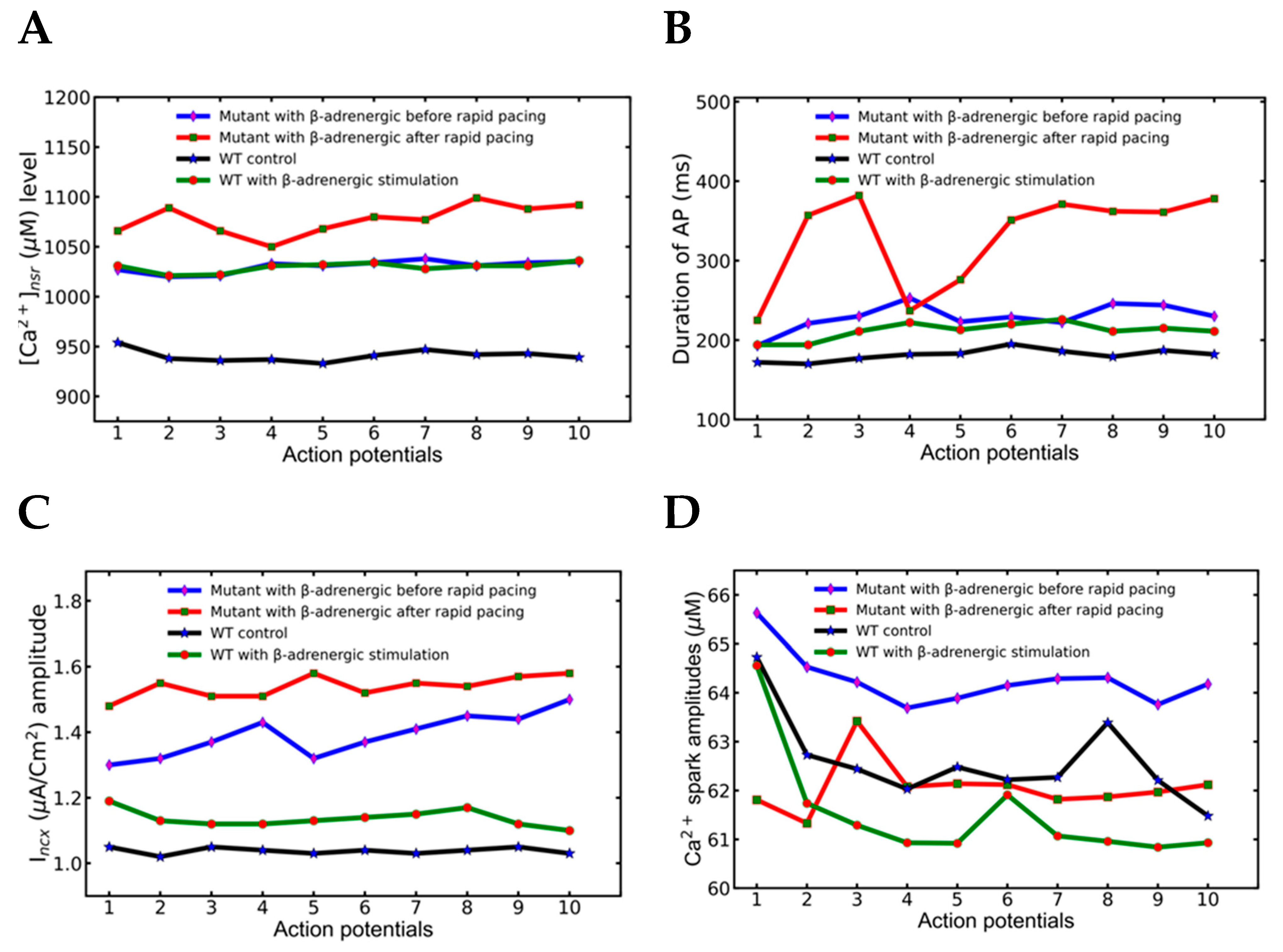
3.4. Action Potential and Ionic Currents during Rapid Pacing
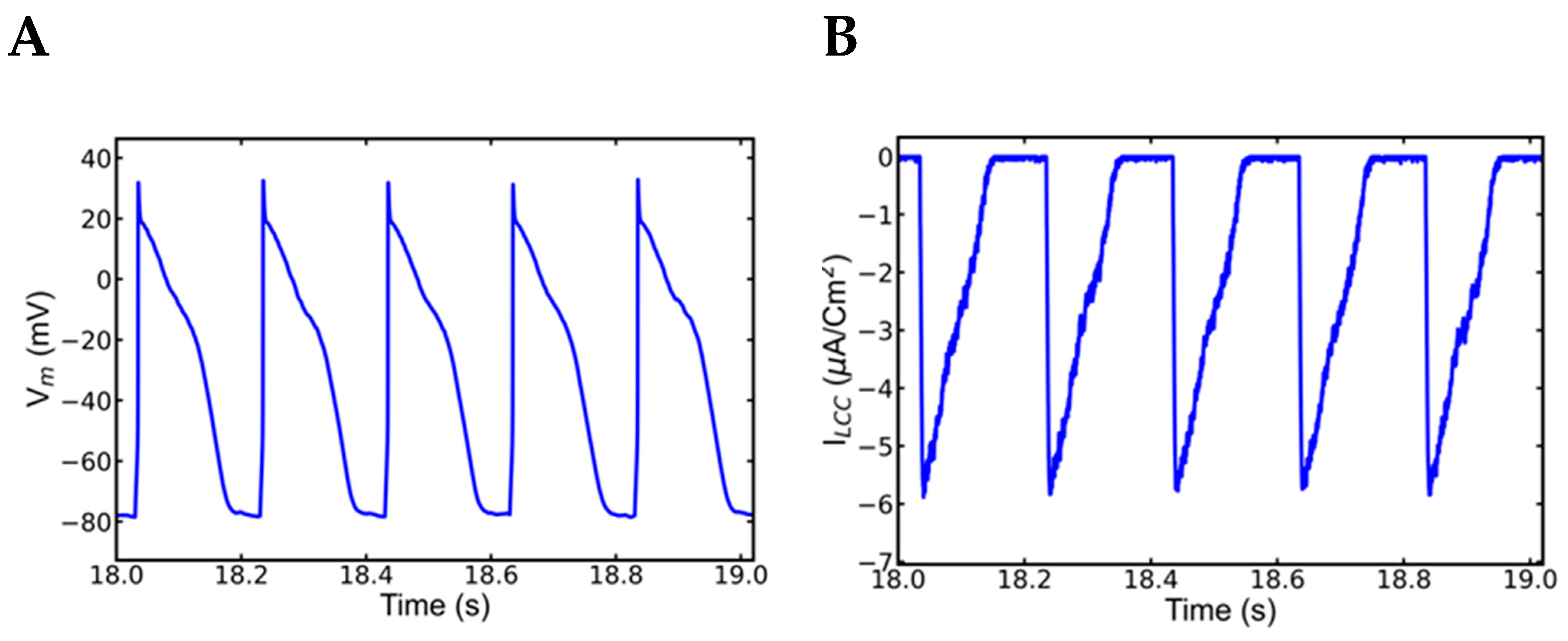
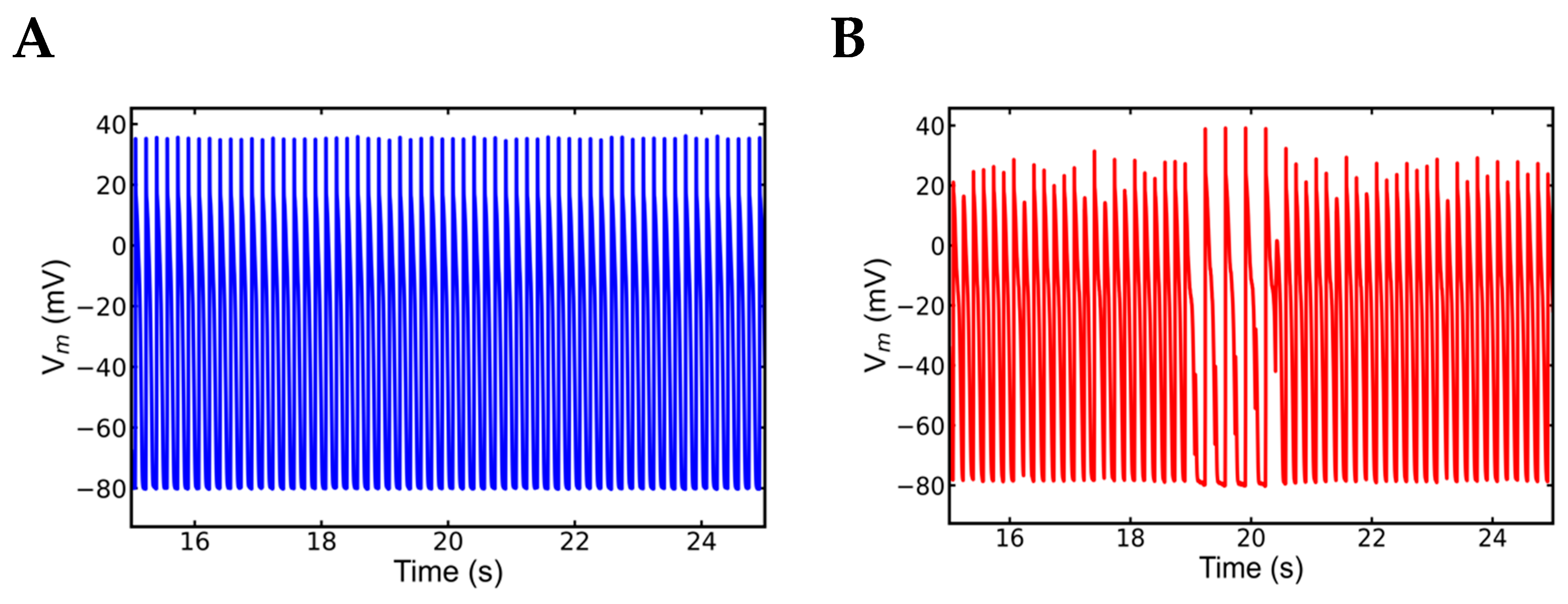
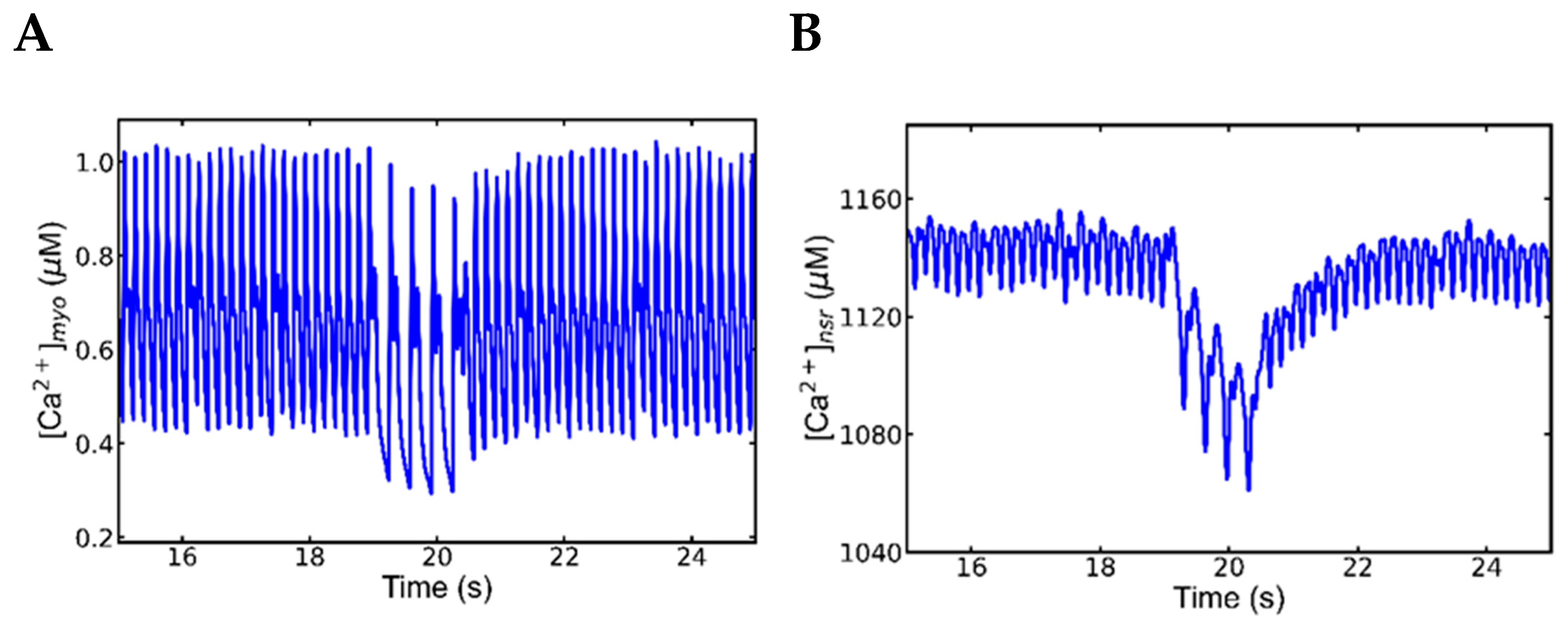
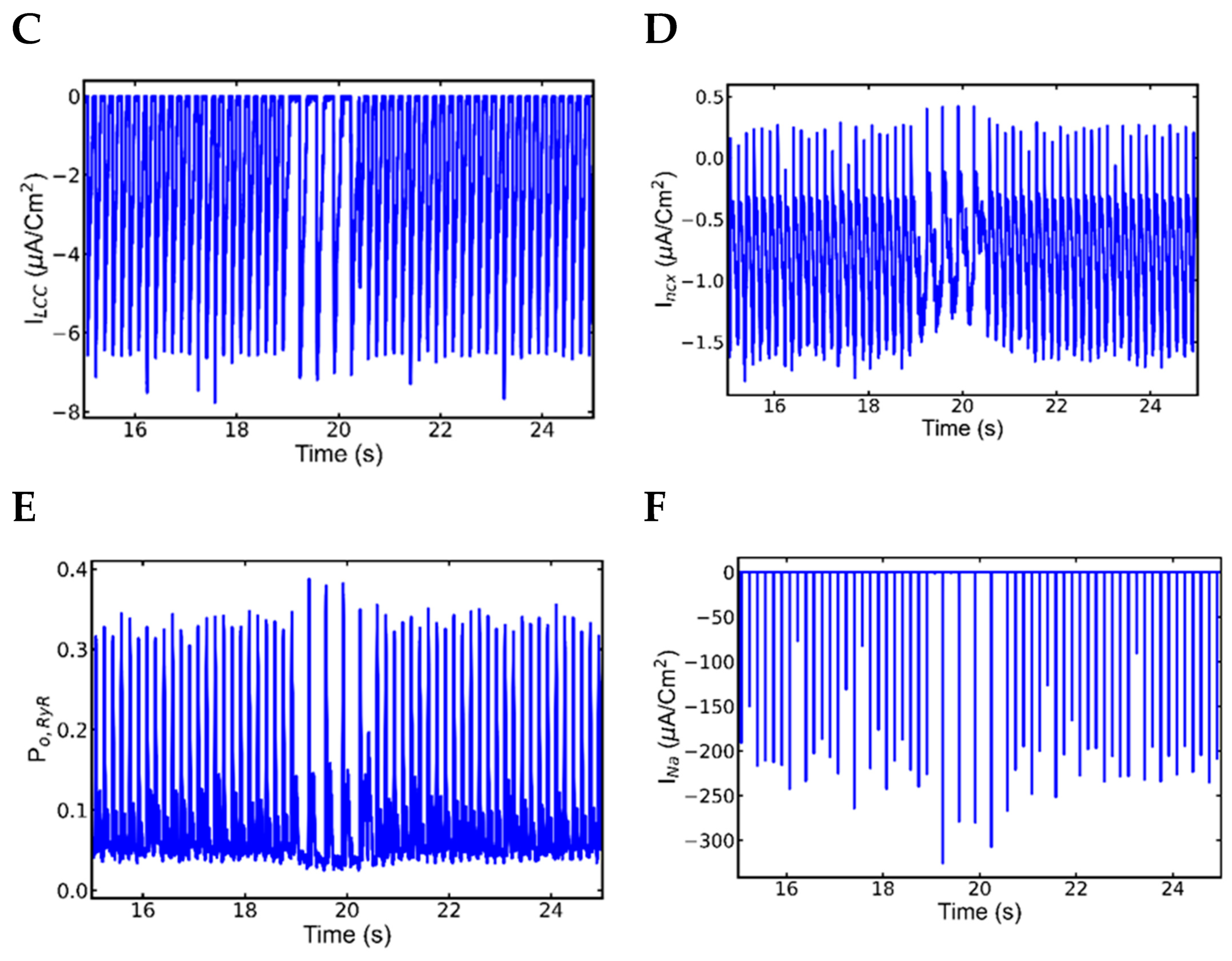
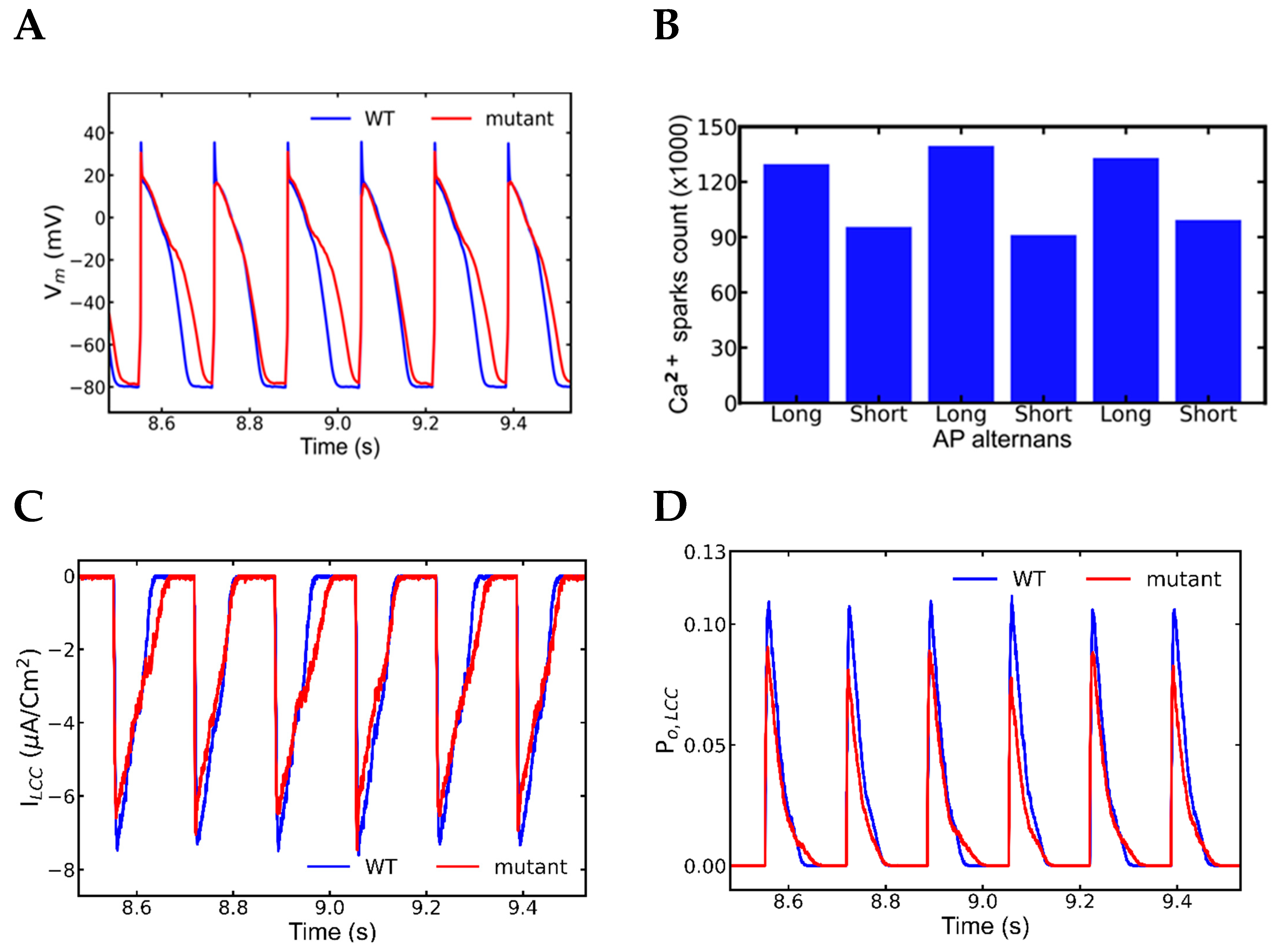
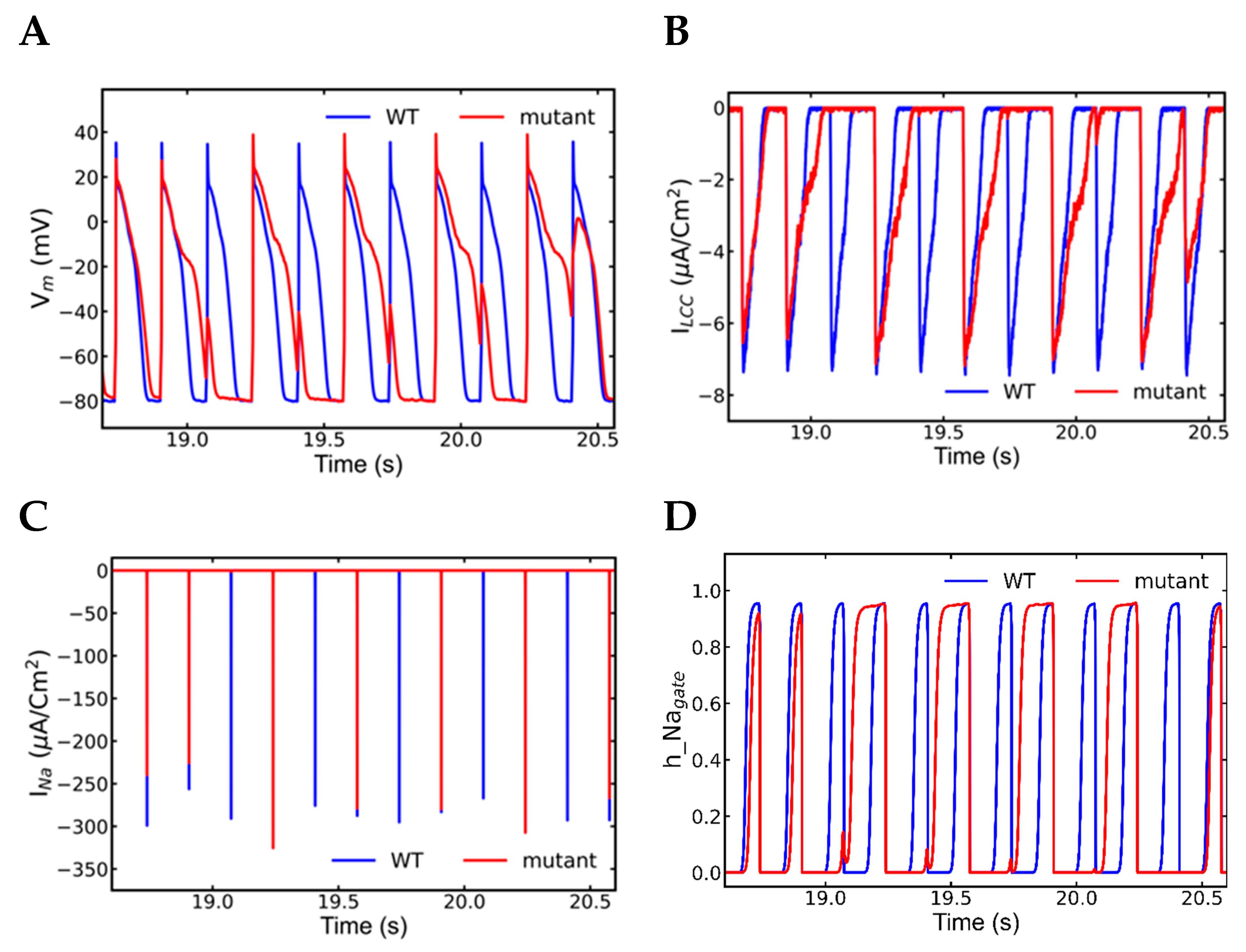
3.5. Alternans and Alternately Skipping Beats
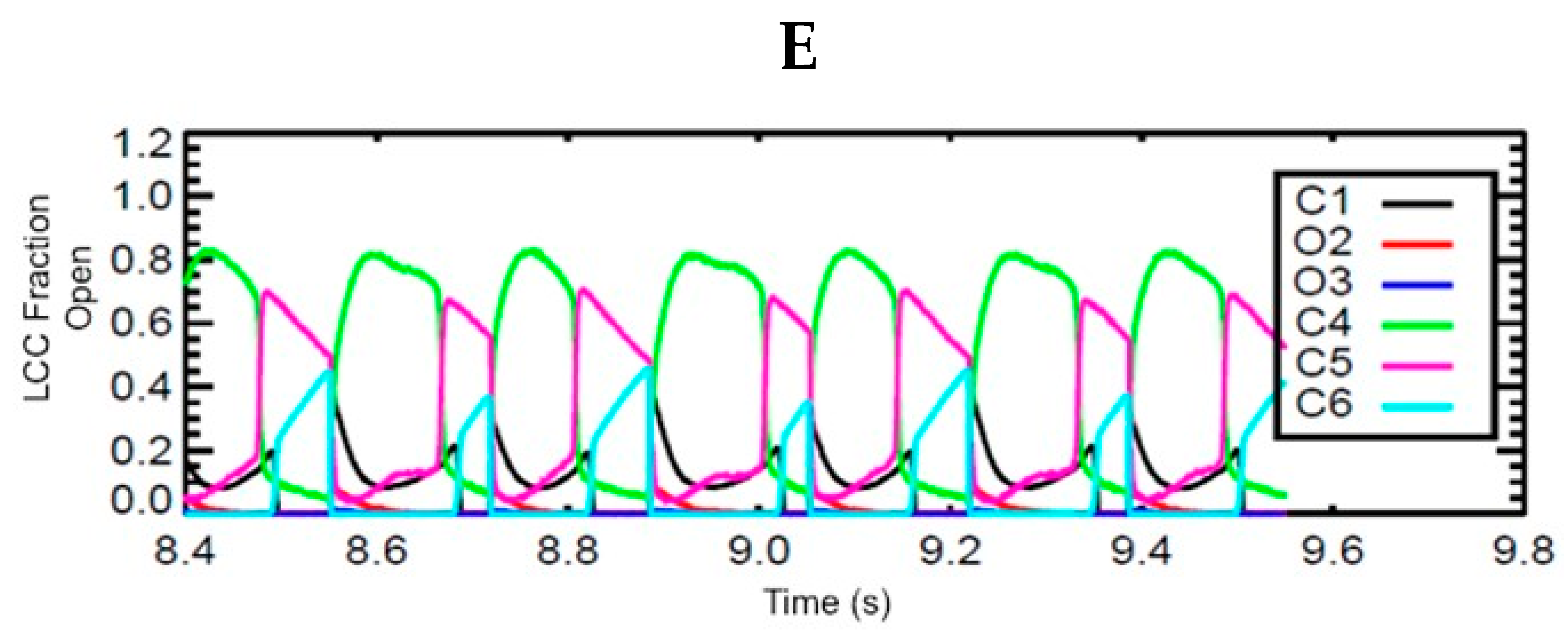
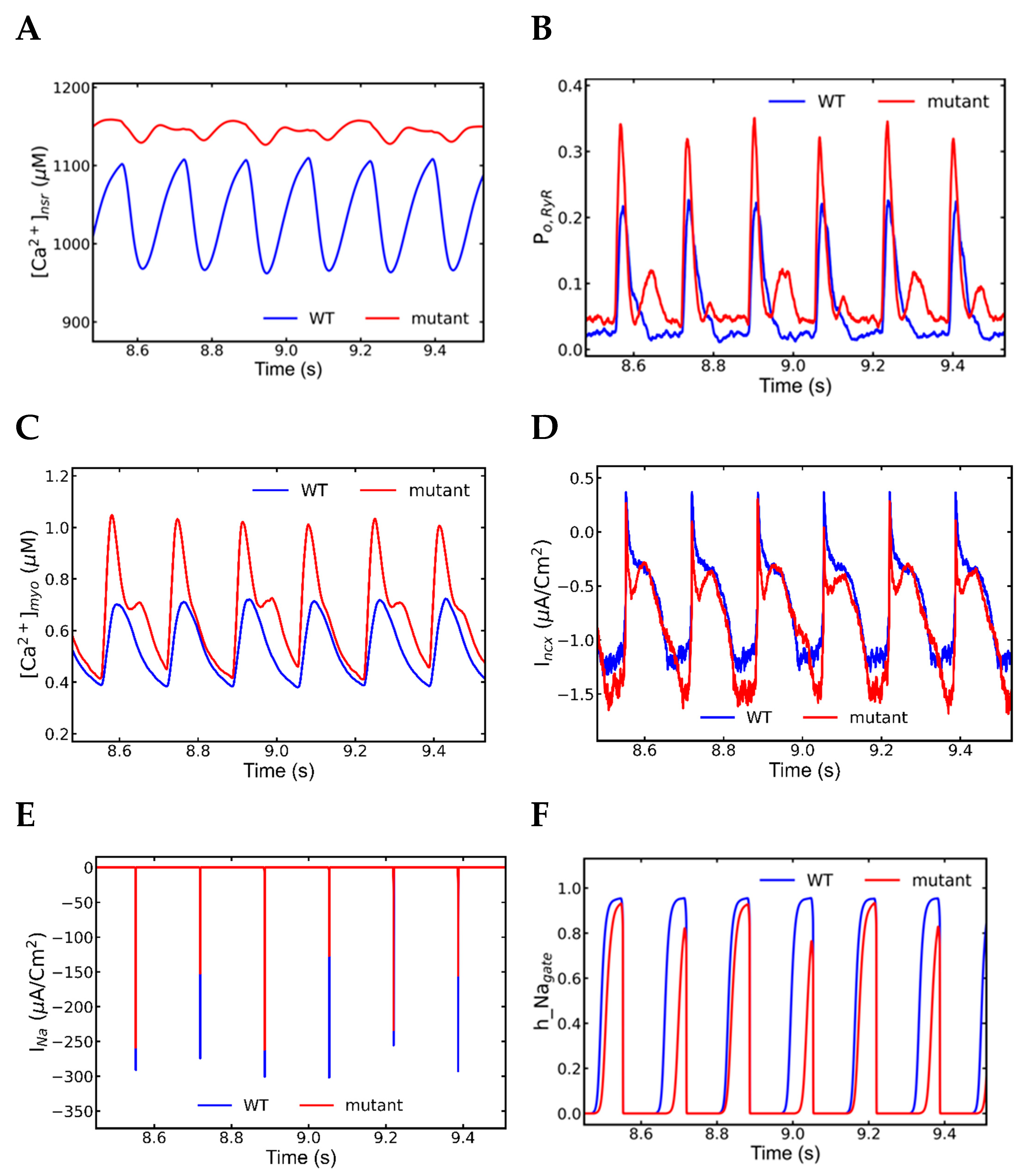
3.6. Mechanism of Alternans
3.7. Nonrecovery of Sodium Channels Results into Alternate Beat Skipping
4. Discussion
4.1. Alternans
- INa plays major role in the action potential amplitude alternans and SR Ca2+ has the main role in the APD alternans.
- Na+ channel alternation has negative feedback on the l-type current alternation.
- Intracellular Ca2+ has negative feedback to extracellular Ca2+ in SR overload.
- Very short diastolic interval for the refilling of Ca2+ after a beat with longer APD.
- Higher fractions of L-type channels exhibit Ca2+-dependent inactivation and voltage-dependent inactivation.
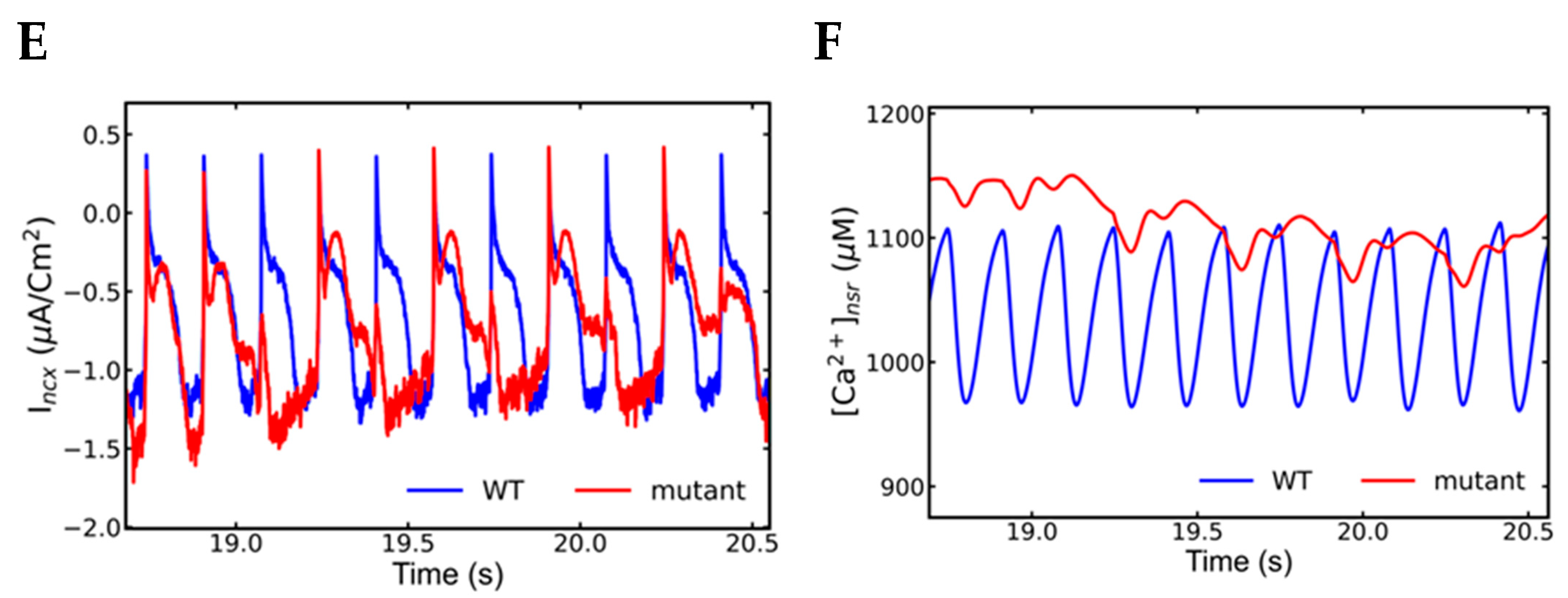
- Increased NCX current (Incx) increases the APD.
4.2. Early Afterdepolarization (EAD)
4.3. Clinical Correlations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bhuiyan, Z.A.; Berg, M.P.; Tintelen, J.P.; Bink-Boelkens, M.T.; Wisefield, A.C.; Alders, M.; Postma, A.V.; Langen, I.; Mannens, M.M.; Wilde, A.A. Expanding spectrum of human RYR2-related disease: New electrocardiographic, structural and genetic features. Circulation 2007, 116, 1569–1576. [Google Scholar] [CrossRef] [PubMed]
- Doste, R.; Bueno-Orovio, A. Multiscale Modelling of β-Adrenergic Stimulation in Cardiac Electromechanical Function. Mathematics 2021, 9, 1785. [Google Scholar] [CrossRef]
- Baltogiannis, G.G.; Lysitsas, D.N.; di Giovanni, G.; Ciconte, G.; Sieira, J.; Conte, G.; Kolettis, T.M.; Chierchia, G.B.; de Asmundis, C.; Brugada, P. CPVT: Arrhythmogenesis, Therapeutic Management, and Future Perspectives. A Brief Review of the Literature. Front. Cardiovasc. Med. 2019, 6, 92. [Google Scholar] [CrossRef] [PubMed]
- Peltenburg, P.J.; Kallas, D.; Bos, J.M.; Lieve, K.V.V.; Franciosi, S.; Roston, T.M.; Denjoy, I.; Sorensen, K.B.; Ohno, S.; Roses-Noguer, F.; et al. An International Multicenter Cohort Study on β-Blockers for the Treatment of Symptomatic Children with Catecholaminergic Polymorphic Ventricular Tachycardia. Circulation 2022, 145, 333–344. [Google Scholar] [CrossRef] [PubMed]
- Coumel, P.; Fidelle, J.; Lucet, V.; Attuel, P.; Bouvrain, Y. Catecholaminergic-induced severe ventricular arrhythmia with Adams-Stokes syndrome in children: Report of four cases. Br. Heart J. 1978, 40, 28–37. [Google Scholar]
- Medeiros-Domingo, A.; Bhuiyan, Z.A.; Tester, D.J.; Hofman, N.; Bikker, H.; Tintelen, J.; Mannens, M.; Wilde, A.; Ackerman, M.J. Comprehensive open reading frame mutational analysis of the RyR2-encoded ryanodine receptor/calcium channel in patients diagnosed previously with either catecholaminergic polymorphic ventricular tachycardia or genotype negative, exercise-induced long QT syndrome: A comprehensive open reading frame mutational analysis. J. Am. Coll. Cardiol. 2009, 54, 2065–2074. [Google Scholar] [PubMed]
- Refaat, M.M.; Aouizerat, B.E.; Pullinger, C.R.; Malloy, M.; Kane, J.; Tseng, Z.H. Association of CASQ2 polymorphism with sudden cardiac arrest and heart failure in patients with coronary Artery disease. Heart Rhythm 2014, 11, 646–652. [Google Scholar] [CrossRef]
- Faggioni, M.; Krystal, D.O.; Knollmann, B.C. Calsequestrin mutations and catecholaminergic polymorphic ventricular tachycardia. Pediatr. Cardiol. 2012, 33, 959–967. [Google Scholar] [CrossRef]
- Lahat, H.; Pras, E.; Eldar, M. A missense mutation in CASQ2 is associated with autosomal recessive catecholamine-induced polymorphic ventricular tachycardia in Bedouin families from Israel. Ann. Med. 2004, 36, 87–91. [Google Scholar] [CrossRef]
- Wang, S.; Trumble, W.R.; Liao, H.; Wesson, C.R.; Dunker, A.K.; Kang, C.H. Crystal structure of calsequestrin from rabbit skeletal muscle sarcoplasmic reticulum. Nat. Struct. Biol. 1998, 5, 476–483. [Google Scholar] [CrossRef]
- Handhle, A.; Ormonde, C.E.; Thomas, N.L.; Bralesford, C.; Williams, A.J.; Lai, F.A.; Zissimopoulos, S. Calsequestrin interacts directly with the cardiac ryanodine receptor luminal domain. J. Cell Sci. 2016, 129, 3983–3988. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.A.D.H.; Beard, N.A.; Dulhunty, A.F. Unique isoform-specific properties of calsequestrin in the heart and sketal muscle. Cell Calcium 2009, 45, 474–484. [Google Scholar] [CrossRef] [PubMed]
- Titus, E.W.; Deiter, F.H.; Shi, C.; Wojciak, J.; Scheinman, M.; Jura, N.; Deo, R.C. The structure of a calsequestrin filament reveals mechanisms of familial arrhythmia. Nat. Struct. Mol. Biol. 2020, 27, 1142–1151. [Google Scholar] [CrossRef] [PubMed]
- Novak, P.; Soukup, T. Calsequestrin distribution, structure and function, it’s role in normal and pathological situations and the effect of thyroid hormones. Physiol. Res. 2011, 60, 439–452. [Google Scholar] [CrossRef]
- di Barletta, M.F.; Viatchenko-Karpinski, S.; Nori, A.; Memmi, M.; Terentyev, D.; Turcato, F.; Valle, G.; Rizzi, N.; Napolitano, C.; Gyroke, S.; et al. Clinical phenotype and functional characterization of CASQ2 mutations associated with catecholaminergic polymorphic ventricular tachycardia. Circulation 2006, 114, 1012–1019. [Google Scholar] [CrossRef]
- Williams, G.S.; Chikando, A.C.; Tuan, H.T.; Sobie, E.A.; Lederer, W.J.; Jafri, M.S. Dynamics of Calcium sparks and calcium leak in the heart. Biophys. J. 2011, 101, 1287–1296. [Google Scholar] [CrossRef]
- Jafri, M.S.; Rice, J.J.; Winslow, R.L. Cardiac Ca2+ Dynamics: The roles of ryanodine receptor adaptation and sarcoplasmic reticulum load. Biophys. J. 1998, 74, 1149–1168. [Google Scholar] [CrossRef]
- Paudel, R.; Jafri, M.S.; Ullah, A. The Role of Ca2+ Sparks in Force Frequency Relationships in the Guinea Pig Ventricular Myocytes. Biomolecules 2022, 12, 1577. [Google Scholar] [CrossRef]
- McCoy, M.D.; Ullah, A.; Lederer, W.J.; Jafri, M.S. Understanding Calmodulin Variants Affecting Calcium Dependent Inactivation of L-type Calcium Channels through Whole-Cell Simulation of the Cardiac Ventricular Myocyte. Biomolecules 2022, in press.
- Hoang-Trong, T.M.; Ullah, A.; Lederer, W.J.; Jafri, M.S. A Stochastic Spatiotemporal Model of Rat Ventricular Myocyte Calcium Dynamics Demonstrated Necessary Features for Calcium Wave Propagation. Membranes 2021, 11, 989. [Google Scholar] [CrossRef]
- Hoang-Trong, M.T.; Ullah, A.; Lederer, W.J.; Jafri, M.S. Cardiac Alternans Occurs through the Synergy of Voltage- and Calcium-Dependent Mechanisms. Membranes 2021, 11, 794. [Google Scholar] [CrossRef] [PubMed]
- Ullah, A.; Hoang-Trong, M.T.; Lederer, W.J.; Winslow, R.L.; Jafri, M.S. Critical Requirements for the Initiation of a Cardiac Arrhythmia in Rat Ventricle: How Many Myocytes? Cells 2022, 11, 1878. [Google Scholar] [CrossRef] [PubMed]
- Luo, C.H.Y.R. A model of the ventricular cardiac action potential. Depolarization, repolarization, and their interaction. Circ. Res. 1991, 68, 1501–1526. [Google Scholar] [CrossRef] [PubMed]
- Luo, C.H.; Rudy, Y. A dynamic model of the ventricular cardiac action potential. I. Simulations of ionic current and concentration changes. Circ. Res. 1994, 74, 1070–1096. [Google Scholar] [CrossRef]
- Luo, C.H.Y.R. A dynamic model of the cardiac ventricular action potential. II. Afterdepolarizations, triggred activity, and potentiation. Circ. Res. 1994, 74, 1097–1113. [Google Scholar] [CrossRef] [PubMed]
- Wagner, E.; Lauterbach, M.A.; Kohl, T.; Westphal, V.; Williams, G.S.B.; Steinbrecher, J.H.; Strreich, J.-H.; Korff, B.; Tuan, H.-T.M.; Hagen, B.; et al. Stimulation emission depletion live-cell super-resolution imaging shows proliferative remodeling of T-tubule membrane structures after myocardial infraction. Circ. Res. 2012, 111, 402–414. [Google Scholar] [CrossRef]
- Sun, L.; Fan, J.-S.; Clark, J.W.; Palade, P.T. A model of the L-type Ca2+ channel in rat ventricular myocytes: Ion selectivity and inactivation mechanisms. J. Physiol. 2000, 15529, 139–158. [Google Scholar] [CrossRef]
- Morotti, S.; Grandi, E.; Summa, A.; Ginsburg, K.S.; Bers, D.M. Theoretical study of L-type Ca2+ current inactivation kinetics during action potential repolarization and early afterdepolarizations. J. Physiol. 2012, 590, 4465–4481. [Google Scholar] [CrossRef]
- Terentydev, D.; Viatchenko-Karpinski, S.; Valdivia, H.H.; Escobar, A.L.; Gyroke, S. Luminal Ca2+ controls termination and refractory behavior of Ca2+-induced Ca2+ release in cardiac myocytes. Circ. Res. 2002, 91, 414–420. [Google Scholar] [CrossRef]
- Kornyeyev, D.; Petrosky, A.D.; Zepeda, B.; Ferreiro, M.; Knollmann, B.; Escobar, A.L. Calsequestrin deletion shortens the refractoriness of Ca2+ release and reduces rate_dependent Ca2+ -alternans in intact mouse hearts. J. Mol. Cell. Cardiol. 2011, 52, 21–31. [Google Scholar] [CrossRef]
- Song, L.; Alcalai, R.; Arad, M.; Wolf, C.M.; Toka, O.; Conner, D.A.; Berul, C.I.; Eldar, M.; Seidman, C.E.; Seidman, J.G. Calsequestrin 2 (CASQ2) mutations increase expression of calreticulin and ryanodine receptors, causing catecholaminergic polymorphic ventricular tachucardia. J. Clin. Investig. 2007, 17, 1814–1823. [Google Scholar] [CrossRef] [PubMed]
- Knollmann, B.C.; Chopra, N.; Hlaing, T.; Akin, B.; Yang, T.; Ettensohn, K.; Knollmann, B.E.C.; Horton, K.D.; Weissman, N.J.; Holinstat, I.; et al. Casq2 deletion causes sarcoplasmic reticulum volume increase, premature Ca2+ release, and catecholaminergic polymorphic ventricular tachycardia. J. Clin. Investig. 2006, 116, 2510–2520. [Google Scholar] [CrossRef] [PubMed]
- Ginsburg, K.S.D.M.B. Modulation of excitation-contraction coupling by isoproterenol in cardiomyocytes with controlled SR Ca2+ load and Ca2+ trigger. J. Physiol. 2004, 556, 463–480. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Lou, Q.; Smith, H.; Velez-Cortes, F.; Dillmann, W.H.; Knollmann, B.C.; Armoundas, A.A.; Györke, S. Conditional Up-Regulation of SERCA2a Exacerbates RyR2-Dependent Ventricular and Atrial Arrhythmias. Int. J. Mol. Sci. 2020, 21, 2535. [Google Scholar] [CrossRef] [PubMed]
- Györke, I.S.G. Regulation of the cardiac ryanodine receptor channel by luminal Ca2+ involves luminal Ca2+ sensing sites. Biophys. J. 1998, 75, 2801–2810. [Google Scholar] [CrossRef]
- Kashimura, T.; Briston, S.J.; Trafford, A.W.; Napolitano, C.; Priori, S.G.; Eisner, D.A.; Venetucci, L.A. In the RyR2R4496C mouse model of CPVT, β-adrenergic stimulation induces Ca waves by increasing SR Ca content and not by decreasing the threshold Ca waves. Circ. Res. 2010, 107, 1483–1489. [Google Scholar] [CrossRef]
- Bers, D.M. Calcium fluxes involved in control of cardiac myocyte concentration. Circ. Res. 2000, 87, 275–281. [Google Scholar] [CrossRef]
- Jafri, M.S.; Hoang-Trong, M.T.; Williams, G.S.B. Method and System for Utilizing Markov Chain Monte Carlo Simulations. US Patent US9009095B1, 14 April 2012. [Google Scholar]
- Guo, T.; Gillespie, D.; Fill, M. Ryanodine receptor current amplitude controls Ca2+ sparks in cardiac muscle. Circ. Res. 2012, 111, 28–36. [Google Scholar] [CrossRef]
- Karagueuzian, H.S.; Pezhouman, A.; Angelini, M.; Olcese, R. Enhanced late Na and Ca currents as effective antiarrhythmic drug test. Front. Pharmacol. 2017, 8, 1–17. [Google Scholar] [CrossRef]
- Sipido, K.R.; Bito, C.; Antoons, G.; Volders, P.G.; Vos, M.A. Na/Ca exchange and cardiac ventrucular arrhythmias. Ann. N. Y. Acad. Sci. 2007, 1099, 339–348. [Google Scholar] [CrossRef]
- Gizzi, A.; Cherry, E.; Gilmour, R.; Luther, S.; Filippi, S.; Fenton, F. Effects of Pacing Site and Stimulation History on Alternans Dynamics and the Development of Complex Spatiotemporal Patterns in Cardiac Tissue. Front. Physiol. 2013, 4. [Google Scholar] [CrossRef] [PubMed]
- Györke, I.; Hester, N.; Jones, L.R.; Györke, S. The role of calsequestrin, triadin, and junctin in conferring cardiac ryanodine receptor responsiveness to luminal calcium. Biophys. J. 2004, 86, 2121–2128. [Google Scholar] [CrossRef] [PubMed]
- Diaz, M.; O’Neil, S.C.; Eisner, D.A. Sarcoplasmic reticulum calcium content fluctuation is the key to cardiac alternans. Circ. Res. 2004, 94, 650–656. [Google Scholar] [CrossRef] [PubMed]
- Kubalova, Z. Inactivation of l-type calcium channels in cardiomyocytes. Experimental and theoretical approaches. Gen. Physiol. Biophys. 2003, 22, 441–454. [Google Scholar] [PubMed]
- Adachi-Akahane, S.; Cleemann, L. Cross-signaling between L-type Ca2+ channels and ryanodine receptors in rat ventricuular myocytes. J. Gen. Physiol. 1996, 108, 435–454. [Google Scholar] [CrossRef] [PubMed]
- Saitoh, H.; Bailey, J.C.; Surawicz, B. Action potential duration alternans in dog purkinje and ventricular muscle fibres. Further evidence in support of two different mechanisms. Circulation 1989, 80, 1421–1431. [Google Scholar] [CrossRef]
- Euler, D.E. Cardiac alternans: Mechanisms and physiological significance. Cardiovasc. Res. 1999, 42, 583–590. [Google Scholar] [CrossRef]
- Tse, G.; Wong, S.T.; Tse, V.; Lee, Y.T.; Lin, H.Y.; Yeo, J.M. Cardiac dynamics: Alternans and arrhythmogenesis. J. Arrhythm. 2016, 32, 411–417. [Google Scholar] [CrossRef]
- Armoundas, A.A.; Hobai, I.A.; Tomaselli, G.F.; Winslow, R.L.; O’Rourke, B. Role of sodium-calcium exchanger in modulating the action potential of ventricular myocytes from normal and failing hearts. Circ. Res. 2003, 93, 46–53. [Google Scholar] [CrossRef]
- Sigg, D.C.; Laizzo, P.A.; Xiao, Y.-F.; He, B. Cardaic Electrophysiology Methods and Models; Springer: New York, NY, USA, 2010. [Google Scholar]
- Mangold, K.E.; Brumback, B.; Angsutararux, P.; Volker, T.L.; Zhu, W.; Kang, P.; Moreno, J.; Silva, J.R. Mechanism and models of cardiac sodium channel inactivation. Channels 2017, 11, 517–533. [Google Scholar] [CrossRef]
- Noble, D.P.J.N. Late sodium current in the pathophysiology of cardiovascular disease: Consequences of sodium-calcium overload. Heart 2006, 92, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Marks, A.R. Ryanodine receptors/calcium release channels in heart failure and sudden cardiac death. J. Mol. Cell Cardiol. 2001, 33, 615–624. [Google Scholar] [CrossRef] [PubMed]
- Burashnikov, A.C.A. Acceleration-induced action potential prolongation and early afterdepolarizations. J. Cardiovasc. Electrophysiol. 1998, 9, 934–948. [Google Scholar] [CrossRef] [PubMed]
- Weiss, J.M.; Garfinkel, A.; Karagueuzian, H.S.; Chen, P.-S.; Qu, Z. Early afterdepolarization and cardiac arrhythmia. Heart Rhythm 2010, 7, 1891–1899. [Google Scholar] [CrossRef] [PubMed]
- Iyer, V.; Hajjar, R.J.; Armoundas, A.A. Mechanisms of abnormal calcium homeostasis in mutations responsible for catecholaminergic polymorphic ventricular tachycardia. Circ. Res. 2007, 100, e22–e31. [Google Scholar] [CrossRef]
- January, C.T.; Riddle, J.M. Early afterdepolarizations: Mechanism of induction and block. A role for L-type Ca2+ current. Circ. Res. 1989, 64, 977–990. [Google Scholar] [CrossRef]
- Antoons, G.; Volders, P.G.; Stankovicova, T.; Bito, V.; Stengl, M.; Vos, M.A.; Sipido, K.R. Window Ca2+ current and its modulation by Ca2+ release in hypertrophied cardiac myocytes from dogs with chronic atrioventricular block. J. Physiol. 2007, 579, 147–160. [Google Scholar] [CrossRef]
- Janvier, N.C.; Harrison, S.M.; Boyett, M.R. The role of inward Na(+)-Ca2+ exchange current in the ferret ventricular action potential. J. Physiol. 1997, 498, 611–625. [Google Scholar] [CrossRef]
- Schouten, V.J.; ter Keurs, H.E.; Quaegebeur, J.M. Influence of electrogenic Na/Ca exchange on the action potential in human heart muscle. Cardiovasc. Res. 1990, 24, 758–767. [Google Scholar] [CrossRef]
- Volders, P.G.; Vos, M.A.; Szabo, B.; Sipido, K.R.; de Groot, S.H.; Gorgels, A.P.; Wellens, H.J.; Lazzara, R. Progress in the understanding of cardiac early afterdepolarizations and torsades de pointes: Time to revise current concepts. Cardiovasc. Res. 2000, 46, 376–392. [Google Scholar] [CrossRef]
- Priori, S.G.; Napolitano, C.; Tiso, N.; Memmi, M.; Viganti, G.; Bloise, R.; Sorrentino, V.; Danieli, G.A. Mutations in the cardiac ryanodine receptor gene(hRyR2) underlie catecholaminergig polymorphic ventricular tachycardia. Circulation 2001, 103, 196–200. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Grandi, E.; Puglisi, J.L.; Sato, D.; Bers, D.M. β-Adrenergic stimulation activates early afterdepolarization transiently via kinetic mismatch of PKA targets. J. Mol. Cell. Cardiol. 2013, 58, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Leenhardt, A.; Denjoy, I.; Guicheney, P. Catecholaminergic Polymorphic Ventricular Tachycardia. Circ. Arrhythm. Electrophysiol. 2012, 5, 1044–1052. [Google Scholar] [CrossRef] [PubMed]
- Khokhlova, A.; Balakina-Vikulova, N.; Katsnelson, L.; Iribe, G.; Solovyova, O. Transmural cellular heterogeneity in myocardial electromechanics. J. Physiol. Sci. 2018, 68, 387–413. [Google Scholar] [CrossRef]
- Colli Franzone, P.; Pavarino, L.F.; Scacchi, S.; Taccardi, B. Modeling ventricular repolarization: Effects of transmural and apex-to-base heterogeneities in action potential durations. Math. Biosci. 2008, 214, 140–152. [Google Scholar] [CrossRef]
- Rosso, R.; Kalman, J.M.; Rogowski, O.; Diamant, S.; Birger, A.; Biner, S.; Belhassen, B.; Viskin, S. Calcium channel blockers and β-blockers versus β-blockers alone for preventing exercise-induced arrhythmias in catecholaminergic polymorphic ventricular tachycardia. Heart Rhythm 2007, 4, 1149–1154. [Google Scholar] [CrossRef]
- Salvage, S.C.; Chandrasekharan, K.H.; Jeevaratnam, K.; Dulhunty, A.F.; Thompson, A.J.; Jackson, A.P.; Huang, C.L. Multiple targets for flecainide action: Implications for cardiac arrhythmogenesis. Br. J. Pharmacol. 2018, 175, 1260–1278. [Google Scholar] [CrossRef]
- Bannister, M.L.; Thomas, N.L.; Sikkel, M.B.; Mukherjee, S.; Maxwell, C.; MacLeod, K.T.; George, C.H.; Williams, A.J. The Mechanism of Flecainide Action in CPVT Does Not Involve a Direct Effect on RyR2. Circ. Res. 2015, 116, 1324–1335. [Google Scholar] [CrossRef]
- Györke, S. Molecular basis of catecholaminergic polymorphic ventricular tachycardia. Heart Rhythm 2009, 6, 123–129. [Google Scholar] [CrossRef]
- Wleklinski, M.J.; Kryshtal, D.O.; Kim, K.; Parikh, S.S.; Blackwell, D.J.; Marty, I.; Iyer, V.R.; Knollmann, B.C. Impaired Dynamic Sarcoplasmic Reticulum Ca Buffering in Autosomal Dominant CPVT2. Circ. Res. 2022, 131, 673–686. [Google Scholar] [CrossRef]
- You, T.; Luo, C.; Zhang, K.; Zhang, H. Electrophysiological Mechanisms Underlying T-Wave Alternans and Their Role in Arrhythmogenesis. Front. Physiol. 2021, 12, 614946. [Google Scholar] [CrossRef] [PubMed]
- Bao, M.; Zhang, J.; Huang, C.; Jiang, H.; Liu, J.; Zhao, D. Abnormal intracellular calcium handling underlying T-wave alternans and its hysteresis. Cardiology 2007, 108, 147–156. [Google Scholar] [CrossRef] [PubMed]


















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | Change (%) | Experimental Change (%) |
|---|---|---|
| Ca2+ regulation coefficient | 90 | Adjusted to match [32] |
| SERCA Pump (Ap) | 50 | 25–50 [32,34] |
| JSR Volume | 50 | 50 [32] |
| NSR Volume | 50 | 50 [32] |
| CASQ Buffer | (−95) | −98 [32] |
| Simulations | Wild-Type Myocyte | Mutant Myocyte |
|---|---|---|
| Control Simulation | Original model parameters with a change in the pacing frequency | Morphological changes (SR Volume Increase, SR Ca2+ buffering diminished) |
| Β-adrenergic stimulation | Increase L-type activity, SERCA activity, and luminal Ca2+ | Morphological changes plus β-adrenergic stimulation |
| Simulations | Wild-Type Myocyte | Mutant Myocyte | Wild-Type Myocyte with β-Adrenergic Stimulation | Mutant Myocyte with β-Adrenergic Stimulation |
| Ca2+ transient maximum (µM) | 0.6889 ± 0.0076 | 0.7368 ± 0.0066 * | 0.8417 ± 0.0271 * | 0.8167 ± 0.0082 * |
| Diastolic [Ca2+] (µM) | 0.1273 ± 0.0017 | 0.1230 ± 0.0017 * | 0.1561 ± 0.0036 * | 0.2194 ± 0.2709 * |
| Experiment | Wild-Type Myocyte | Mutant Myocyte | Wild-Type Myocyte with β-Adrenergic Stimulation | Mutant Myocyte with β-Adrenergic Stimulation |
| Ca2+ transient Maximum (Fratio) | 2.23 ± 0.36 | 2.42 ± 0.37 * | 3.22 ± 0.57 | 3.04 ± 0.57 * |
| Diastolic [Ca2+] (Fratio) | 1.50 ± 017 | 1.70 ± 0.24 | 1.49 ± 0.16 | 1.67 ± 0.22 |
| Myocyte | APD (Beat) 1 | Ca2+ Sparks/s 2 | Ca2+ Sparks/Beat 1 | Spark Amp. (Beat) 1 |
|---|---|---|---|---|
| WT Control | 181.3 ± 6.96 | 60,951 ± 2439 | 50,695 ± 1986 | 62.6 ± 0.85 |
| WT β-adrenergic | 211.7 ± 10.1 | 156,302 ± 9592 | 112,388 ± 5480 | 61.52 ± 10.7 |
| Mutant First slow phase with β-adrenergic | 229.1 ± 15.93 | 155,082 ± 8638 | 123,847 ± 8638 | 64.27 ± 0.52 |
| Mutant Second slow phase with β-adrenergic | 330 ± 56.95 | 368,841 ± 26,994 | 247,392 ± 68,966 | 62.07 ± 0.51 |
| AP | Beat 1 | Beat 2 | Beat 3 | Beat 4 | Beat 5 | Beat 6 | St. Dev. |
|---|---|---|---|---|---|---|---|
| Duration (ms) | 157 | 131 | 156 | 132 | 156 | 139 | 11.45 |
| Amplitude (mV) | 31.15 | 17.23 | 31.51 | 16.83 | 31.51 | 17.31 | 7.14 |
| Spark Count | 129,638 | 92,570 | 139,542 | 91,152 | 132,970 | 99,334 | 20,219 |
| Currents/Transients | Beat (n − 1) | Beat (n) | Beat (n + 1) |
|---|---|---|---|
| INa | Longer | Shorter | Longer |
| ICa | Shorter | Longer | Shorter |
| AP | Longer | Shorter | Longer |
| [Ca2+]nsr | Longer | Shorter | Longer |
| Incx | Longer | Shorter | Longer |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paudel, R.; Jafri, M.S.; Ullah, A. Pacing Dynamics Determines the Arrhythmogenic Mechanism of the CPVT2-Causing CASQ2G112+5X Mutation in a Guinea Pig Ventricular Myocyte Computational Model. Genes 2023, 14, 23. https://doi.org/10.3390/genes14010023
Paudel R, Jafri MS, Ullah A. Pacing Dynamics Determines the Arrhythmogenic Mechanism of the CPVT2-Causing CASQ2G112+5X Mutation in a Guinea Pig Ventricular Myocyte Computational Model. Genes. 2023; 14(1):23. https://doi.org/10.3390/genes14010023
Chicago/Turabian StylePaudel, Roshan, Mohsin Saleet Jafri, and Aman Ullah. 2023. "Pacing Dynamics Determines the Arrhythmogenic Mechanism of the CPVT2-Causing CASQ2G112+5X Mutation in a Guinea Pig Ventricular Myocyte Computational Model" Genes 14, no. 1: 23. https://doi.org/10.3390/genes14010023
APA StylePaudel, R., Jafri, M. S., & Ullah, A. (2023). Pacing Dynamics Determines the Arrhythmogenic Mechanism of the CPVT2-Causing CASQ2G112+5X Mutation in a Guinea Pig Ventricular Myocyte Computational Model. Genes, 14(1), 23. https://doi.org/10.3390/genes14010023