
Indoloquinoline-Mediated Targeted Downregulation of KRAS through Selective Stabilization of the Mid-Promoter G-Quadruplex Structure

 , , and
, , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Chemicals, Compounds, and Oligonucleotides
2.2. Chemical Synthesis
2.3. FRET Melt/FRET Melt2
2.4. Electronic Crcular Dichroism (ECD)
2.5. Luciferase Assay
2.6. Cell Culture, Cytotoxicity and qPCR
2.7. Three-Dimensional Cell Culture, Cytotoxicity, and Combination Chemotherapy Cytotoxicity
2.8. Statistics
3. Results
3.1. NSC 317605 as a G4 Stabilizer
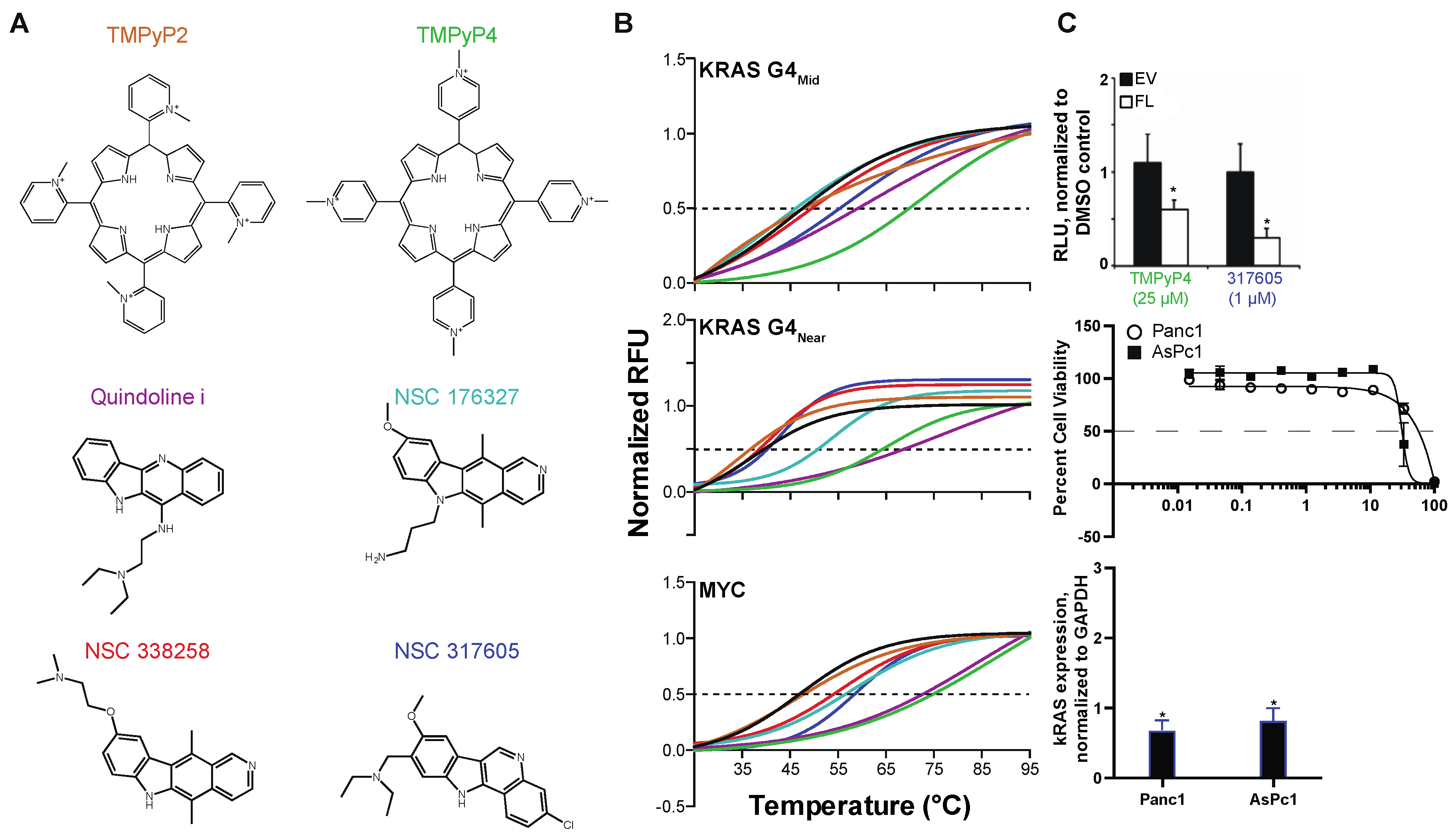
3.1.1. Screening the NCI Diversity Set III
3.1.2. Identification of Pan-KRAS G4-Stabilizing Compounds, and those Selective for G4near or G4mid
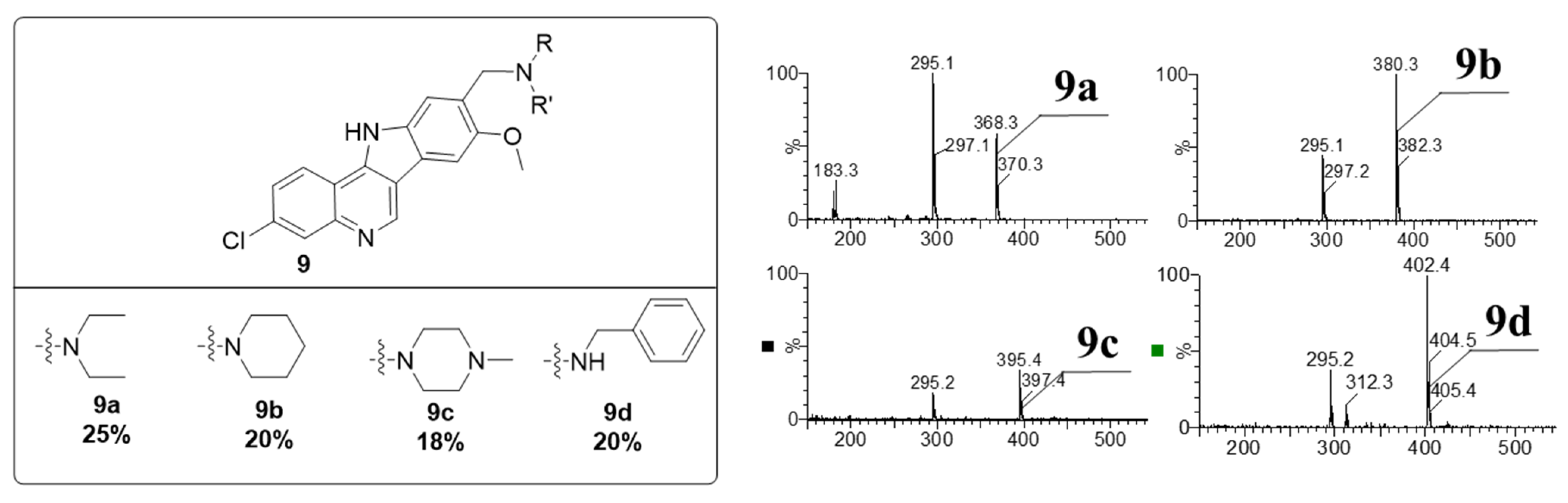
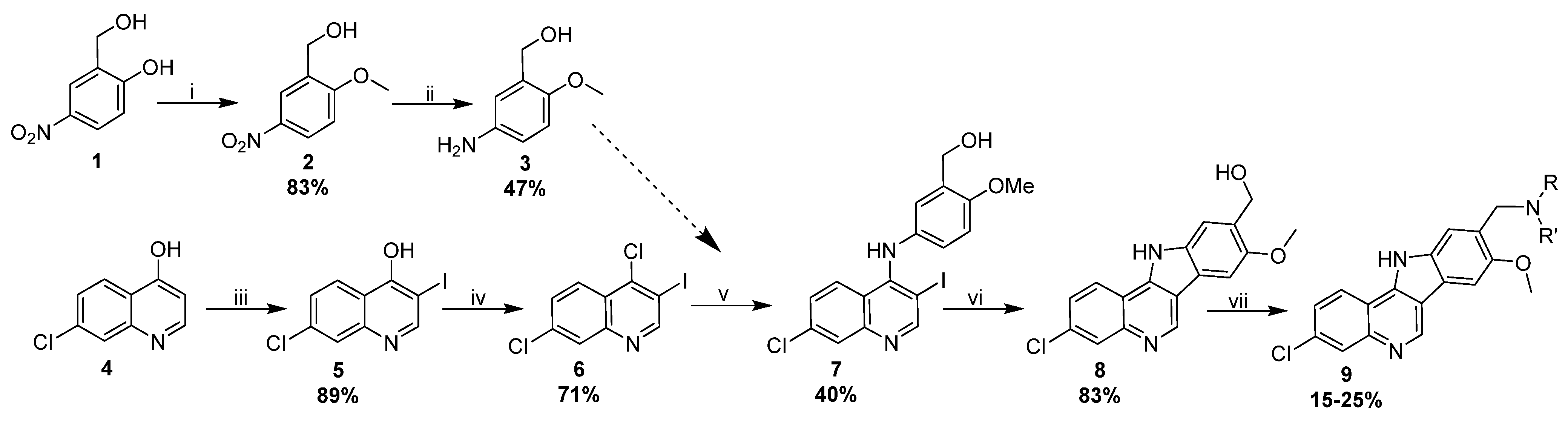
3.2. Synthesis and Characterization of 317605 and Novel Indoloquinolines
3.2.1. Synthesis of Indoloquinolines
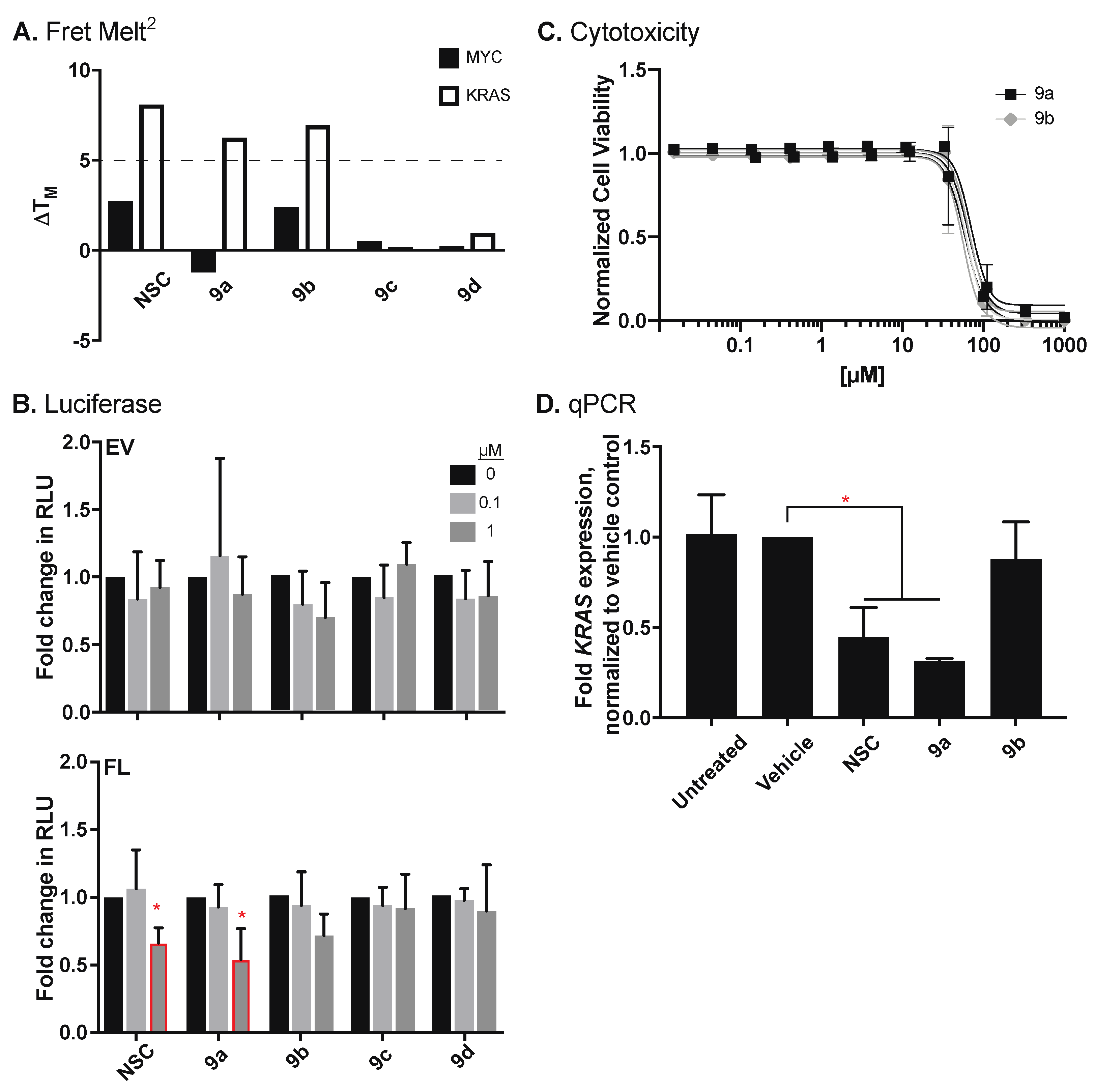
3.2.2. Characterization of Indoloquinolines
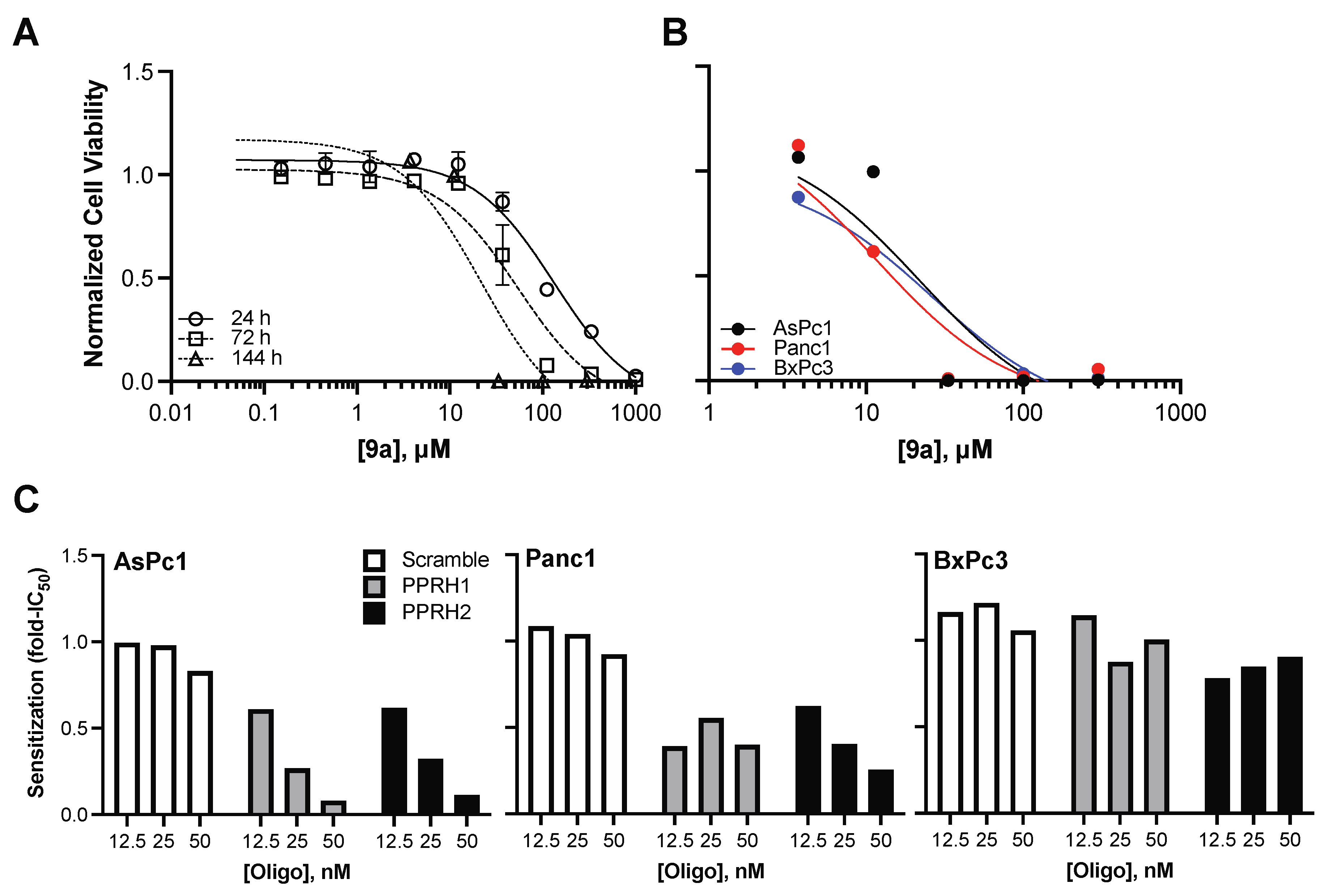
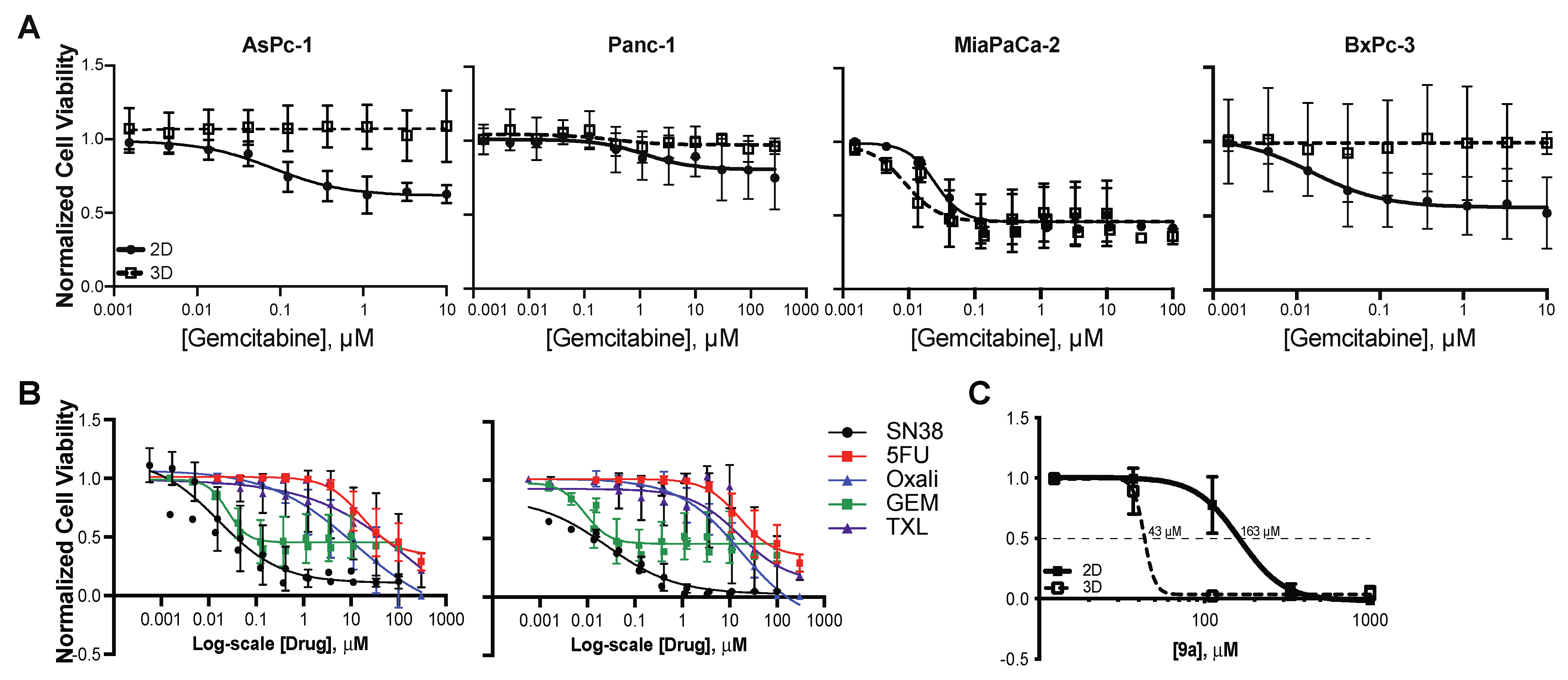
3.3. Activity of Compound 9a in Pancreatic Cancer Cell Lines
3.3.1. Compound 9a Cytotoxicity Is Enhanced by G4mid-Stabilizing PPRH Oligonucleotides in KRAS-Dependent Pancreatic Cell Lines
3.3.2. Compound 9a Demonstrates Enhanced Cytotoxicity in a Three-Dimensional Morphology of MiaPaCa2 Pancreatic Cancer Cells
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Adjei, A.A. Blocking oncogenic Ras signaling for cancer therapy. J. Natl. Cancer Inst. 2001, 93, 1062–1074. [Google Scholar] [CrossRef] [PubMed]
- Colicelli, J. Human RAS superfamily proteins and related GTPases. Sci. STKE 2004, 2004, RE13. [Google Scholar] [CrossRef]
- Friday, B.B.; Adjei, A.A. K-ras as a target for cancer therapy. Biochim. Biophys. Acta 2005, 1756, 127–144. [Google Scholar] [CrossRef] [PubMed]
- Young, A.; Lyons, J.; Miller, A.L.; Phan, V.T.; Alarcon, I.R.; McCormick, F. Ras signaling and therapies. Adv. Cancer Res. 2009, 102, 1–17. [Google Scholar] [PubMed]
- Saxena, N.; Lahiri, S.S.; Hambarde, S.; Tripathi, R.P. RAS: Target for cancer therapy. Cancer Invest 2008, 26, 948–955. [Google Scholar] [CrossRef]
- Hingorani, S.R.; Petricoin, E.F.; Maitra, A.; Rajapakse, V.; King, C.; Jacobetz, M.A.; Ross, S.; Conrads, T.P.; Veenstra, T.D.; Hitt, B.A.; et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell 2003, 4, 437–450. [Google Scholar] [CrossRef]
- Birkeland, E.; Wik, E.; Mjos, S.; Hoivik, E.A.; Trovik, J.; Werner, H.M.; Kusonmano, K.; Petersen, K.; Raeder, M.B.; Holst, F.; et al. KRAS gene amplification and overexpression but not mutation associates with aggressive and metastatic endometrial cancer. Br. J. Cancer 2012, 107, 1997–2004. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609–615. [Google Scholar] [CrossRef]
- Chen, Y.; Mc Gee, J.; Chen, X.; Doman, T.N.; Gong, X.; Zhang, Y.; Hamm, N.; Ma, X.; Higgs, R.E.; Bhagwat, S.V.; et al. Identification of druggable cancer driver genes amplified across TCGA datasets. PLoS ONE 2014, 9, e98293. [Google Scholar] [CrossRef]
- Giltnane, J.M.; Balko, J.M. Rationale for targeting the Ras/MAPK pathway in triple-negative breast cancer. Discov. Med. 2014, 17, 275–283. [Google Scholar]
- Goddard, N.C.; McIntyre, A.; Summersgill, B.; Gilbert, D.; Kitazawa, S.; Shipley, J. KIT and RAS signalling pathways in testicular germ cell tumours: New data and a review of the literature. Int. J. Androl. 2007, 30, 337–348. [Google Scholar] [CrossRef]
- Mackenzie, R.; Kommoss, S.; Winterhoff, B.J.; Kipp, B.R.; Garcia, J.J.; Voss, J.; Halling, K.; Karnezis, A.; Senz, J.; Yang, W.; et al. Targeted deep sequencing of mucinous ovarian tumors reveals multiple overlapping RAS-pathway activating mutations in borderline and cancerous neoplasms. BMC Cancer 2015, 15, 415. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.T.; Nakayama, K.; Rahman, M.; Katagiri, H.; Katagiri, A.; Ishibashi, T.; Ishikawa, M.; Sato, E.; Iida, K.; Nakayama, N.; et al. KRAS and MAPK1 gene amplification in type II ovarian carcinomas. Int. J. Mol. Sci. 2013, 14, 13748–13762. [Google Scholar] [CrossRef] [PubMed]
- Sotorasib Edges Closer to Approval. Cancer Discov. 2021, 11, OF2. [CrossRef]
- Hong, D.S.; Fakih, M.G.; Strickler, J.H.; Desai, J.; Durm, G.A.; Shapiro, G.I.; Falchook, G.S.; Price, T.J.; Sacher, A.; Denlinger, C.S.; et al. KRAS(G12C) Inhibition with Sotorasib in Advanced Solid Tumors. N. Engl. J. Med. 2020, 383, 1207–1217. [Google Scholar] [CrossRef] [PubMed]
- Luo, J. KRAS mutation in pancreatic cancer. Semin. Oncol. 2021, 48, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Meng, M.; Zhong, K.; Jiang, T.; Liu, Z.; Kwan, H.Y.; Su, T. The current understanding on the impact of KRAS on colorectal cancer. Biomed. Pharm. 2021, 140, 111717. [Google Scholar] [CrossRef]
- Duursma, A.M.; Agami, R. Ras interference as cancer therapy. Semin. Cancer Biol. 2003, 13, 267–273. [Google Scholar] [CrossRef]
- Wickstrom, E. Oligonucleotide treatment of ras-induced tumors in nude mice. Mol. Biotechnol. 2001, 18, 35–55. [Google Scholar] [CrossRef]
- Ali, S.; Ahmad, A.; Aboukameel, A.; Bao, B.; Padhye, S.; Philip, P.A.; Sarkar, F.H. RETRACTED: Increased Ras GTPase activity is regulated by miRNAs that can be attenuated by CDF treatment in pancreatic cancer cells. Cancer Lett. 2012, 319, 173–181. [Google Scholar] [CrossRef]
- Hu, Y.; Ou, Y.; Wu, K.; Chen, Y.; Sun, W. miR-143 inhibits the metastasis of pancreatic cancer and an associated signaling pathway. Tumour Biol. 2012, 33, 1863–1870. [Google Scholar] [CrossRef] [PubMed]
- Brooks, T.A.; Kendrick, S.; Hurley, L. Making sense of G-quadruplex and i-motif functions in oncogene promoters. FEBS J. 2010, 277, 3459–3469. [Google Scholar] [CrossRef] [PubMed]
- Hansel-Hertsch, R.; Beraldi, D.; Lensing, S.V.; Marsico, G.; Zyner, K.; Parry, A.; Di Antonio, M.; Pike, J.; Kimura, H.; Narita, M.; et al. G-quadruplex structures mark human regulatory chromatin. Nat. Genet. 2016, 48, 1267–1272. [Google Scholar] [CrossRef] [PubMed]
- Hansel-Hertsch, R.; Di Antonio, M.; Balasubramanian, S. DNA G-quadruplexes in the human genome: Detection, functions and therapeutic potential. Nat. Rev. Mol. Cell Biol. 2017, 18, 279–284. [Google Scholar] [CrossRef] [PubMed]
- Hansel-Hertsch, R.; Spiegel, J.; Marsico, G.; Tannahill, D.; Balasubramanian, S. Genome-wide mapping of endogenous G-quadruplex DNA structures by chromatin immunoprecipitation and high-throughput sequencing. Nat. Protoc. 2018, 13, 551–564. [Google Scholar] [CrossRef] [PubMed]
- Krafcikova, M.; Dzatko, S.; Caron, C.; Granzhan, A.; Fiala, R.; Loja, T.; Teulade-Fichou, M.P.; Fessl, T.; Hansel-Hertsch, R.; Mergny, J.L.; et al. Monitoring DNA-Ligand Interactions in Living Human Cells Using NMR Spectroscopy. J. Am. Chem. Soc. 2019, 141, 13281–13285. [Google Scholar] [CrossRef]
- Spiegel, J.; Cuesta, S.M.; Adhikari, S.; Hansel-Hertsch, R.; Tannahill, D.; Balasubramanian, S. G-quadruplexes are transcription factor binding hubs in human chromatin. Genome Biol. 2021, 22, 117. [Google Scholar] [CrossRef]
- Balasubramanian, S.; Hurley, L.H.; Neidle, S. Targeting G-quadruplexes in gene promoters: A novel anticancer strategy? Nat. Rev. Drug Discov. 2011, 10, 261–275. [Google Scholar] [CrossRef]
- Hansel-Hertsch, R.; Simeone, A.; Shea, A.; Hui, W.W.I.; Zyner, K.G.; Marsico, G.; Rueda, O.M.; Bruna, A.; Martin, A.; Zhang, X.; et al. Landscape of G-quadruplex DNA structural regions in breast cancer. Nat. Genet. 2020, 52, 878–883. [Google Scholar] [CrossRef]
- Shukla, V.; Samaniego-Castruita, D.; Dong, Z.; Gonzalez-Avalos, E.; Yan, Q.; Sarma, K.; Rao, A. TET deficiency perturbs mature B cell homeostasis and promotes oncogenesis associated with accumulation of G-quadruplex and R-loop structures. Nat. Immunol. 2022, 23, 99–108. [Google Scholar] [CrossRef]
- Jordano, J.; Perucho, M. Chromatin structure of the promoter region of the human c-K-ras gene. Nucleic Acids Res. 1986, 14, 7361–7378. [Google Scholar] [CrossRef] [PubMed]
- Jordano, J.; Perucho, M. Initial characterization of a potential transcriptional enhancer for the human c-K-ras gene. Oncogene 1988, 2, 359–366. [Google Scholar] [PubMed]
- Yamamoto, F.; Perucho, M. Characterization of the human c-K-ras gene promoter. Oncogene Res. 1988, 3, 125–130. [Google Scholar] [PubMed]
- Cogoi, S.; Paramasivam, M.; Filichev, V.; Geci, I.; Pedersen, E.B.; Xodo, L.E. Identification of a new G-quadruplex motif in the KRAS promoter and design of pyrene-modified G4-decoys with antiproliferative activity in pancreatic cancer cells. J. Med. Chem. 2009, 52, 564–568. [Google Scholar] [CrossRef] [PubMed]
- Cogoi, S.; Paramasivam, M.; Spolaore, B.; Xodo, L.E. Structural polymorphism within a regulatory element of the human KRAS promoter: Formation of G4-DNA recognized by nuclear proteins. Nucleic Acids Res. 2008, 36, 3765–3780. [Google Scholar] [CrossRef]
- Cogoi, S.; Xodo, L.E. G-quadruplex formation within the promoter of the KRAS proto-oncogene and its effect on transcription. Nucleic Acids Res. 2006, 34, 2536–2549. [Google Scholar] [CrossRef] [PubMed]
- Membrino, A.; Paramasivam, M.; Cogoi, S.; Alzeer, J.L.; Luedtke, N.W.; Xodo, L.E. Cellular uptake and binding of guanidine-modified phthalocyanines to KRAS/HRAS G-quadruplexes. Chem. Commun. (Camb.) 2010, 46, 625–627. [Google Scholar] [CrossRef] [PubMed]
- Morgan, R.K.; Batra, H.; Gaerig, V.C.; Hockings, J.; Brooks, T.A. Identification and characterization of a new G-quadruplex forming region within the kRAS promoter as a transcriptional regulator. Biochim. Et Biophys. Acta 2016, 1859, 235–245. [Google Scholar] [CrossRef]
- De Cian, A.; Guittat, L.; Kaiser, M.; Sacca, B.; Amrane, S.; Bourdoncle, A.; Alberti, P.; Teulade-Fichou, M.P.; Lacroix, L.; Mergny, J.L. Fluorescence-based melting assays for studying quadruplex ligands. Methods 2007, 42, 183–195. [Google Scholar] [CrossRef] [PubMed]
- Morgan, R.K.; Psaras, A.M.; Lassiter, Q.; Raymer, K.; Brooks, T.A. G-quadruplex deconvolution with physiological mimicry enhances primary screening: Optimizing the FRET Melt(2) assay. Biochim. Biophys. Acta. Gene Regul. Mech. 2020, 1863, 194478. [Google Scholar] [CrossRef] [PubMed]
- Boddupally, P.V.; Hahn, S.; Beman, C.; De, B.; Brooks, T.A.; Gokhale, V.; Hurley, L.H. Anticancer activity and cellular repression of c-MYC by the G-quadruplex-stabilizing 11-piperazinylquindoline is not dependent on direct targeting of the G-quadruplex in the c-MYC promoter. J. Med. Chem. 2012, 55, 6076–6086. [Google Scholar] [CrossRef] [PubMed]
- Ou, T.M.; Lu, Y.J.; Zhang, C.; Huang, Z.S.; Wang, X.D.; Tan, J.H.; Chen, Y.; Ma, D.L.; Wong, K.Y.; Tang, J.C.; et al. Stabilization of G-quadruplex DNA and down-regulation of oncogene c-myc by quindoline derivatives. J. Med. Chem. 2007, 50, 1465–1474. [Google Scholar] [CrossRef] [PubMed]
- Brown, R.V.; Danford, F.L.; Gokhale, V.; Hurley, L.H.; Brooks, T.A. Demonstration that drug-targeted down-regulation of MYC in non-Hodgkins lymphoma is directly mediated through the promoter G-quadruplex. J. Biol. Chem. 2011, 286, 41018–41027. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, E.S.; Montgomerie, A.M.; Sneddon, A.H.; Proctor, G.R.; Green, B. Synthesis of Indolo [3,2-C] Quinolines and Indolo [3,2-D] Benzazepines and Their Interaction with DNA. Eur. J. Med. Chem. 1988, 23, 183–188. [Google Scholar] [CrossRef]
- Psaras, A.M.; Valiuska, S.; Noe, V.; Ciudad, C.J.; Brooks, T.A. Targeting KRAS Regulation with PolyPurine Reverse Hoogsteen Oligonucleotides. Int. J. Mol. Sci. 2022, 23, 2097. [Google Scholar] [CrossRef] [PubMed]
- Kokaji, E.; Shimomura, A.; Minamisaka, T.; Nakajima, T.; Miwa, S.; Hatta, H.; Nishida, T.; Kiya, C.; Imura, J. Endoglin (CD105) and SMAD4 regulate spheroid formation and the suppression of the invasive ability of human pancreatic cancer cells. Int. J. Oncol. 2018, 52, 892–900. [Google Scholar] [CrossRef] [PubMed]
- Feng, H.; Ou, B.C.; Zhao, J.K.; Yin, S.; Lu, A.G.; Oechsle, E.; Thasler, W.E. Homogeneous pancreatic cancer spheroids mimic growth pattern of circulating tumor cell clusters and macrometastases: Displaying heterogeneity and crater-like structure on inner layer. J. Cancer Res. Clin. Oncol. 2017, 143, 1771–1786. [Google Scholar] [CrossRef]
- Ryu, N.E.; Lee, S.H.; Park, H. Spheroid Culture System Methods and Applications for Mesenchymal Stem Cells. Cells 2019, 8, 1620. [Google Scholar] [CrossRef]
- Kainuma, O.; Asano, T.; Hasegawa, M.; Kenmochi, T.; Nakagohri, T.; Tokoro, Y.; Isono, K. Inhibition of growth and invasive activity of human pancreatic cancer cells by a farnesyltransferase inhibitor, manumycin. Pancreas 1997, 15, 379–383. [Google Scholar] [CrossRef]
- Rachagani, S.; Senapati, S.; Chakraborty, S.; Ponnusamy, M.P.; Kumar, S.; Smith, L.M.; Jain, M.; Batra, S.K. Activated KrasG (1) (2) D is associated with invasion and metastasis of pancreatic cancer cells through inhibition of E-cadherin. Br. J. Cancer 2011, 104, 1038–1048. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Singh, A.; Greninger, P.; Rhodes, D.; Koopman, L.; Violette, S.; Bardeesy, N.; Settleman, J. A gene expression signature associated with “K-Ras addiction” reveals regulators of EMT and tumor cell survival. Cancer Cell 2009, 15, 489–500. [Google Scholar] [CrossRef]
- Lavrado, J.; Brito, H.; Borralho, P.M.; Ohnmacht, S.A.; Kim, N.S.; Leitao, C.; Pisco, S.; Gunaratnam, M.; Rodrigues, C.M.; Moreira, R.; et al. KRAS oncogene repression in colon cancer cell lines by G-quadruplex binding indolo [3,2-c] quinolines. Sci. Rep. 2015, 5, 9696. [Google Scholar] [CrossRef]
- Morgan, R.K.; Brooks, T.A. Targeting Promoter G-quadruplexes for Transcriptional Control. In Small-molecule Transcription Factor Inhibitors in Oncology; Rahman, M.T.D., Ed.; RSC Drug Discovery: London, UK, 2018. [Google Scholar]
- Chen, P.Y.; Muzumdar, M.D.; Dorans, K.J.; Robbins, R.; Bhutkar, A.; Del Rosario, A.; Mertins, P.; Qiao, J.; Schafer, A.C.; Gertler, F.; et al. Adaptive and Reversible Resistance to Kras Inhibition in Pancreatic Cancer Cells. Cancer Res. 2018, 78, 985–1002. [Google Scholar] [CrossRef] [PubMed]
- Dai, J.; Carver, M.; Mathad, R.; Hurley, L.H.; Yang, D. Structure of 2:1 quindoline–MYC G-quadruplex: Ligand-induced reorientation and insights into drug design. Nat. Chem. 2011, 133(44), 17673–17680, submitted. [Google Scholar]
- Avino, A.; Eritja, R.; Ciudad, C.J.; Noe, V. Parallel Clamps and Polypurine Hairpins (PPRH) for Gene Silencing and Triplex-Affinity Capture: Design, Synthesis, and Use. Curr. Protoc. Nucleic Acid Chem. 2019, 77, e78. [Google Scholar] [CrossRef]
- Ciudad, C.J.; Rodriguez, L.; Villalobos, X.; Felix, A.J.; Noe, V. Polypurine Reverse Hoogsteen Hairpins as a Gene Silencing Tool for Cancer. Curr. Med. Chem. 2017, 24, 2809–2826. [Google Scholar] [CrossRef]
- Noe, V.; Aubets, E.; Felix, A.J.; Ciudad, C.J. Nucleic acids therapeutics using PolyPurine Reverse Hoogsteen hairpins. Biochem. Pharm. 2020, 189, 114371. [Google Scholar] [CrossRef] [PubMed]
- Noe, V.; Ciudad, C.J. Polypurine Reverse-Hoogsteen Hairpins as a Tool for Exon Skipping at the Genomic Level in Mammalian Cells. Int. J. Mol. Sci. 2021, 22, 3784. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, L.; Villalobos, X.; Sole, A.; Lliberos, C.; Ciudad, C.J.; Noe, V. Improved design of PPRHs for gene silencing. Mol. Pharm. 2015, 12, 867–877. [Google Scholar] [CrossRef]
- Sole, A.; Delagoutte, E.; Ciudad, C.J.; Noe, V.; Alberti, P. Polypurine reverse-Hoogsteen (PPRH) oligonucleotides can form triplexes with their target sequences even under conditions where they fold into G-quadruplexes. Sci. Rep. 2017, 7, 39898. [Google Scholar] [CrossRef]
- Villalobos, X.; Rodriguez, L.; Sole, A.; Lliberos, C.; Mencia, N.; Ciudad, C.J.; Noe, V. Effect of Polypurine Reverse Hoogsteen Hairpins on Relevant Cancer Target Genes in Different Human Cell Lines. Nucleic Acid Ther. 2015, 25, 198–208. [Google Scholar] [CrossRef] [PubMed]
- Paramasivam, M.; Membrino, A.; Cogoi, S.; Fukuda, H.; Nakagama, H.; Xodo, L.E. Protein hnRNP A1 and its derivative Up1 unfold quadruplex DNA in the human KRAS promoter: Implications for transcription. Nucleic Acids Res. 2009, 37, 2841–2853. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Wang, Y.; Li, X. Targeting the untargetable KRAS in cancer therapy. Acta Pharm. Sin. B 2019, 9, 871–879. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, T.; Roberts-Thomson, R.; Broadbridge, V.; Price, T. Targeting Mutated KRAS Genes to Treat Solid Tumours. Mol. Diagn. Ther. 2022, 26, 39–49. [Google Scholar] [CrossRef]
- Nagasaka, M.; Potugari, B.; Nguyen, A.; Sukari, A.; Azmi, A.S.; Ou, S.I. KRAS Inhibitors- yes but what next? Direct targeting of KRAS- vaccines, adoptive T cell therapy and beyond. Cancer Treat. Rev. 2021, 101, 102309. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Sequence (5′-3′) |
|---|---|
| MYC | GCGCTTATGGGGAGGGTGGGGAGGGTGGGGAAGGTGGGGAGGAGAC |
| KRAS G4near | AGGGCGGTGTGGGAAGAGGGAAGAGGGGGAGG |
| KRAS G4mid | GCGGGGAGAAGGAGGGGGCCGGGCCGGGCCGGCGGGGGAGGAGCGGGGGCCGGGCC |
| Scramble | AAGAAGAAGAAGAGAAGAATTTTAAGAAGAGAAGAAGAAGAA |
| PPRH1 | GGCGGGGGAGGAGCGGGGGTTTTGGGGGCGAGGAGGGGGCGG |
| PPRH2 | GGGGAGAAGGAGGGGGCCGGGTTTTGGGCCGGGGGAGGAAGAGGGG |
| Compound | KRAS G4mid | KRAS G4near | MYC |
|---|---|---|---|
| Control | 47 | 37 | 46 |
| TMPyP2 | 47 | 34 | 46 |
| TMPyP4 | 69 | 65 | 75 |
| Quindoline i | 58 | 77 | 71 |
| NSC 176327 | 46 | 54 | 55 |
| NSC 338258 | 49 | 37 | 54 |
| NSC 317605 | 54 | 38 | 57 |
| Chemotherapy | 2D | 3D |
|---|---|---|
| SN38 | 0.02 ± 0.01 | 0.02 ± 0.01 |
| 5FU | 19 ± 7 | 19 ± 7 |
| Oxali | 5 ± 3 | 11 ± 3 2 |
| GEM | 0.02 ± 0.01 1 | 0.01 ± 0.00 1,2 |
| TXL | 14 ± 14 | 13 ± 11 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Psaras, A.M.; Carty, R.K.; Miller, J.T.; Tumey, L.N.; Brooks, T.A. Indoloquinoline-Mediated Targeted Downregulation of KRAS through Selective Stabilization of the Mid-Promoter G-Quadruplex Structure. Genes 2022, 13, 1440. https://doi.org/10.3390/genes13081440
Psaras AM, Carty RK, Miller JT, Tumey LN, Brooks TA. Indoloquinoline-Mediated Targeted Downregulation of KRAS through Selective Stabilization of the Mid-Promoter G-Quadruplex Structure. Genes. 2022; 13(8):1440. https://doi.org/10.3390/genes13081440
Chicago/Turabian StylePsaras, Alexandra Maria, Rhianna K. Carty, Jared T. Miller, L. Nathan Tumey, and Tracy A. Brooks. 2022. "Indoloquinoline-Mediated Targeted Downregulation of KRAS through Selective Stabilization of the Mid-Promoter G-Quadruplex Structure" Genes 13, no. 8: 1440. https://doi.org/10.3390/genes13081440
APA StylePsaras, A. M., Carty, R. K., Miller, J. T., Tumey, L. N., & Brooks, T. A. (2022). Indoloquinoline-Mediated Targeted Downregulation of KRAS through Selective Stabilization of the Mid-Promoter G-Quadruplex Structure. Genes, 13(8), 1440. https://doi.org/10.3390/genes13081440