
Comparative Transcriptome Profiling Reveals Potential Candidate Genes, Transcription Factors, and Biosynthetic Pathways for Phosphite Response in Potato (Solanum tuberosum L.)

 ,
, 
 ,
,  ,
,  ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Planting Materials and In Vitro Propagation Conditions
Experimental Design and Treatments
2.2. Isolation of RNA, Library Construction, and RNA Sequencing
2.3. Sequencing, Reads Mapping, and Differential Expression Analysis Identification
2.4. Identification of Transcription Factors in the Differentially Expressed Genes
2.5. Validation of DEGs by qRT-PCR
2.6. Analysis of Data
3. Results
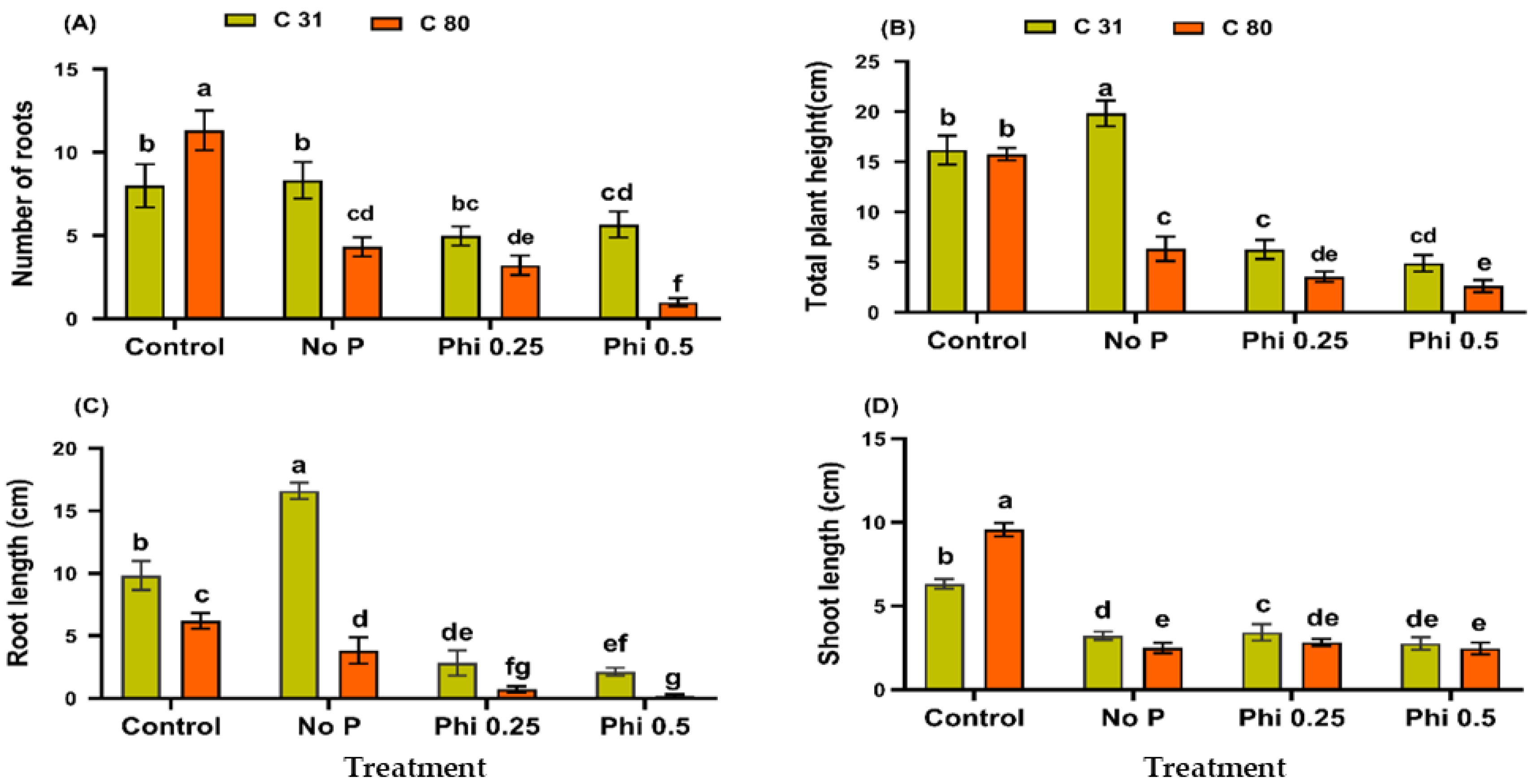
3.1. Phi Impedes Physiological Growth and Development of the Potato in a Medium without Pi
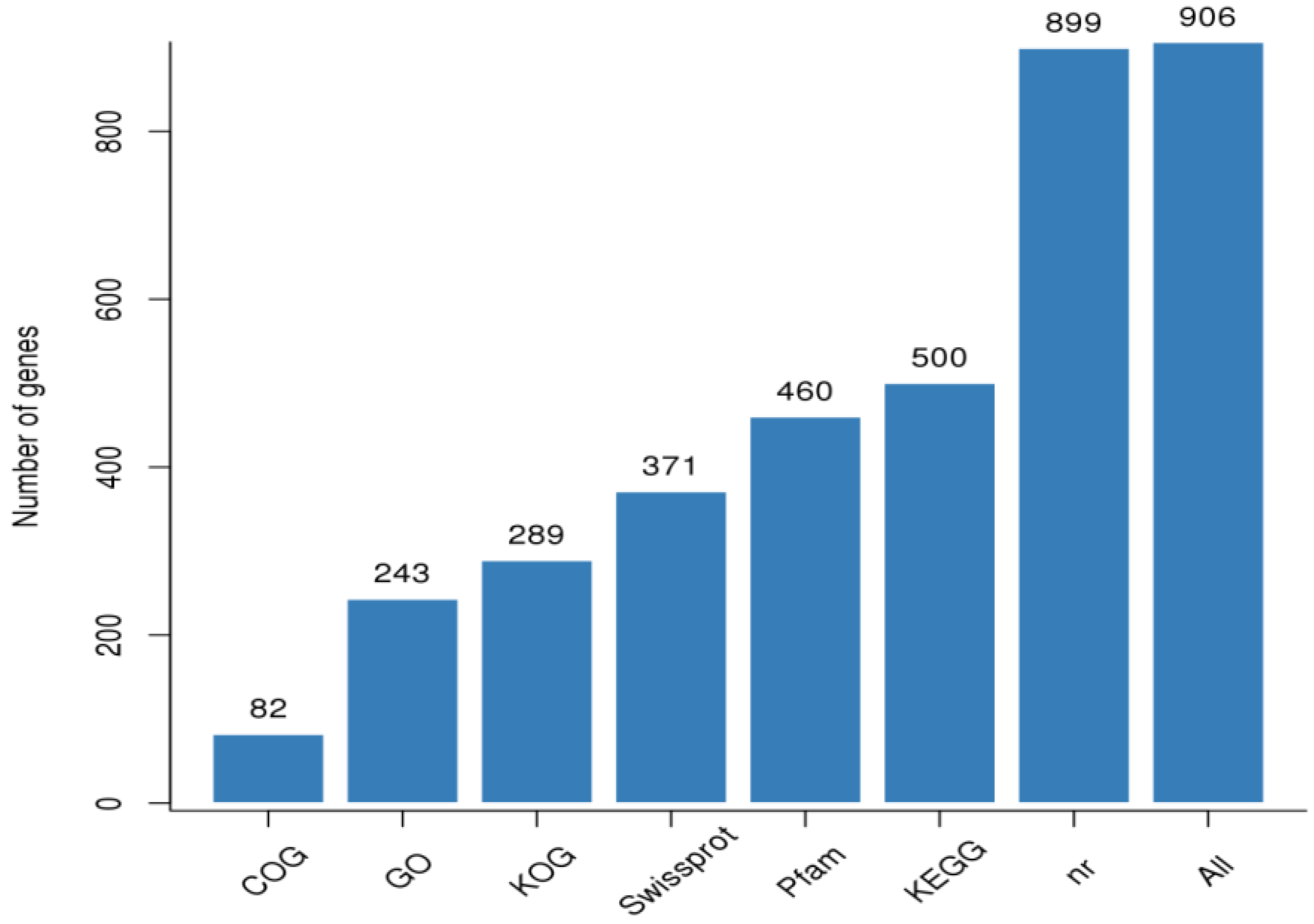
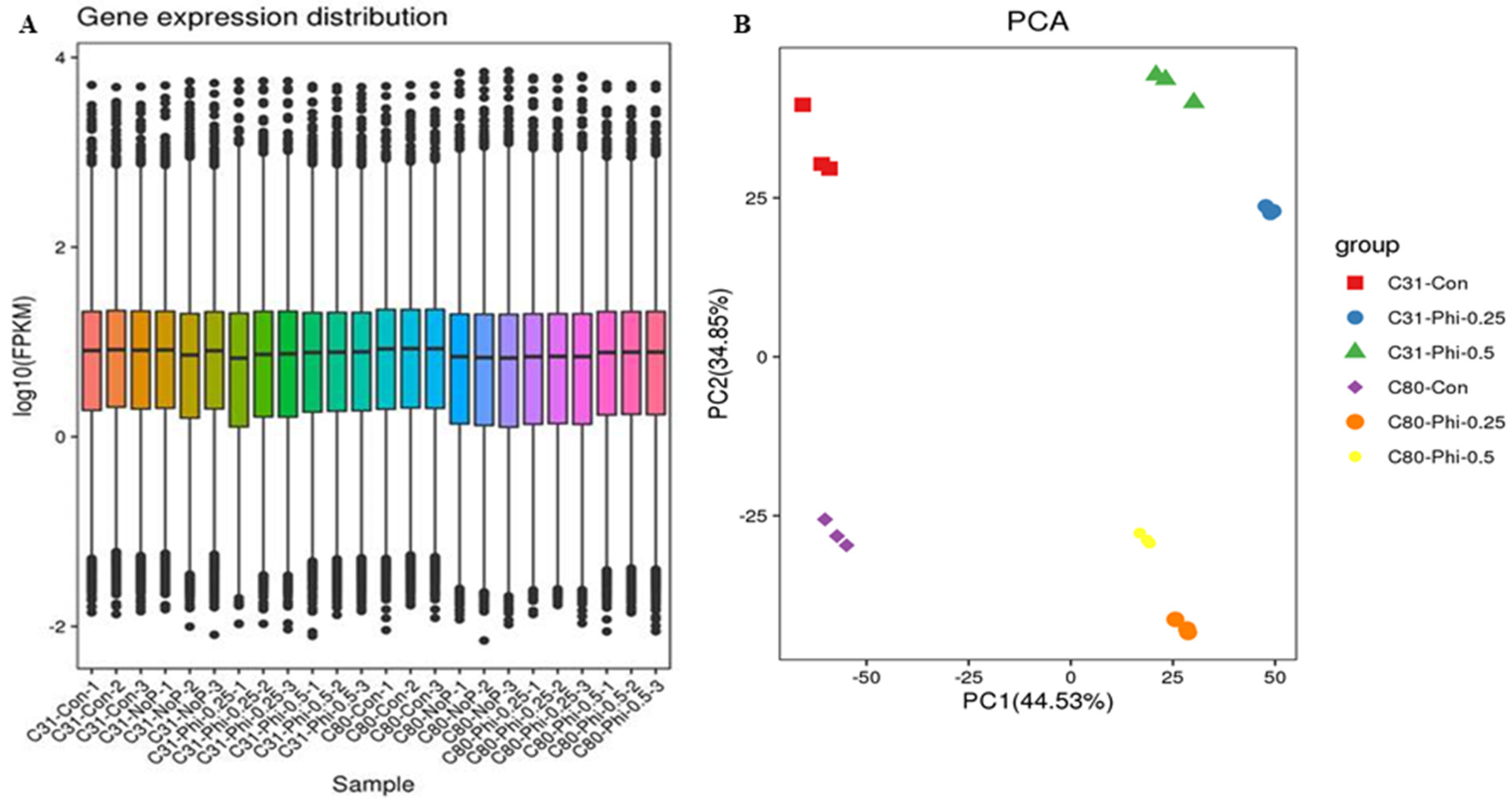
3.2. RNA Sample Sequencing, Quality Control, and Functional Annotation
Gene Annotation and Functional Classification of DEGs
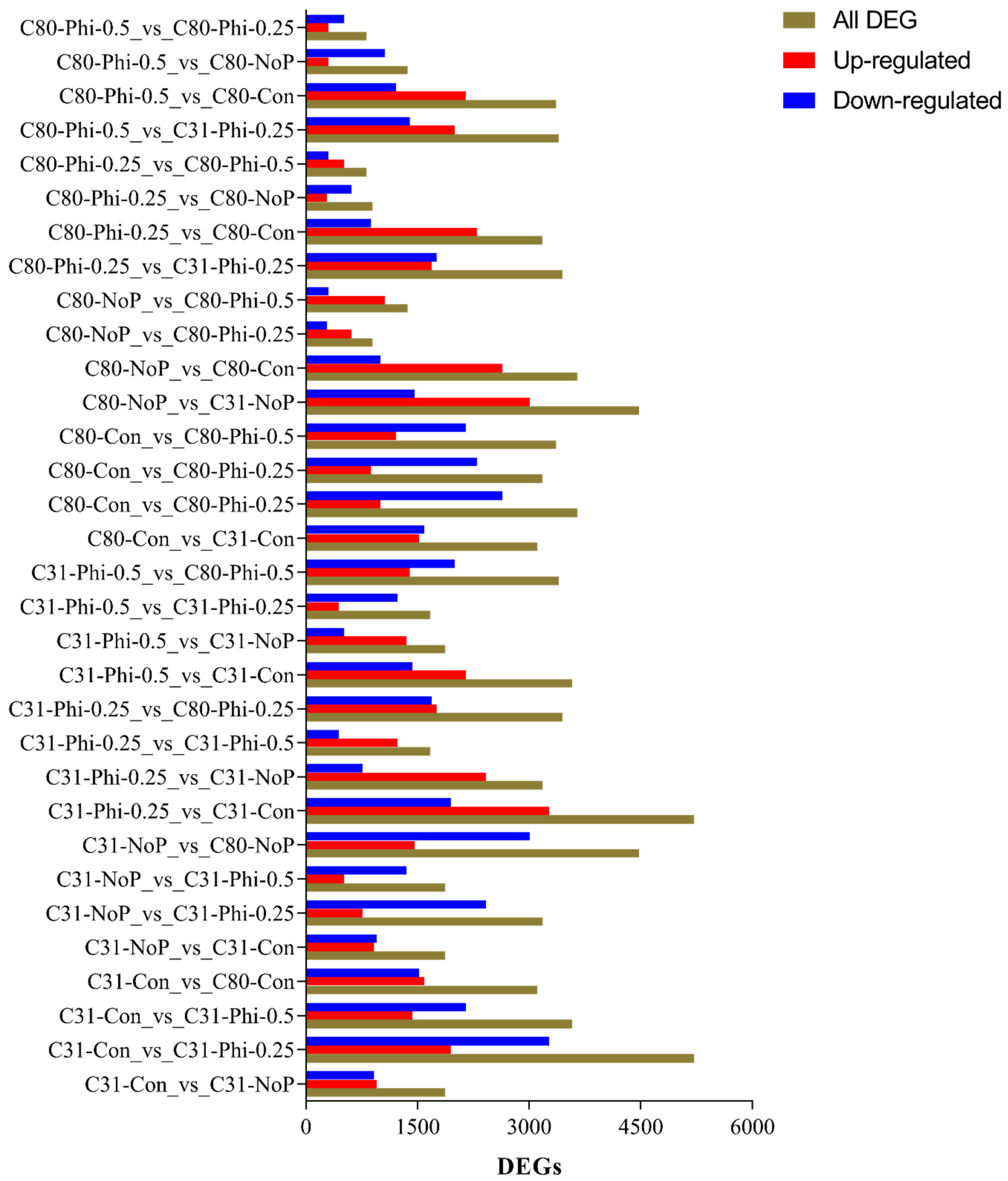
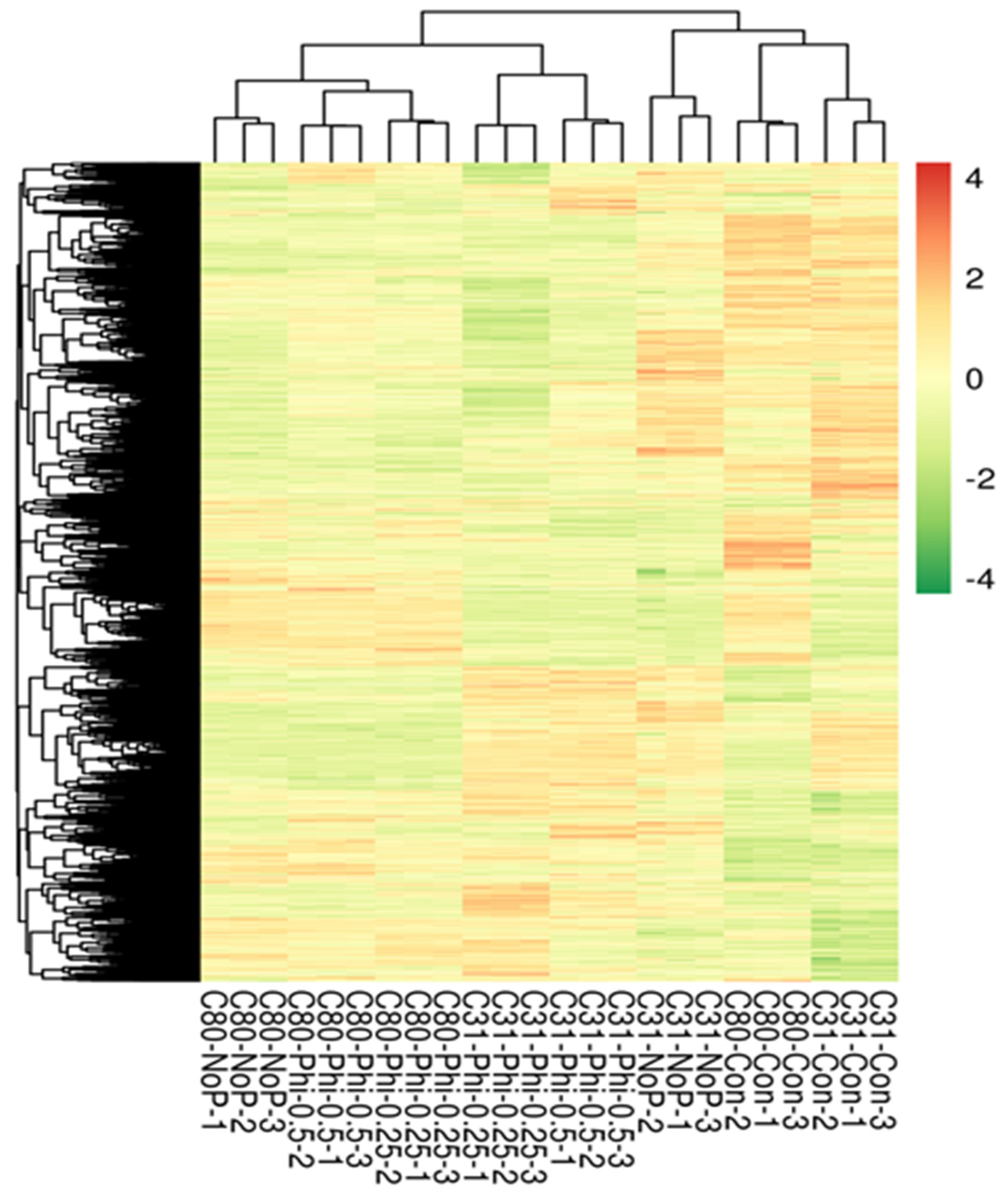
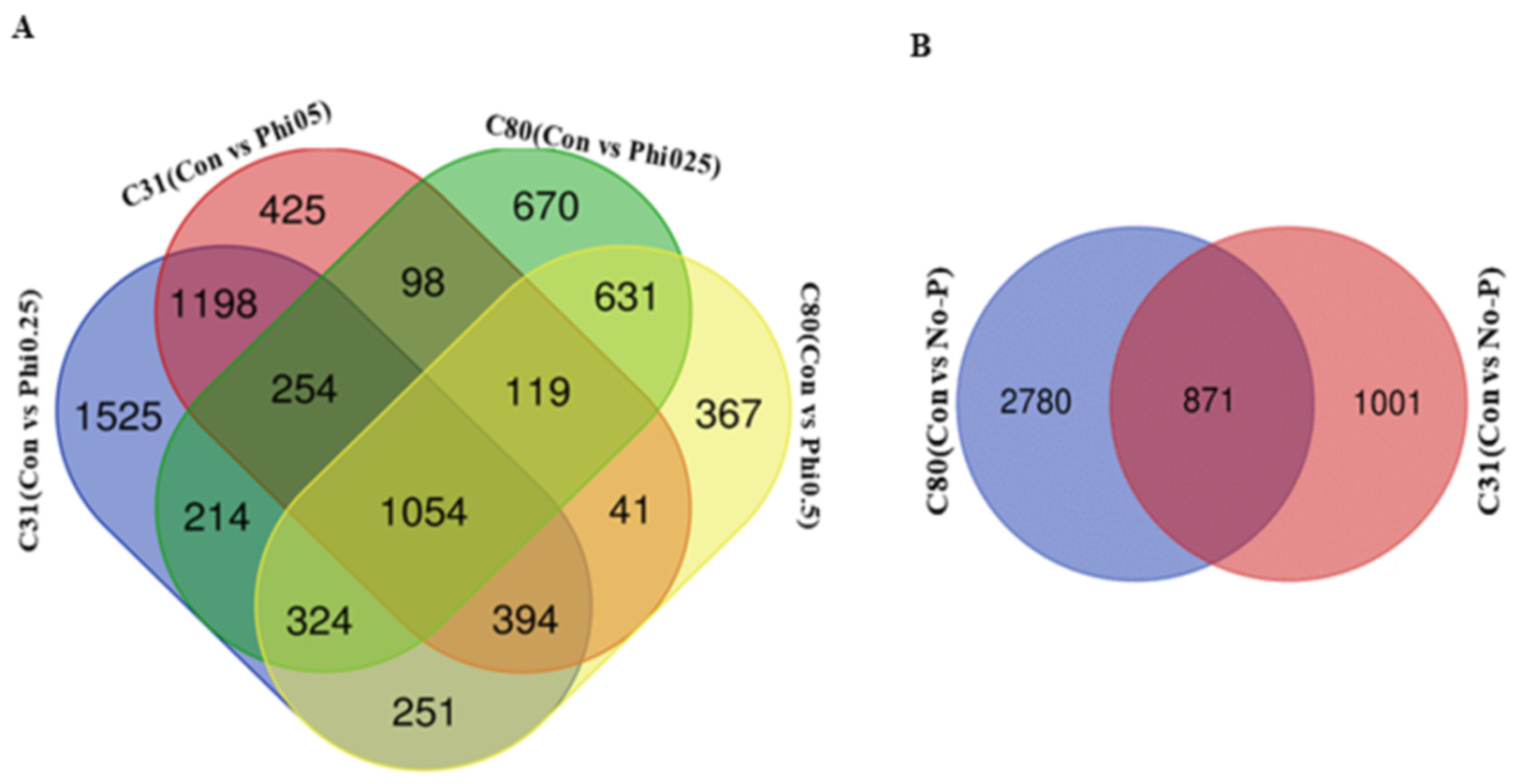
3.3. Identification of Differentially Expressed Genes in Pairwise Comparison between the Six Categories
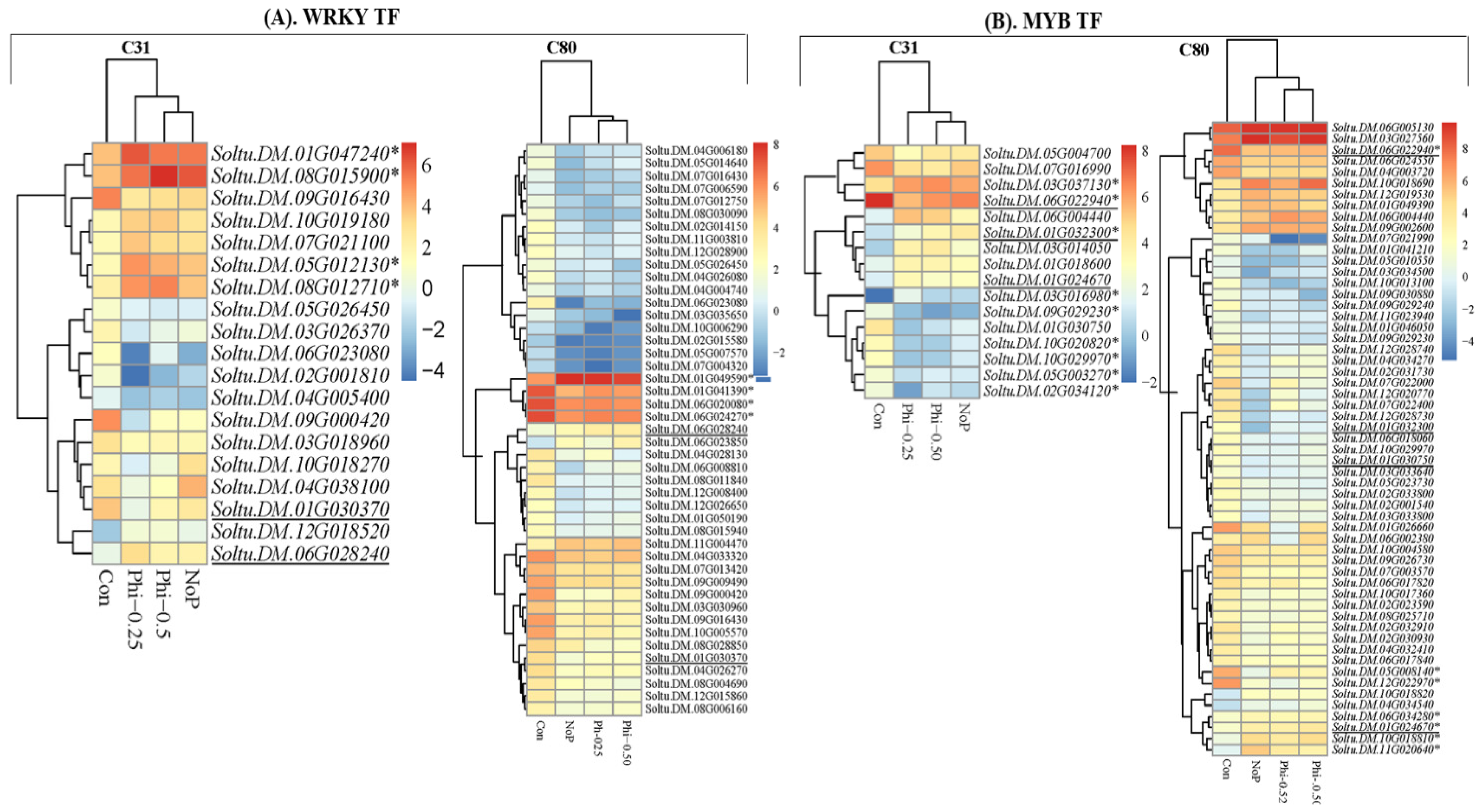
3.4. 6 DEGs in the Context of Transcription Factors
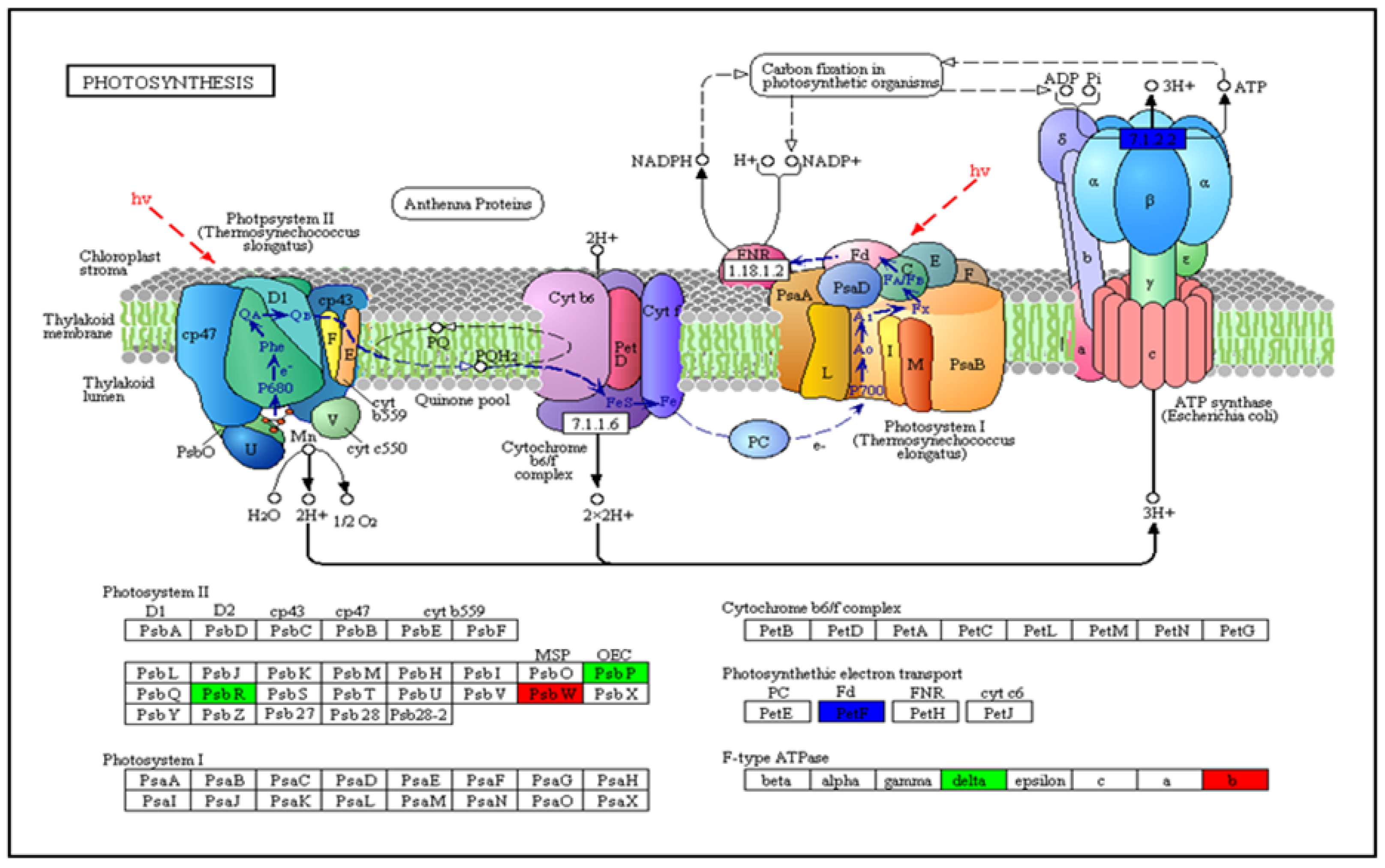
3.5. Analysis of COG Enhancement and KEGG Pathway DEGs
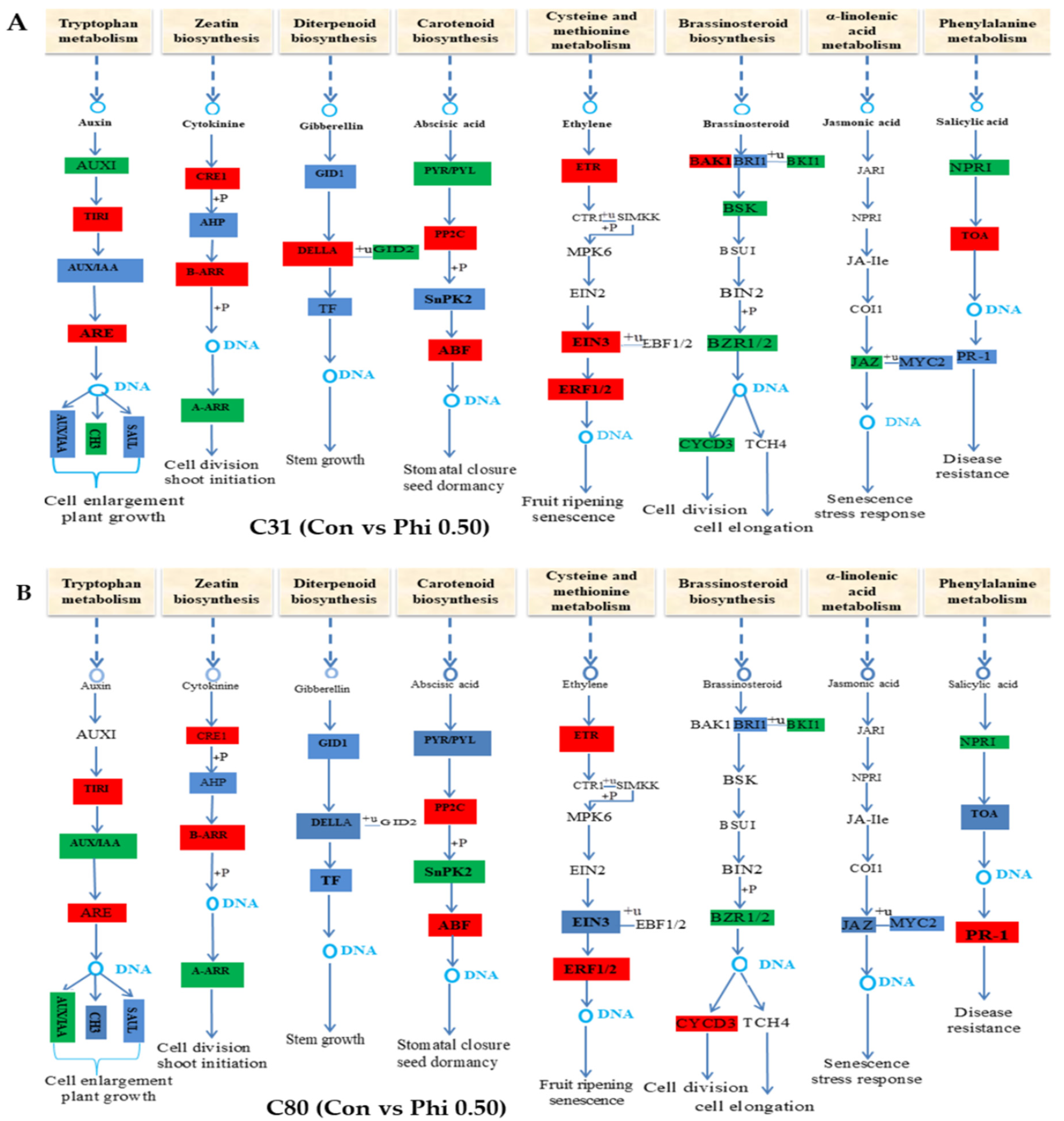
3.6. Genes Involved in Hormone Signaling in Plants Have Been Discovered for Differential Expression Analysis
3.7. Expression Patterns of 18 Different Genes in Response to Phi Stress by qRT-PCR
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Marschner, P.; Rengel, Z. Nutrient availability in soils. In Marschner’s Mineral Nutrition of Higher Plants; Elsevier: Amsterdam, The Netherlands, 2012; pp. 315–330. [Google Scholar]
- Raghothama, K. Phosphate acquisition. Annu. Rev. Plant Biol. 1999, 50, 665–693. [Google Scholar] [CrossRef] [PubMed]
- López-Arredondo, D.L.; Leyva-González, M.A.; González-Morales, S.I.; López-Bucio, J.; Herrera-Estrella, L. Phosphate nutrition: Improving low-phosphate tolerance in crops. Annu. Rev. Plant Biol. 2014, 65, 95–123. [Google Scholar] [CrossRef] [PubMed]
- Misson, J.; Raghothama, K.G.; Jain, A.; Jouhet, J.; Block, M.A.; Bligny, R.; Ortet, P.; Creff, A.; Somerville, S.; Rolland, N. A genome-wide transcriptional analysis using Arabidopsis thaliana Affymetrix gene chips determined plant responses to phosphate deprivation. Proc. Natl. Acad. Sci. USA 2005, 102, 11934–11939. [Google Scholar] [CrossRef] [PubMed]
- Bustos, R.; Castrillo, G.; Linhares, F.; Puga, M.I.; Rubio, V.; Pérez-Pérez, J.; Solano, R.; Leyva, A.; Paz-Ares, J. A central regulatory system largely controls transcriptional activation and repression responses to phosphate starvation in Arabidopsis. PLoS Genet. 2010, 6, e1001102. [Google Scholar] [CrossRef]
- Secco, D.; Jabnoune, M.; Walker, H.; Shou, H.; Wu, P.; Poirier, Y.; Whelan, J. Spatio-temporal transcript profiling of rice roots and shoots in response to phosphate starvation and recovery. Plant Cell 2013, 25, 4285–4304. [Google Scholar] [CrossRef]
- Liu, Y.; Cao, X. Characteristics and significance of the pre-metastatic niche. Cancer Cell 2016, 30, 668–681. [Google Scholar] [CrossRef]
- Raghothama, K. Phosphate transport and signaling. Curr. Opin. Plant Biol. 2000, 3, 182–187. [Google Scholar] [CrossRef]
- Ullrich-Eberius, C.I.; Novacky, A.; Fischer, E.; Lüttge, U. Relationship between energy-dependent phosphate uptake and the electrical membrane potential in Lemna gibba G1. Plant Physiol. 1981, 67, 797–801. [Google Scholar] [CrossRef]
- Guest, D.; Grant, B. The complex action of phosphonates as antifungal agents. Biol. Rev. 1991, 66, 159–187. [Google Scholar] [CrossRef]
- Carswell, M.C.; Grant, B.R.; Plaxton, W.C. Disruption of the phosphate-starvation response of oilseed rape suspension cells by the fungicide phosphonate. Planta 1997, 203, 67–74. [Google Scholar] [CrossRef]
- Ouimette, D.; Coffey, M. Symplastic entry and phloem translocation of phosphonate. Pestic. Biochem. Physiol. 1990, 38, 18–25. [Google Scholar] [CrossRef]
- MacIntire, W.; Winterberg, S.; Harbin, L. Fertilizer evaluation of certain phosphorus, phosporous and phosphoric materials by means of pot cultures. Agron. J. 1950, 42, 543–549. [Google Scholar] [CrossRef]
- Sukarno, N.; Smith, S.; Scott, E. The effect of fungicides on vesicular–arbuscular mycorrhizal symbiosis: I. The effects on vesicular–arbuscular mycorrhizal fungi and plant growth. New Phytol. 1993, 125, 139–147. [Google Scholar] [CrossRef]
- Ticconi, C.A.; Delatorre, C.A.; Abel, S. Attenuation of phosphate starvation responses by phosphite in Arabidopsis. Plant Physiol. 2001, 127, 963–972. [Google Scholar] [CrossRef] [PubMed]
- Ohtake, H.; Wu, H.; Imazu, K.; Anbe, Y.; Kato, J.; Kuroda, A. Bacterial phosphonate degradation, phosphite oxidation and polyphosphate accumulation. Resour. Conserv. Recycl. 1996, 18, 125–134. [Google Scholar] [CrossRef]
- Fang, E.F.; Kassahun, H.; Croteau, D.L.; Scheibye-Knudsen, M.; Marosi, K.; Lu, H.; Shamanna, R.A.; Kalyanasundaram, S.; Bollineni, R.C.; Wilson, M.A. NAD+ replenishment improves lifespan and healthspan in ataxia telangiectasia models via mitophagy and DNA repair. Cell Metab. 2016, 24, 566–581. [Google Scholar] [CrossRef]
- Conesa, A.; Madrigal, P.; Tarazona, S.; Gomez-Cabrero, D.; Cervera, A.; McPherson, A.; Szcześniak, M.W.; Gaffney, D.J.; Elo, L.L.; Zhang, X. A survey of best practices for RNA-seq data analysis. Genome Biol. 2016, 17, 13. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Yao, X.; Ren, H.; Chang, J.; Wang, K. RNA-Seq reveals flavonoid biosynthesis-related genes in pecan (Carya illinoinensis) kernels. J. Agric. Food Chem. 2018, 67, 148–158. [Google Scholar] [CrossRef] [PubMed]
- Effah, Z.; Li, L.; Xie, J.; Karikari, B.; Liu, C.; Xu, A.; Zeng, M. Transcriptome profiling reveals major structural genes, transcription factors and biosynthetic pathways involved in leaf senescence and nitrogen remobilization in rainfed spring wheat under different nitrogen fertilization rates. Genomics 2022, 114, 110271. [Google Scholar] [CrossRef]
- Bhardwaj, V.; Gumber, D.; Abbot, V.; Dhiman, S.; Sharma, P. Pyrrole: A resourceful small molecule in key medicinal hetero-aromatics. RSC Adv. 2015, 5, 15233–15266. [Google Scholar] [CrossRef]
- Garg, R.; Shankar, R.; Thakkar, B.; Kudapa, H.; Krishnamurthy, L.; Mantri, N.; Varshney, R.K.; Bhatia, S.; Jain, M. Transcriptome analyses reveal genotype-and developmental stage-specific molecular responses to drought and salinity stresses in chickpea. Sci. Rep. 2016, 6, 19228. [Google Scholar] [CrossRef] [PubMed]
- Hammond, J.P.; Bennett, M.J.; Bowen, H.C.; Broadley, M.R.; Eastwood, D.C.; May, S.T.; Rahn, C.; Swarup, R.; Woolaway, K.E.; White, P.J. Changes in gene expression in Arabidopsis shoots during phosphate starvation and the potential for developing smart plants. Plant Physiol. 2003, 132, 578–596. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.; Ma, L.; Hou, X.; Wang, M.; Wu, Y.; Liu, F.; Deng, X.W. Phosphate starvation triggers distinct alterations of genome expression in Arabidopsis roots and leaves. Plant Physiol. 2003, 132, 1260–1271. [Google Scholar] [CrossRef] [PubMed]
- Morcuende, R.; Bari, R.; Gibon, Y.; Zheng, W.; Pant, B.D.; Bläsing, O.; Usadel, B.; Czechowski, T.; Udvardi, M.K.; Stitt, M. Genome-wide reprogramming of metabolism and regulatory networks of Arabidopsis in response to phosphorus. Plant Cell Environ. 2007, 30, 85–112. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.Y.; Roessner, U.; Eickmeier, I.; Genc, Y.; Callahan, D.L.; Shirley, N.; Langridge, P.; Bacic, A. Metabolite profiling reveals distinct changes in carbon and nitrogen metabolism in phosphate-deficient barley plants (Hordeum vulgare L.). Plant Cell Physiol. 2008, 49, 691–703. [Google Scholar] [CrossRef]
- Rae, A.L.; Cybinski, D.H.; Jarmey, J.M.; Smith, F.W. Characterization of two phosphate transporters from barley; evidence for diverse function and kinetic properties among members of the Pht1 family. Plant Mol. Biol. 2003, 53, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Preuss, C.P.; Huang, C.Y.; Gilliham, M.; Tyerman, S.D. Channel-like characteristics of the low-affinity barley phosphate transporter PHT1; 6 when expressed in Xenopus oocytes. Plant Physiol. 2010, 152, 1431–1441. [Google Scholar] [CrossRef] [PubMed]
- Nussaume, L.; Kanno, S.; Javot, H.; Marin, E.; Pochon, N.; Ayadi, A.; Nakanishi, T.M.; Thibaud, M.-C. Phosphate import in plants: Focus on the PHT1 transporters. Front. Plant Sci. 2011, 2, 83. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Cai, S.; Zhang, G.; Ruan, S. Transcriptome-Based Analysis of Phosphite-Induced Resistance against Pathogens in Rice. Plants 2020, 9, 1334. [Google Scholar] [CrossRef]
- Coquoz, J.; Buchala, A.; Meuwly, P.; Metraux, J. Arachidonic acid induces local but not systemic synthesis of salicylic acid and confers systemic resistance in potato plants to Phytophthora infestans and Alternaria solani. Phytopathology 1995, 85, 1219–1224. [Google Scholar] [CrossRef]
- Dalio, R.J.; Fleischmann, F.; Humez, M.; Osswald, W. Phosphite protects Fagus sylvatica seedlings towards Phytophthora plurivora via local toxicity, priming and facilitation of pathogen recognition. PLoS ONE 2014, 9, e87860. [Google Scholar] [CrossRef] [PubMed]
- Gill, U.S.; Sun, L.; Rustgi, S.; Tang, Y.; von Wettstein, D.; Mysore, K.S. Transcriptome-based analyses of phosphite-mediated suppression of rust pathogens Puccinia emaculata and Phakopsora pachyrhizi and functional characterization of selected fungal target genes. Plant J. 2018, 93, 894–904. [Google Scholar] [CrossRef]
- Zhou, J.; Jiao, F.; Wu, Z.; Li, Y.; Wang, X.; He, X.; Zhong, W.; Wu, P. OsPHR2 is involved in phosphate-starvation signaling and excessive phosphate accumulation in shoots of plants. Plant Physiol. 2008, 146, 1673–1686. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Gao, Q.; Liu, Y.; He, C.; Zhang, X.; Zhang, J. Overexpression of transcription factor ZmPTF1 improves low phosphate tolerance of maize by regulating carbon metabolism and root growth. Planta 2011, 233, 1129–1143. [Google Scholar] [CrossRef] [PubMed]
- Devaiah, B.N.; Karthikeyan, A.S.; Raghothama, K.G. WRKY75 transcription factor is a modulator of phosphate acquisition and root development in Arabidopsis. Plant Physiol. 2007, 143, 1789–1801. [Google Scholar] [CrossRef]
- Yi, K.; Wu, Z.; Zhou, J.; Du, L.; Guo, L.; Wu, Y.; Wu, P. OsPTF1, a novel transcription factor involved in tolerance to phosphate starvation in rice. Plant Physiol. 2005, 138, 2087–2096. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.; Scossa, F.; Bolger, M.E.; Lanz, C.; Maumus, F.; Tohge, T.; Quesneville, H.; Alseekh, S.; Sørensen, I.; Lichtenstein, G. The genome of the stress-tolerant wild tomato species Solanum pennellii. Nat. Genet. 2014, 46, 1034–1038. [Google Scholar] [CrossRef]
- Murashige, T.; Skoog, F. A revised medium for rapid growth and bio assays with tobacco tissue cultures. Physiol. Plant. 1962, 15, 473–497. [Google Scholar] [CrossRef]
- Dormatey, R.; Sun, C.; Ali, K.; Qin, T.; Xu, D.; Bi, Z.; Bai, J. Influence of Phosphite Supply in the MS Medium on Root Morphological Characteristics, Fresh Biomass and Enzymatic Behavior in Five Genotypes of Potato (Solanum tuberosum L.). Horticulturae 2021, 7, 265. [Google Scholar] [CrossRef]
- Iqbal, A.; Gui, H.; Zhang, H.; Wang, X.; Pang, N.; Dong, Q.; Song, M. Genotypic variation in cotton genotypes for phosphorus-use efficiency. Agronomy 2019, 9, 689. [Google Scholar] [CrossRef]
- Wei, K.; Zhang, Y.; Wu, L.; Li, H.; Ruan, L.; Bai, P.; Zhang, C.; Zhang, F.; Xu, L.; Wang, L. Gene expression analysis of bud and leaf color in tea. Plant Physiol. Biochem. 2016, 107, 310–318. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2013. [Google Scholar]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; Van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [PubMed]
- Guo, B.; Guo, Y.; Hong, H.; Qiu, L.-J. Identification of genomic insertion and flanking sequence of G2-EPSPS and GAT transgenes in soybean using whole genome sequencing method. Front. Plant Sci. 2016, 7, 1009. [Google Scholar] [CrossRef] [PubMed]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef]
- Deng, W.; Zhu, X.; Skogerbø, G.; Zhao, Y.; Fu, Z.; Wang, Y.; He, H.; Cai, L.; Sun, H.; Liu, C. Organization of the Caenorhabditis elegans small non-coding transcriptome: Genomic features, biogenesis, and expression. Genome Res. 2006, 16, 20–29. [Google Scholar] [CrossRef]
- Finn, R.D.; Bateman, A.; Clements, J.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Heger, A.; Hetherington, K.; Holm, L.; Mistry, J. Pfam: The protein families database. Nucleic Acids Res. 2014, 42, D222–D230. [Google Scholar] [CrossRef] [PubMed]
- Tatusov, R.L.; Galperin, M.Y.; Natale, D.A.; Koonin, E.V. The COG database: A tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 2000, 28, 33–36. [Google Scholar] [CrossRef] [PubMed]
- Koonin, E.V.; Fedorova, N.D.; Jackson, J.D.; Jacobs, A.R.; Krylov, D.M.; Makarova, K.S.; Mazumder, R.; Mekhedov, S.L.; Nikolskaya, A.N.; Rao, B.S. A comprehensive evolutionary classification of proteins encoded in complete eukaryotic genomes. Genome Biol. 2004, 5, R7. [Google Scholar] [CrossRef]
- Boeckmann, B.; Bairoch, A.; Apweiler, R.; Blatter, M.-C.; Estreicher, A.; Gasteiger, E.; Martin, M.J.; Michoud, K.; O’Donovan, C.; Phan, I. The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Res. 2003, 31, 365–370. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T. Gene ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Yue, J.; Liu, J.; Tang, W.; Wu, Y.Q.; Tang, X.; Li, W.; Yang, Y.; Wang, L.; Huang, S.; Fang, C. Kiwifruit Genome Database (KGD): A comprehensive resource for kiwifruit genomics. Hortic. Res. 2020, 7, 117. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.; Chai, Z.; Yao, W.; Chen, B.; Zhang, M. Interactions between ScNAC23 and ScGAI regulate GA-mediated flowering and senescence in sugarcane. Plant Sci. 2021, 304, 110806. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B (Methodol.) 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Zheng, Y.; Jiao, C.; Sun, H.; Rosli, H.; Pombo, M.A.; Zhang, P.; Banf, M.; Dai, X.; Martin, G.B.; Giovannoni, J.J. iTAK: A program for genome-wide prediction and classification of plant transcription factors, transcriptional regulators, and protein kinases. Mol. Plant 2016, 9, 1667–1670. [Google Scholar] [CrossRef] [PubMed]
- Mistry, J.; Finn, R.D.; Eddy, S.R.; Bateman, A.; Punta, M. Challenges in homology search: HMMER3 and convergent evolution of coiled-coil regions. Nucleic Acids Res. 2013, 41, e121. [Google Scholar] [CrossRef]
- Zhang, H.-M.; Chen, H.; Liu, W.; Liu, H.; Gong, J.; Wang, H.; Guo, A.-Y. AnimalTFDB: A comprehensive animal transcription factor database. Nucleic Acids Res. 2012, 40, D144–D149. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2− ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Gao, Y.; Liu, C.; Li, X.; Xu, H.; Liang, Y.; Ma, N.; Fei, Z.; Gao, J.; Jiang, C.-Z.; Ma, C. Transcriptome profiling of petal abscission zone and functional analysis of an Aux/IAA family gene RhIAA16 involved in petal shedding in rose. Front. Plant Sci. 2016, 7, 1375. [Google Scholar] [CrossRef]
- Tran, L.-S.P.; Nishiyama, R.; Yamaguchi-Shinozaki, K.; Shinozaki, K. Potential utilization of NAC transcription factors to enhance abiotic stress tolerance in plants by biotechnological approach. GM Crops 2010, 1, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Erpen, L.; Devi, H.S.; Grosser, J.W.; Dutt, M. Potential use of the DREB/ERF, MYB, NAC and WRKY transcription factors to improve abiotic and biotic stress in transgenic plants. Plant Cell Tissue Organ Cult. (PCTOC) 2018, 132, 1–25. [Google Scholar] [CrossRef]
- Shen, N.; Hou, S.; Tu, G.; Lan, W.; Jing, Y. Transcription factor WRKY33 mediates the phosphate deficiency-induced remodeling of root architecture by modulating iron homeostasis in Arabidopsis roots. Int. J. Mol. Sci. 2021, 22, 9275. [Google Scholar] [CrossRef] [PubMed]
- Cai, J.; Cai, W.; Huang, X.; Yang, S.; Wen, J.; Xia, X.; Yang, F.; Shi, Y.; Guan, D.; He, S. Ca14-3-3 interacts with cawrky58 to positively modulate pepper response to low-phosphorus starvation. Front. Plant Sci. 2021, 2208, 607878. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Li, K.; Liu, X.; Ruan, H.; Zheng, M.; Yu, Z.; Gai, J.; Yang, S. Transcription factor GmWRKY46 enhanced phosphate starvation tolerance and root development in transgenic plants. Front. Plant Sci. 2021, 1984, 700651. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Wang, Y.; Zhang, W.-H. OsWRKY74, a WRKY transcription factor, modulates tolerance to phosphate starvation in rice. J. Exp. Bot. 2016, 67, 947–960. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Wang, Y.; Yang, A.; Zhang, W.-H. OsMYB2P-1, an R2R3 MYB transcription factor, is involved in the regulation of phosphate-starvation responses and root architecture in rice. Plant Physiol. 2012, 159, 169–183. [Google Scholar] [CrossRef] [PubMed]
- Chutia, R.; Scharfenberg, S.; Neumann, S.; Abel, S.; Ziegler, J. Modulation of phosphate deficiency-induced metabolic changes by iron availability in Arabidopsis thaliana. Int. J. Mol. Sci. 2021, 22, 7609. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Buylla, E.R.; García-Ponce, B.; Sánchez, M.d.l.P.; Espinosa-Soto, C.; García-Gómez, M.L.; Piñeyro-Nelson, A.; Garay-Arroyo, A. MADS-box genes underground becoming mainstream: Plant root developmental mechanisms. New Phytol. 2019, 223, 1143–1158. [Google Scholar] [CrossRef]
- Nuruzzaman, M.; Sharoni, A.M.; Kikuchi, S. Roles of NAC transcription factors in the regulation of biotic and abiotic stress responses in plants. Front. Microbiol. 2013, 4, 248. [Google Scholar] [CrossRef]
- Ryu, H.; Cho, Y.-G. Plant hormones in salt stress tolerance. J. Plant Biol. 2015, 58, 147–155. [Google Scholar] [CrossRef]
- Dormatey, R.; Sun, C.; Ali, K.; Fiaz, S.; Xu, D.; Calderón-Urrea, A.; Bi, Z.; Zhang, J.; Bai, J. ptxD/Phi as alternative selectable marker system for genetic transformation for bio-safety concerns: A review. PeerJ 2021, 9, e11809. [Google Scholar] [CrossRef] [PubMed]
- Vinas, M.; Mendez, J.C.; Jiménez, V.M. Effect of foliar applications of phosphites on growth, nutritional status and defense responses in tomato plants. Sci. Hortic. 2020, 265, 109200. [Google Scholar] [CrossRef]
- Bielecka, M.; Watanabe, M.; Morcuende, R.; Scheible, W.-R.; Hawkesford, M.J.; Hesse, H.; Hoefgen, R. Transcriptome and metabolome analysis of plant sulfate starvation and resupply provides novel information on transcriptional regulation of metabolism associated with sulfur, nitrogen and phosphorus nutritional responses in Arabidopsis. Front. Plant Sci. 2015, 5, 805. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhou, Z.; Gao, J.; Wu, Y.; Xia, Z.; Zhang, H.; Wu, J. The mechanisms of maize resistance to Fusarium verticillioides by comprehensive analysis of RNA-seq data. Front. Plant Sci. 2016, 7, 1654. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.-Z.; Liu, J.-H. Transcriptional profiling of canker-resistant transgenic sweet orange (Citrus sinensis Osbeck) constitutively overexpressing a spermidine synthase gene. BioMed Res. Int. 2013, 2013, 918136. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Tian, Q.; Pang, T.; Jiang, L.; Wu, R.; Xia, X.; Yin, W. Deep-sequencing transcriptome analysis of low temperature perception in a desert tree, Populus euphratica. BMC Genom. 2014, 15, 326. [Google Scholar] [CrossRef] [PubMed]
- Wei, T.; Deng, K.; Wang, H.; Zhang, L.; Wang, C.; Song, W.; Zhang, Y.; Chen, C. Comparative transcriptome analyses reveal potential mechanisms of enhanced drought tolerance in transgenic Salvia miltiorrhiza plants expressing AtDREB1A from Arabidopsis. Int. J. Mol. Sci. 2018, 19, 827. [Google Scholar] [CrossRef] [PubMed]
- Hirst, J.; Robinson, M.S. Clathrin and adaptors. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 1998, 1404, 173–193. [Google Scholar] [CrossRef]
- Holstein, S.E. Molecular dissection of the clathrin-endocytosis machinery in plants. In Plant Endocytosis; Springer: Berlin/Heidelberg, Germany, 2005; pp. 83–101. [Google Scholar]
- Paul, M.J.; Frigerio, L. Coated vesicles in plant cells. Semin. Cell Dev. Biol. 2007, 18, 471–478. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Q.; Niu, F.; Sun, X.; Hu, Z.; Li, X.; Ma, Y.; Zhang, H. RNA-seq analysis of unintended effects in transgenic wheat overexpressing the transcription factor GmDREB1. Crop J. 2017, 5, 207–218. [Google Scholar] [CrossRef]
- Javid, M.G.; Sorooshzadeh, A.; Moradi, F.; Modarres Sanavy, S.A.M.; Allahdadi, I. The role of phytohormones in alleviating salt stress in crop plants. Aust. J. Crop Sci. 2011, 5, 726–734. [Google Scholar]
- Gamalero, E.; Glick, B.R. Ethylene and abiotic stress tolerance in plants. In Environmental Adaptations and Stress Tolerance of Plants in the Era of Climate Change; Springer: Berlin/Heidelberg, Germany, 2012; pp. 395–412. [Google Scholar]
- Wilkinson, S.; Davies, W.J. Drought, ozone, ABA and ethylene: New insights from cell to plant to community. Plant Cell Environ. 2010, 33, 510–525. [Google Scholar] [CrossRef] [PubMed]
- Mao, C.; Lu, S.; Lv, B.; Zhang, B.; Shen, J.; He, J.; Luo, L.; Xi, D.; Chen, X.; Ming, F. A Rice NAC Transcription Factor Promotes Leaf Senescence via ABA Biosynthesis. Plant Physiol. 2017, 174, 1747–1763. [Google Scholar] [CrossRef]
- Song, Y.; Xiang, F.; Zhang, G.; Miao, Y.; Miao, C.; Song, C.-P. Abscisic acid as an internal integrator of multiple physiological processes modulates leaf senescence onset in Arabidopsis thaliana. Front. Plant Sci. 2016, 7, 181. [Google Scholar] [CrossRef] [PubMed]
- Tan, B.C.; Joseph, L.M.; Deng, W.T.; Liu, L.; Li, Q.B.; Cline, K.; McCarty, D.R. Molecular characterization of the Arabidopsis 9-cis epoxycarotenoid dioxygenase gene family. Plant J. 2003, 35, 44–56. [Google Scholar] [CrossRef] [PubMed]
- Liang, C.; Wang, Y.; Zhu, Y.; Tang, J.; Hu, B.; Liu, L.; Ou, S.; Wu, H.; Sun, X.; Chu, J. OsNAP connects abscisic acid and leaf senescence by fine-tuning abscisic acid biosynthesis and directly targeting senescence-associated genes in rice. Proc. Natl. Acad. Sci. USA 2014, 111, 10013–10018. [Google Scholar] [CrossRef] [PubMed]
- Woo, H.R.; Kim, H.J.; Lim, P.O.; Nam, H.G. Leaf senescence: Systems and dynamics aspects. Annu. Rev. Plant Biol. 2019, 70, 347–376. [Google Scholar] [CrossRef] [PubMed]
- Cabello, J.V.; Lodeyro, A.F.; Zurbriggen, M.D. Novel perspectives for the engineering of abiotic stress tolerance in plants. Curr. Opin. Biotechnol. 2014, 26, 62–70. [Google Scholar] [CrossRef] [PubMed]
- Koyama, T. A hidden link between leaf development and senescence. Plant Sci. 2018, 276, 105–110. [Google Scholar] [CrossRef]
- Nur-Athirah, A.-H.; Muhammad-Izzat, A.-F.; Zamri, Z.; Ismanizan, I. Diverse and dynamic roles of F-box proteins in plant biology. Planta 2020, 251, 68. [Google Scholar]
- Li, N.; Han, X.; Feng, D.; Yuan, D.; Huang, L.-J. Signaling crosstalk between salicylic acid and ethylene/jasmonate in plant defense: Do we understand what they are whispering? Int. J. Mol. Sci. 2019, 20, 671. [Google Scholar] [CrossRef] [PubMed]
- Wan, L.; Li, B.; Pandey, M.K.; Wu, Y.; Lei, Y.; Yan, L.; Dai, X.; Jiang, H.; Zhang, J.; Wei, G. Transcriptome analysis of a new peanut seed coat mutant for the physiological regulatory mechanism involved in seed coat cracking and pigmentation. Front. Plant Sci. 2016, 7, 1491. [Google Scholar] [CrossRef]
- Li, Z.; Hu, G.; Liu, X.; Zhou, Y.; Li, Y.; Zhang, X.; Yuan, X.; Zhang, Q.; Yang, D.; Wang, T. Transcriptome sequencing identified genes and gene ontologies associated with early freezing tolerance in maize. Front. Plant Sci. 2016, 7, 1477. [Google Scholar] [CrossRef]
- Singh, D.; Laxmi, A. Transcriptional regulation of drought response: A tortuous network of transcriptional factors. Front. Plant Sci. 2015, 6, 895. [Google Scholar] [CrossRef]
- Ham, B.-K.; Chen, J.; Yan, Y.; Lucas, W.J. Insights into plant phosphate sensing and signaling. Curr. Opin. Biotechnol. 2018, 49, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Bakshi, M.; Oelmüller, R. WRKY transcription factors: Jack of many trades in plants. Plant Signal. Behav. 2014, 9, e27700. [Google Scholar] [CrossRef]
- Wang, Y.; Tao, X.; Tang, X.-M.; Xiao, L.; Sun, J.-l.; Yan, X.-F.; Li, D.; Deng, H.-Y.; Ma, X.-R. Comparative transcriptome analysis of tomato (Solanum lycopersicum) in response to exogenous abscisic acid. BMC Genom. 2013, 14, 1–14. [Google Scholar] [CrossRef]
- Fan, F.; Wang, Q.; Li, H.; Ding, G.; Wen, X. Transcriptome-wide identification and expression profiles of masson pine WRKY transcription factors in response to low phosphorus stress. Plant Mol. Biol. Report. 2021, 39, 814. [Google Scholar] [CrossRef]
- Nilsson, L.; Müller, R.; Nielsen, T.H. Dissecting the plant transcriptome and the regulatory responses to phosphate deprivation. Physiol. Plant. 2010, 139, 129–143. [Google Scholar] [CrossRef]
- Li, C.; Ng, C.K.-Y.; Fan, L.-M. MYB transcription factors, active players in abiotic stress signaling. Environ. Exp. Bot. 2015, 114, 80–91. [Google Scholar] [CrossRef]
- Liu, Y.; Cui, S.; Wu, F.; Yan, S.; Lin, X.; Du, X.; Chong, K.; Schilling, S.; Theißen, G.; Meng, Z. Functional conservation of MIKC*-Type MADS box genes in Arabidopsis and rice pollen maturation. Plant Cell 2013, 25, 1288–1303. [Google Scholar] [CrossRef] [PubMed]
- Sahito, J.H.; Zheng, F.; Tang, H.; He, X.; Luo, B.; Zhang, X.; Ma, P.; Ding, X.; Liu, D.; Wu, L. Identification, association, and expression analysis of ZmNAC134 gene response to phosphorus deficiency tolerance traits in maize at seedling stage. Euphytica 2020, 216, 100. [Google Scholar] [CrossRef]
- Shi, S.-y.; Zhang, F.-f.; Si, G.; Kai, X. Expression pattern and function analyses of the MADS thranscription factor genes in wheat (Triticum aestivum L.) under phosphorus-starvation condition. J. Integr. Agric. 2016, 15, 1703–1715. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, J.; Wang, X.; Liu, S.; Wang, G.; Zhou, J.; Yuan, Y.; Chen, T.; Jiang, C.; Zha, L. Validation of suitable reference genes for assessing gene expression of microRNAs in Lonicera japonica. Front. Plant Sci. 2016, 7, 1101. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | Raw Reads (bp) | Reads Mapped (bp) | Unique Mapped (bp) | GC (%) | ≥Q30 (%) |
|---|---|---|---|---|---|
| C31-Con | 42,225,842 | 29,434,183 | 29,367,816 | 41.38 | 94.95 |
| C31No-P | 42,043,470 | 29,486,431 | 29,421,504 | 42.22 | 94.88 |
| C31Phi0.25 | 42,635,159 | 29,718,175 | 29,643,683 | 41.98 | 94.50 |
| C31Phi0.5 | 41,941,310 | 29,174,524 | 29,104,150 | 41.69 | 94.77 |
| C80-Con | 41,401,177 | 28,685,887 | 28,618,083 | 41.52 | 94.57 |
| C80No-P | 41,101,662 | 28,165,842 | 28,087,967 | 41.78 | 94.46 |
| C80Phi0.25 | 39,716,517 | 27,377,396 | 27,303,171 | 42.02 | 94.48 |
| C80Phi0.5 | 41,855,121 | 29,440,480 | 29,365,884 | 42.10 | 94.57 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dormatey, R.; Qin, T.; Wang, Y.; Karikari, B.; Dekomah, S.D.; Fan, Y.; Bi, Z.; Yao, P.; Ali, K.; Sun, C.; et al. Comparative Transcriptome Profiling Reveals Potential Candidate Genes, Transcription Factors, and Biosynthetic Pathways for Phosphite Response in Potato (Solanum tuberosum L.). Genes 2022, 13, 1379. https://doi.org/10.3390/genes13081379
Dormatey R, Qin T, Wang Y, Karikari B, Dekomah SD, Fan Y, Bi Z, Yao P, Ali K, Sun C, et al. Comparative Transcriptome Profiling Reveals Potential Candidate Genes, Transcription Factors, and Biosynthetic Pathways for Phosphite Response in Potato (Solanum tuberosum L.). Genes. 2022; 13(8):1379. https://doi.org/10.3390/genes13081379
Chicago/Turabian StyleDormatey, Richard, Tianyuan Qin, Yihao Wang, Benjamin Karikari, Simon Dontoro Dekomah, Youfang Fan, Zhenzhen Bi, Panfeng Yao, Kazim Ali, Chao Sun, and et al. 2022. "Comparative Transcriptome Profiling Reveals Potential Candidate Genes, Transcription Factors, and Biosynthetic Pathways for Phosphite Response in Potato (Solanum tuberosum L.)" Genes 13, no. 8: 1379. https://doi.org/10.3390/genes13081379
APA StyleDormatey, R., Qin, T., Wang, Y., Karikari, B., Dekomah, S. D., Fan, Y., Bi, Z., Yao, P., Ali, K., Sun, C., & Bai, J. (2022). Comparative Transcriptome Profiling Reveals Potential Candidate Genes, Transcription Factors, and Biosynthetic Pathways for Phosphite Response in Potato (Solanum tuberosum L.). Genes, 13(8), 1379. https://doi.org/10.3390/genes13081379