
The Epitranscriptome in miRNAs: Crosstalk, Detection, and Function in Cancer

 ,
,  ,
, 

Abstract
1. Introduction
2. The Epitranscriptome in RNA
2.1. N6-Methyladenosine (m6A)
m6A and miRNAs
2.2. 2′-O-Methyl
2′-O-Methyl in miRNAs
2.3. 5-Methylcytosine (m5C)
m5C in miRNAs
2.4. 7-Methylguanosine (m7G)
7-m7G in miRNAs
2.5. 3′ Poly-Uridine (Poly-U)
Poly-U in miRNAs
2.6. A-to-I Editing
A-to-I Editing of miRNAs
3. Crosstalk between Epitranscriptomic Modifications
3.1. Crosstalk between m6A and A-to-I Editing
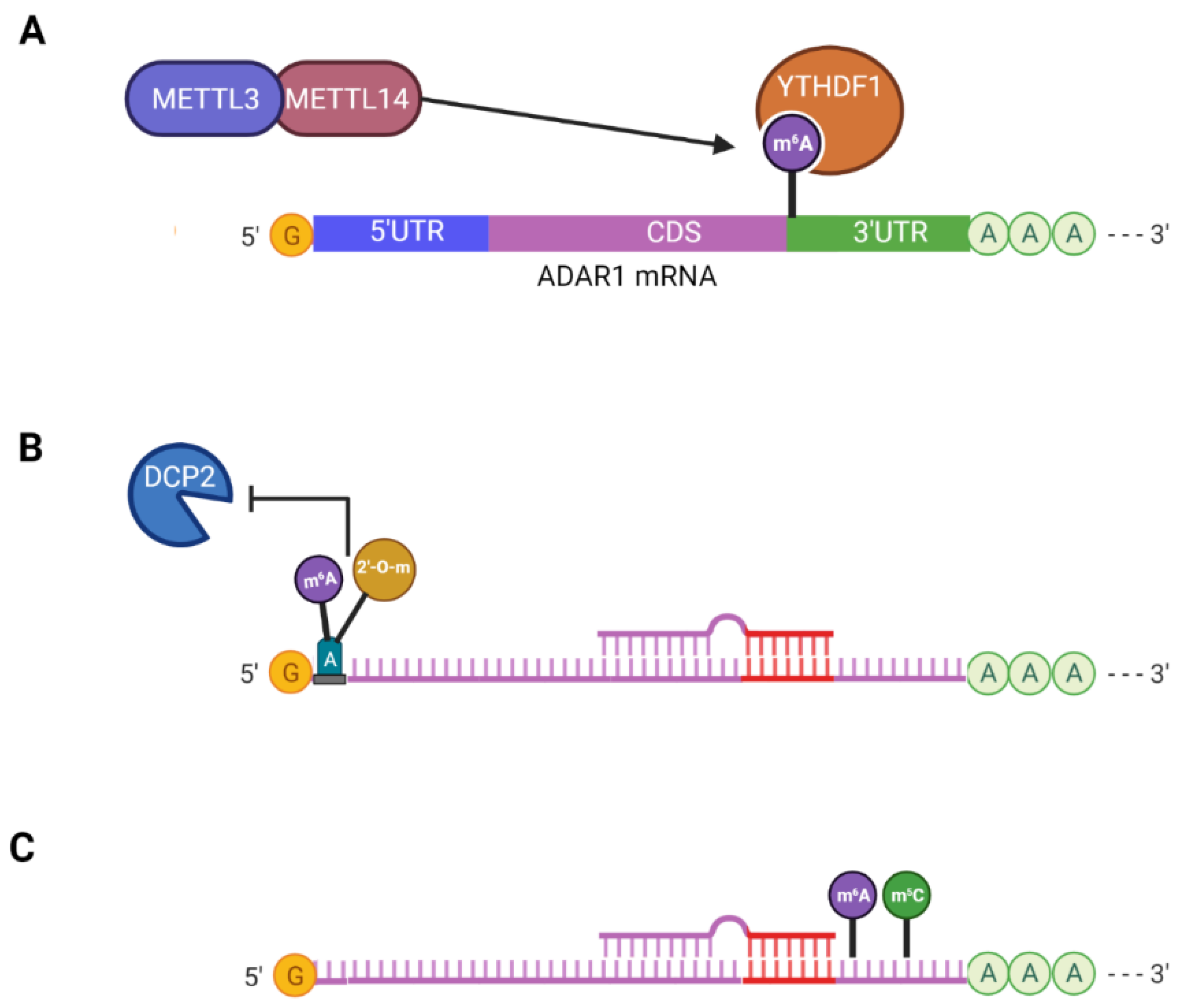
3.2. N6,2′-O-Dimethyladenosine (m6Am)
3.3. Cooperative Interaction between m6A and m5C
4. Methods for Detecting Epitranscriptomic Modifications
4.1. Thin-Layer Chromatography (TLC)
4.2. Liquid Chromatography–Mass Spectrometry (LC-MS)
4.3. RNA Sequencing
4.3.1. Methylated RNA Immunoprecipitation Coupled with High-Throughput Sequencing (MeRIP-seq) and m5C-RIP-seq
4.3.2. RNA Crosslinking and Immunoprecipitation (CLIP) Methods
4.3.3. m6A-Level and Isoform-Characterization Sequencing (m6A-LAIC-seq)
4.3.4. 5-Azacytidine-Mediated RNA Immunoprecipitation (Aza-IP-seq)
4.3.5. RNA Bisulfite Sequencing Technology (RNA-BisSeq)
4.3.6. 2′-O-Methyl Sequencing (2′-O-Me-Seq)
4.3.7. Ribose Methylation Sequencing (RiboMeth-Seq)
4.3.8. Ribose Oxidation Sequencing (RibOxi-Seq)
4.3.9. Nm-Seq
4.3.10. TAIL-Seq
4.3.11. Borohydride Reduction (BoRed-Seq)
4.3.12. Inosine Chemical Erasing Sequencing (ICE-seq)
4.3.13. Detecting A-to-I Editing in RNAseq Data
4.3.14. Nanopore Sequencing
5. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hoerter, J.E.; Ellis, S.R. Biochemistry, Protein Synthesis; StatPearls: Treasure Island, FL, USA, 2022. [Google Scholar]
- Lian, H.; Wang, Q.H.; Zhu, C.B.; Ma, J.; Jin, W.L. Deciphering the Epitranscriptome in Cancer. Trends Cancer 2018, 4, 207–221. [Google Scholar] [CrossRef] [PubMed]
- Kadumuri, R.V.; Janga, S.C. Epitranscriptomic Code and Its Alterations in Human Disease. Trends Mol. Med. 2018, 24, 886–903. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Yang, X.; Qi, Z.; Sang, Y.; Liu, Y.; Xu, B.; Liu, W.; Xu, Z.; Deng, Y. The role of mRNA m(6)A methylation in the nervous system. Cell Biosci. 2019, 9, 66. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Zhai, Y.; Zhang, S.; Dai, X.; Li, Z. Roles of N6-Methyladenosine (m(6)A) in Stem Cell Fate Decisions and Early Embryonic Development in Mammals. Front. Cell Dev. Biol. 2020, 8, 782. [Google Scholar] [CrossRef]
- Fustin, J.M.; Doi, M.; Yamaguchi, Y.; Hida, H.; Nishimura, S.; Yoshida, M.; Isagawa, T.; Morioka, M.S.; Kakeya, H.; Manabe, I.; et al. RNA-methylation-dependent RNA processing controls the speed of the circadian clock. Cell 2013, 155, 793–806. [Google Scholar] [CrossRef]
- Zhou, J.; Wan, J.; Gao, X.; Zhang, X.; Jaffrey, S.R.; Qian, S.B. Dynamic m(6)A mRNA methylation directs translational control of heat shock response. Nature 2015, 526, 591–594. [Google Scholar] [CrossRef]
- Nachtergaele, S.; He, C. The emerging biology of RNA post-transcriptional modifications. RNA Biol. 2017, 14, 156–163. [Google Scholar] [CrossRef]
- De Paolis, V.; Lorefice, E.; Orecchini, E.; Carissimi, C.; Laudadio, I.; Fulci, V. Epitranscriptomics: A New Layer of microRNA Regulation in Cancer. Cancers 2021, 13, 3372. [Google Scholar] [CrossRef]
- Lee, C.T.; Risom, T.; Strauss, W.M. Evolutionary conservation of microRNA regulatory circuits: An examination of microRNA gene complexity and conserved microRNA-target interactions through metazoan phylogeny. DNA Cell Biol. 2007, 26, 209–218. [Google Scholar] [CrossRef]
- Lin, S.; Gregory, R.I. MicroRNA biogenesis pathways in cancer. Nat. Rev. Cancer 2015, 15, 321–333. [Google Scholar] [CrossRef]
- Kim, V.N. MicroRNA biogenesis: Coordinated cropping and dicing. Nat. Rev. Mol. Cell Biol. 2005, 6, 376–385. [Google Scholar] [CrossRef] [PubMed]
- Chendrimada, T.P.; Gregory, R.I.; Kumaraswamy, E.; Norman, J.; Cooch, N.; Nishikura, K.; Shiekhattar, R. TRBP recruits the Dicer complex to Ago2 for microRNA processing and gene silencing. Nature 2005, 436, 740–744. [Google Scholar] [CrossRef] [PubMed]
- Grimson, A.; Farh, K.K.; Johnston, W.K.; Garrett-Engele, P.; Lim, L.P.; Bartel, D.P. MicroRNA targeting specificity in mammals: Determinants beyond seed pairing. Mol. Cell 2007, 27, 91–105. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Croce, C.M. The role of MicroRNAs in human cancer. Signal Transduct. Target Ther. 2016, 1, 15004. [Google Scholar] [CrossRef] [PubMed]
- Cho, W.C. OncomiRs: The discovery and progress of microRNAs in cancers. Mol. Cancer 2007, 6, 60. [Google Scholar] [CrossRef]
- Zhao, Y.; Chen, Y.; Jin, M.; Wang, J. The crosstalk between m(6)A RNA methylation and other epigenetic regulators: A novel perspective in epigenetic remodeling. Theranostics 2021, 11, 4549–4566. [Google Scholar] [CrossRef]
- Boccaletto, P.; Stefaniak, F.; Ray, A.; Cappannini, A.; Mukherjee, S.; Purta, E.; Kurkowska, M.; Shirvanizadeh, N.; Destefanis, E.; Groza, P.; et al. MODOMICS: A database of RNA modification pathways. 2021 update. Nucleic Acids Res. 2022, 50, D231–D235. [Google Scholar] [CrossRef]
- Perry, R.P.; Kelley, D.E. Existence of methylated messenger RNA in mouse L cells. Cell 1974, 1, 37–42. [Google Scholar] [CrossRef]
- Wei, C.M.; Gershowitz, A.; Moss, B. Methylated nucleotides block 5′ terminus of HeLa cell messenger RNA. Cell 1975, 4, 379–386. [Google Scholar] [CrossRef]
- Grozhik, A.V.; Linder, B.; Olarerin-George, A.O.; Jaffrey, S.R. Mapping m6A at Individual-Nucleotide Resolution Using Crosslinking and Immunoprecipitation (miCLIP). Methods Mol. Biol. 2017, 1562, 55–78. [Google Scholar] [CrossRef]
- Meyer, K.D.; Saletore, Y.; Zumbo, P.; Elemento, O.; Mason, C.E.; Jaffrey, S.R. Comprehensive analysis of mRNA methylation reveals enrichment in 3′ UTRs and near stop codons. Cell 2012, 149, 1635–1646. [Google Scholar] [CrossRef] [PubMed]
- Louloupi, A.; Ntini, E.; Conrad, T.; Orom, U.A.V. Transient N-6-Methyladenosine Transcriptome Sequencing Reveals a Regulatory Role of m6A in Splicing Efficiency. Cell Rep. 2018, 23, 3429–3437. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Ieong, K.W.; Demirci, H.; Chen, J.; Petrov, A.; Prabhakar, A.; O’Leary, S.E.; Dominissini, D.; Rechavi, G.; Soltis, S.M.; et al. N(6)-methyladenosine in mRNA disrupts tRNA selection and translation-elongation dynamics. Nat. Struct. Mol. Biol. 2016, 23, 110–115. [Google Scholar] [CrossRef] [PubMed]
- Mao, Y.; Dong, L.; Liu, X.M.; Guo, J.; Ma, H.; Shen, B.; Qian, S.B. m(6)A in mRNA coding regions promotes translation via the RNA helicase-containing YTHDC2. Nat. Commun. 2019, 10, 5332. [Google Scholar] [CrossRef]
- Meyer, K.D.; Patil, D.P.; Zhou, J.; Zinoviev, A.; Skabkin, M.A.; Elemento, O.; Pestova, T.V.; Qian, S.B.; Jaffrey, S.R. 5′ UTR m(6)A Promotes Cap-Independent Translation. Cell 2015, 163, 999–1010. [Google Scholar] [CrossRef]
- Liu, J.; Yue, Y.; Han, D.; Wang, X.; Fu, Y.; Zhang, L.; Jia, G.; Yu, M.; Lu, Z.; Deng, X.; et al. A METTL3-METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat. Chem. Biol. 2014, 10, 93–95. [Google Scholar] [CrossRef]
- van Tran, N.; Ernst, F.G.M.; Hawley, B.R.; Zorbas, C.; Ulryck, N.; Hackert, P.; Bohnsack, K.E.; Bohnsack, M.T.; Jaffrey, S.R.; Graille, M.; et al. The human 18S rRNA m6A methyltransferase METTL5 is stabilized by TRMT112. Nucleic Acids Res. 2019, 47, 7719–7733. [Google Scholar] [CrossRef]
- Su, R.; Dong, L.; Li, Y.; Gao, M.; He, P.C.; Liu, W.; Wei, J.; Zhao, Z.; Gao, L.; Han, L.; et al. METTL16 exerts an m(6)A-independent function to facilitate translation and tumorigenesis. Nat. Cell Biol. 2022, 24, 205–216. [Google Scholar] [CrossRef]
- Brown, J.A.; Kinzig, C.G.; DeGregorio, S.J.; Steitz, J.A. Methyltransferase-like protein 16 binds the 3′-terminal triple helix of MALAT1 long noncoding RNA. Proc. Natl. Acad. Sci. USA 2016, 113, 14013–14018. [Google Scholar] [CrossRef]
- Ping, X.L.; Sun, B.F.; Wang, L.; Xiao, W.; Yang, X.; Wang, W.J.; Adhikari, S.; Shi, Y.; Lv, Y.; Chen, Y.S.; et al. Mammalian WTAP is a regulatory subunit of the RNA N6-methyladenosine methyltransferase. Cell Res. 2014, 24, 177–189. [Google Scholar] [CrossRef]
- Qian, J.Y.; Gao, J.; Sun, X.; Cao, M.D.; Shi, L.; Xia, T.S.; Zhou, W.B.; Wang, S.; Ding, Q.; Wei, J.F. KIAA1429 acts as an oncogenic factor in breast cancer by regulating CDK1 in an N6-methyladenosine-independent manner. Oncogene 2019, 38, 6123–6141. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, S.; Mumbach, M.R.; Jovanovic, M.; Wang, T.; Maciag, K.; Bushkin, G.G.; Mertins, P.; Ter-Ovanesyan, D.; Habib, N.; Cacchiarelli, D.; et al. Perturbation of m6A writers reveals two distinct classes of mRNA methylation at internal and 5′ sites. Cell Rep. 2014, 8, 284–296. [Google Scholar] [CrossRef] [PubMed]
- Knuckles, P.; Lence, T.; Haussmann, I.U.; Jacob, D.; Kreim, N.; Carl, S.H.; Masiello, I.; Hares, T.; Villasenor, R.; Hess, D.; et al. Zc3h13/Flacc is required for adenosine methylation by bridging the mRNA-binding factor Rbm15/Spenito to the m(6)A machinery component Wtap/Fl(2)d. Genes Dev. 2018, 32, 415–429. [Google Scholar] [CrossRef]
- Ruzicka, K.; Zhang, M.; Campilho, A.; Bodi, Z.; Kashif, M.; Saleh, M.; Eeckhout, D.; El-Showk, S.; Li, H.; Zhong, S.; et al. Identification of factors required for m(6) A mRNA methylation in Arabidopsis reveals a role for the conserved E3 ubiquitin ligase HAKAI. New Phytol. 2017, 215, 157–172. [Google Scholar] [CrossRef] [PubMed]
- Patil, D.P.; Chen, C.K.; Pickering, B.F.; Chow, A.; Jackson, C.; Guttman, M.; Jaffrey, S.R. m(6)A RNA methylation promotes XIST-mediated transcriptional repression. Nature 2016, 537, 369–373. [Google Scholar] [CrossRef]
- Liu, J.; Ren, D.; Du, Z.; Wang, H.; Zhang, H.; Jin, Y. m(6)A demethylase FTO facilitates tumor progression in lung squamous cell carcinoma by regulating MZF1 expression. Biochem. Biophys. Res. Commun. 2018, 502, 456–464. [Google Scholar] [CrossRef]
- Zhang, S.; Zhao, B.S.; Zhou, A.; Lin, K.; Zheng, S.; Lu, Z.; Chen, Y.; Sulman, E.P.; Xie, K.; Bogler, O.; et al. m(6)A Demethylase ALKBH5 Maintains Tumorigenicity of Glioblastoma Stem-like Cells by Sustaining FOXM1 Expression and Cell Proliferation Program. Cancer Cell 2017, 31, 591–606.e6. [Google Scholar] [CrossRef]
- Mauer, J.; Luo, X.; Blanjoie, A.; Jiao, X.; Grozhik, A.V.; Patil, D.P.; Linder, B.; Pickering, B.F.; Vasseur, J.J.; Chen, Q.; et al. Reversible methylation of m(6)Am in the 5′ cap controls mRNA stability. Nature 2017, 541, 371–375. [Google Scholar] [CrossRef]
- Zhen, D.; Wu, Y.; Zhang, Y.; Chen, K.; Song, B.; Xu, H.; Tang, Y.; Wei, Z.; Meng, J. m(6)A Reader: Epitranscriptome Target Prediction and Functional Characterization of N (6)-Methyladenosine (m(6)A) Readers. Front. Cell Dev. Biol. 2020, 8, 741. [Google Scholar] [CrossRef]
- Zaccara, S.; Ries, R.J.; Jaffrey, S.R. Reading, writing and erasing mRNA methylation. Nat. Rev. Mol. Cell Biol. 2019, 20, 608–624. [Google Scholar] [CrossRef]
- Yang, C.; Hu, Y.; Zhou, B.; Bao, Y.; Li, Z.; Gong, C.; Yang, H.; Wang, S.; Xiao, Y. The role of m(6)A modification in physiology and disease. Cell Death Dis. 2020, 11, 960. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Yu, C.; Guo, M.; Zheng, X.; Ali, S.; Huang, H.; Zhang, L.; Wang, S.; Huang, Y.; Qie, S.; et al. Down-Regulation of m6A mRNA Methylation Is Involved in Dopaminergic Neuronal Death. ACS Chem. Neurosci. 2019, 10, 2355–2363. [Google Scholar] [CrossRef] [PubMed]
- Shen, G.S.; Zhou, H.B.; Zhang, H.; Chen, B.; Liu, Z.P.; Yuan, Y.; Zhou, X.Z.; Xu, Y.J. The GDF11-FTO-PPARgamma axis controls the shift of osteoporotic MSC fate to adipocyte and inhibits bone formation during osteoporosis. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 3644–3654. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhang, Q.; Cui, G.; Zhao, F.; Tian, X.; Sun, B.F.; Yang, Y.; Li, W. m(6)A Regulates Liver Metabolic Disorders and Hepatogenous Diabetes. Genom. Proteom. Bioinform. 2020, 18, 371–383. [Google Scholar] [CrossRef]
- Wang, T.; Kong, S.; Tao, M.; Ju, S. The potential role of RNA N6-methyladenosine in Cancer progression. Mol. Cancer 2020, 19, 88. [Google Scholar] [CrossRef]
- Geula, S.; Moshitch-Moshkovitz, S.; Dominissini, D.; Mansour, A.A.; Kol, N.; Salmon-Divon, M.; Hershkovitz, V.; Peer, E.; Mor, N.; Manor, Y.S.; et al. Stem cells. m6A mRNA methylation facilitates resolution of naive pluripotency toward differentiation. Science 2015, 347, 1002–1006. [Google Scholar] [CrossRef]
- Batista, P.J.; Molinie, B.; Wang, J.; Qu, K.; Zhang, J.; Li, L.; Bouley, D.M.; Lujan, E.; Haddad, B.; Daneshvar, K.; et al. m(6)A RNA modification controls cell fate transition in mammalian embryonic stem cells. Cell Stem Cell 2014, 15, 707–719. [Google Scholar] [CrossRef]
- Wang, Y.; Li, Y.; Toth, J.I.; Petroski, M.D.; Zhang, Z.; Zhao, J.C. N6-methyladenosine modification destabilizes developmental regulators in embryonic stem cells. Nat. Cell Biol. 2014, 16, 191–198. [Google Scholar] [CrossRef]
- Dorn, L.E.; Lasman, L.; Chen, J.; Xu, X.; Hund, T.J.; Medvedovic, M.; Hanna, J.H.; van Berlo, J.H.; Accornero, F. The N(6)-Methyladenosine mRNA Methylase METTL3 Controls Cardiac Homeostasis and Hypertrophy. Circulation 2019, 139, 533–545. [Google Scholar] [CrossRef]
- Wang, C.X.; Cui, G.S.; Liu, X.; Xu, K.; Wang, M.; Zhang, X.X.; Jiang, L.Y.; Li, A.; Yang, Y.; Lai, W.Y.; et al. METTL3-mediated m6A modification is required for cerebellar development. PLoS Biol. 2018, 16, e2004880. [Google Scholar] [CrossRef]
- Vu, L.P.; Pickering, B.F.; Cheng, Y.; Zaccara, S.; Nguyen, D.; Minuesa, G.; Chou, T.; Chow, A.; Saletore, Y.; MacKay, M.; et al. The N(6)-methyladenosine (m(6)A)-forming enzyme METTL3 controls myeloid differentiation of normal hematopoietic and leukemia cells. Nat. Med. 2017, 23, 1369–1376. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, S.; Zhao, T.; Dang, C. METTL3 mediated m6A modification of Bcl 2 mRNA promotes non small cell lung cancer progression. Oncol. Rep. 2021, 46, 163. [Google Scholar] [CrossRef] [PubMed]
- Alarcon, C.R.; Lee, H.; Goodarzi, H.; Halberg, N.; Tavazoie, S.F. N6-methyladenosine marks primary microRNAs for processing. Nature 2015, 519, 482–485. [Google Scholar] [CrossRef]
- Berulava, T.; Rahmann, S.; Rademacher, K.; Klein-Hitpass, L.; Horsthemke, B. N6-adenosine methylation in MiRNAs. PLoS ONE 2015, 10, e0118438. [Google Scholar] [CrossRef]
- Chen, X.; Xu, M.; Xu, X.; Zeng, K.; Liu, X.; Sun, L.; Pan, B.; He, B.; Pan, Y.; Sun, H.; et al. METTL14 Suppresses CRC Progression via Regulating N6-Methyladenosine-Dependent Primary miR-375 Processing. Mol. Ther. 2020, 28, 599–612. [Google Scholar] [CrossRef] [PubMed]
- Ronkainen, J.; Mondini, E.; Cinti, F.; Cinti, S.; Sebert, S.; Savolainen, M.J.; Salonurmi, T. Fto-Deficiency Affects the Gene and MicroRNA Expression Involved in Brown Adipogenesis and Browning of White Adipose Tissue in Mice. Int. J. Mol. Sci. 2016, 17, 1851. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.Z.; Yang, F.; Zhou, C.C.; Liu, F.; Yuan, J.H.; Wang, F.; Wang, T.T.; Xu, Q.G.; Zhou, W.P.; Sun, S.H. METTL14 suppresses the metastatic potential of hepatocellular carcinoma by modulating N(6)-methyladenosine-dependent primary MicroRNA processing. Hepatology 2017, 65, 529–543. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Guo, J.; Fan, Z. Interactions between m6A modification and miRNAs in malignant tumors. Cell Death Dis. 2021, 12, 598. [Google Scholar] [CrossRef]
- Du, M.; Zhang, Y.; Mao, Y.; Mou, J.; Zhao, J.; Xue, Q.; Wang, D.; Huang, J.; Gao, S.; Gao, Y. MiR-33a suppresses proliferation of NSCLC cells via targeting METTL3 mRNA. Biochem. Biophys. Res. Commun. 2017, 482, 582–589. [Google Scholar] [CrossRef]
- He, H.; Wu, W.; Sun, Z.; Chai, L. MiR-4429 prevented gastric cancer progression through targeting METTL3 to inhibit m(6)A-caused stabilization of SEC62. Biochem. Biophys. Res. Commun. 2019, 517, 581–587. [Google Scholar] [CrossRef]
- Li, J.; Wu, L.; Pei, M.; Zhang, Y. YTHDF2, a protein repressed by miR-145, regulates proliferation, apoptosis, and migration in ovarian cancer cells. J. Ovarian Res. 2020, 13, 111. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.P.; Ling, X.; Lu, H.; Zhou, C.X.; Zhang, J.K.; Yu, Q. Low expression of microRNA-1266 promotes colorectal cancer progression via targeting FTO. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 8220–8226. [Google Scholar] [CrossRef]
- Monaco, P.L.; Marcel, V.; Diaz, J.J.; Catez, F. 2′-O-Methylation of Ribosomal RNA: Towards an Epitranscriptomic Control of Translation? Biomolecules 2018, 8, 106. [Google Scholar] [CrossRef] [PubMed]
- Kirino, Y.; Mourelatos, Z. 2′-O-methyl modification in mouse piRNAs and its methylase. Nucleic Acids Symp. Ser. 2007, 51, 417–418. [Google Scholar] [CrossRef]
- Elliott, B.A.; Ho, H.T.; Ranganathan, S.V.; Vangaveti, S.; Ilkayeva, O.; Abou Assi, H.; Choi, A.K.; Agris, P.F.; Holley, C.L. Modification of messenger RNA by 2′-O-methylation regulates gene expression in vivo. Nat. Commun. 2019, 10, 3401. [Google Scholar] [CrossRef] [PubMed]
- Prusiner, P.; Yathindra, N.; Sundaralingam, M. Effect of ribose O(2′)-methylation on the conformation of nucleosides and nucleotides. Biochim. Biophys. Acta 1974, 366, 115–123. [Google Scholar] [CrossRef]
- Decatur, W.A.; Fournier, M.J. rRNA modifications and ribosome function. Trends Biochem. Sci. 2002, 27, 344–351. [Google Scholar] [CrossRef]
- Polikanov, Y.S.; Melnikov, S.V.; Soll, D.; Steitz, T.A. Structural insights into the role of rRNA modifications in protein synthesis and ribosome assembly. Nat. Struct. Mol. Biol. 2015, 22, 342–344. [Google Scholar] [CrossRef]
- Erales, J.; Marchand, V.; Panthu, B.; Gillot, S.; Belin, S.; Ghayad, S.E.; Garcia, M.; Laforets, F.; Marcel, V.; Baudin-Baillieu, A.; et al. Evidence for rRNA 2′-O-methylation plasticity: Control of intrinsic translational capabilities of human ribosomes. Proc. Natl. Acad. Sci. USA 2017, 114, 12934–12939. [Google Scholar] [CrossRef]
- Dai, Q.; Moshitch-Moshkovitz, S.; Han, D.; Kol, N.; Amariglio, N.; Rechavi, G.; Dominissini, D.; He, C. Nm-seq maps 2′-O-methylation sites in human mRNA with base precision. Nat. Methods 2017, 14, 695–698, Erratum in Nat. Methods 2018, 15, 226–227. [Google Scholar] [CrossRef]
- Belanger, F.; Stepinski, J.; Darzynkiewicz, E.; Pelletier, J. Characterization of hMTr1, a human Cap1 2′-O-ribose methyltransferase. J. Biol. Chem. 2010, 285, 33037–33044. [Google Scholar] [CrossRef] [PubMed]
- Shatkin, A.J. Capping of eucaryotic mRNAs. Cell 1976, 9, 645–653. [Google Scholar] [CrossRef]
- Daffis, S.; Szretter, K.J.; Schriewer, J.; Li, J.; Youn, S.; Errett, J.; Lin, T.Y.; Schneller, S.; Zust, R.; Dong, H.; et al. 2′-O methylation of the viral mRNA cap evades host restriction by IFIT family members. Nature 2010, 468, 452–456. [Google Scholar] [CrossRef] [PubMed]
- Sikorski, P.J.; Warminski, M.; Kubacka, D.; Ratajczak, T.; Nowis, D.; Kowalska, J.; Jemielity, J. The identity and methylation status of the first transcribed nucleotide in eukaryotic mRNA 5′ cap modulates protein expression in living cells. Nucleic Acids Res. 2020, 48, 1607–1626. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Indrisiunaite, G.; DeMirci, H.; Ieong, K.W.; Wang, J.; Petrov, A.; Prabhakar, A.; Rechavi, G.; Dominissini, D.; He, C.; et al. 2′-O-methylation in mRNA disrupts tRNA decoding during translation elongation. Nat. Struct. Mol. Biol. 2018, 25, 208–216. [Google Scholar] [CrossRef]
- Yu, B.; Yang, Z.; Li, J.; Minakhina, S.; Yang, M.; Padgett, R.W.; Steward, R.; Chen, X. Methylation as a crucial step in plant microRNA biogenesis. Science 2005, 307, 932–935. [Google Scholar] [CrossRef]
- Abe, M.; Naqvi, A.; Hendriks, G.J.; Feltzin, V.; Zhu, Y.; Grigoriev, A.; Bonini, N.M. Impact of age-associated increase in 2′-O-methylation of miRNAs on aging and neurodegeneration in Drosophila. Genes Dev. 2014, 28, 44–57. [Google Scholar] [CrossRef]
- Li, J.; Yang, Z.; Yu, B.; Liu, J.; Chen, X. Methylation protects miRNAs and siRNAs from a 3′-end uridylation activity in Arabidopsis. Curr. Biol. 2005, 15, 1501–1507. [Google Scholar] [CrossRef]
- Liang, H.; Jiao, Z.; Rong, W.; Qu, S.; Liao, Z.; Sun, X.; Wei, Y.; Zhao, Q.; Wang, J.; Liu, Y.; et al. 3′-Terminal 2′-O-methylation of lung cancer miR-21-5p enhances its stability and association with Argonaute 2. Nucleic Acids Res. 2020, 48, 7027–7040. [Google Scholar] [CrossRef]
- Desrosiers, R.; Friderici, K.; Rottman, F. Identification of methylated nucleosides in messenger RNA from Novikoff hepatoma cells. Proc. Natl. Acad. Sci. USA 1974, 71, 3971–3975. [Google Scholar] [CrossRef]
- Xue, C.; Zhao, Y.; Li, L. Advances in RNA cytosine-5 methylation: Detection, regulatory mechanisms, biological functions and links to cancer. Biomark. Res. 2020, 8, 43. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.S.; Yang, W.L.; Zhao, Y.L.; Yang, Y.G. Dynamic transcriptomic m(5) C and its regulatory role in RNA processing. Wiley Interdiscip. Rev. RNA 2021, 12, e1639. [Google Scholar] [CrossRef] [PubMed]
- Bohnsack, K.E.; Hobartner, C.; Bohnsack, M.T. Eukaryotic 5-methylcytosine (m(5)C) RNA Methyltransferases: Mechanisms, Cellular Functions, and Links to Disease. Genes 2019, 10, 102. [Google Scholar] [CrossRef] [PubMed]
- Hussain, S.; Sajini, A.A.; Blanco, S.; Dietmann, S.; Lombard, P.; Sugimoto, Y.; Paramor, M.; Gleeson, J.G.; Odom, D.T.; Ule, J.; et al. NSun2-mediated cytosine-5 methylation of vault noncoding RNA determines its processing into regulatory small RNAs. Cell Rep. 2013, 4, 255–261. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Yang, Y.; Sun, B.F.; Chen, Y.S.; Xu, J.W.; Lai, W.Y.; Li, A.; Wang, X.; Bhattarai, D.P.; Xiao, W.; et al. 5-methylcytosine promotes mRNA export—NSUN2 as the methyltransferase and ALYREF as an m(5)C reader. Cell Res. 2017, 27, 606–625. [Google Scholar] [CrossRef] [PubMed]
- Roundtree, I.A.; Evans, M.E.; Pan, T.; He, C. Dynamic RNA Modifications in Gene Expression Regulation. Cell 2017, 169, 1187–1200. [Google Scholar] [CrossRef]
- Goll, M.G.; Kirpekar, F.; Maggert, K.A.; Yoder, J.A.; Hsieh, C.L.; Zhang, X.; Golic, K.G.; Jacobsen, S.E.; Bestor, T.H. Methylation of tRNAAsp by the DNA methyltransferase homolog Dnmt2. Science 2006, 311, 395–398. [Google Scholar] [CrossRef]
- Jeltsch, A.; Ehrenhofer-Murray, A.; Jurkowski, T.P.; Lyko, F.; Reuter, G.; Ankri, S.; Nellen, W.; Schaefer, M.; Helm, M. Mechanism and biological role of Dnmt2 in Nucleic Acid Methylation. RNA Biol. 2017, 14, 1108–1123. [Google Scholar] [CrossRef]
- Shanmugam, R.; Fierer, J.; Kaiser, S.; Helm, M.; Jurkowski, T.P.; Jeltsch, A. Cytosine methylation of tRNA-Asp by DNMT2 has a role in translation of proteins containing poly-Asp sequences. Cell Discov. 2015, 1, 15010. [Google Scholar] [CrossRef]
- Squires, J.E.; Patel, H.R.; Nousch, M.; Sibbritt, T.; Humphreys, D.T.; Parker, B.J.; Suter, C.M.; Preiss, T. Widespread occurrence of 5-methylcytosine in human coding and non-coding RNA. Nucleic Acids Res. 2012, 40, 5023–5033. [Google Scholar] [CrossRef]
- Blanco, S.; Dietmann, S.; Flores, J.V.; Hussain, S.; Kutter, C.; Humphreys, P.; Lukk, M.; Lombard, P.; Treps, L.; Popis, M.; et al. Aberrant methylation of tRNAs links cellular stress to neuro-developmental disorders. EMBO J. 2014, 33, 2020–2039. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.A.; Rafiq, M.A.; Noor, A.; Hussain, S.; Flores, J.V.; Rupp, V.; Vincent, A.K.; Malli, R.; Ali, G.; Khan, F.S.; et al. Mutation in NSUN2, which encodes an RNA methyltransferase, causes autosomal-recessive intellectual disability. Am. J. Hum. Genet. 2012, 90, 856–863. [Google Scholar] [CrossRef] [PubMed]
- Van Haute, L.; Dietmann, S.; Kremer, L.; Hussain, S.; Pearce, S.F.; Powell, C.A.; Rorbach, J.; Lantaff, R.; Blanco, S.; Sauer, S.; et al. Deficient methylation and formylation of mt-tRNA(Met) wobble cytosine in a patient carrying mutations in NSUN3. Nat. Commun. 2016, 7, 12039. [Google Scholar] [CrossRef] [PubMed]
- Harris, T.; Marquez, B.; Suarez, S.; Schimenti, J. Sperm motility defects and infertility in male mice with a mutation in Nsun7, a member of the Sun domain-containing family of putative RNA methyltransferases. Biol. Reprod. 2007, 77, 376–382. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Liu, F.; Chen, W.; Miao, H.; Liang, H.; Liao, Z.; Zhang, Z.; Zhang, B. The role of RNA m(5)C modification in cancer metastasis. Int. J. Biol. Sci. 2021, 17, 3369–3380. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Li, A.; Sun, B.F.; Yang, Y.; Han, Y.N.; Yuan, X.; Chen, R.X.; Wei, W.S.; Liu, Y.; Gao, C.C.; et al. 5-methylcytosine promotes pathogenesis of bladder cancer through stabilizing mRNAs. Nat. Cell Biol. 2019, 21, 978–990. [Google Scholar] [CrossRef] [PubMed]
- Konno, M.; Koseki, J.; Asai, A.; Yamagata, A.; Shimamura, T.; Motooka, D.; Okuzaki, D.; Kawamoto, K.; Mizushima, T.; Eguchi, H.; et al. Distinct methylation levels of mature microRNAs in gastrointestinal cancers. Nat. Commun. 2019, 10, 3888. [Google Scholar] [CrossRef]
- Cheray, M.; Etcheverry, A.; Jacques, C.; Pacaud, R.; Bougras-Cartron, G.; Aubry, M.; Denoual, F.; Peterlongo, P.; Nadaradjane, A.; Briand, J.; et al. Cytosine methylation of mature microRNAs inhibits their functions and is associated with poor prognosis in glioblastoma multiforme. Mol. Cancer 2020, 19, 36. [Google Scholar] [CrossRef]
- Brower, J.V.; Clark, P.A.; Lyon, W.; Kuo, J.S. MicroRNAs in cancer: Glioblastoma and glioblastoma cancer stem cells. Neurochem. Int. 2014, 77, 68–77. [Google Scholar] [CrossRef]
- Li, Y.; Kuscu, C.; Banach, A.; Zhang, Q.; Pulkoski-Gross, A.; Kim, D.; Liu, J.; Roth, E.; Li, E.; Shroyer, K.R.; et al. miR-181a-5p Inhibits Cancer Cell Migration and Angiogenesis via Downregulation of Matrix Metalloproteinase-14. Cancer Res. 2015, 75, 2674–2685. [Google Scholar] [CrossRef]
- Luo, Y.; Yao, Y.; Wu, P.; Zi, X.; Sun, N.; He, J. The potential role of N(7)-methylguanosine (m7G) in cancer. J. Hematol. Oncol. 2022, 15, 63. [Google Scholar] [CrossRef] [PubMed]
- Borden, K.; Culjkovic-Kraljacic, B.; Cowling, V.H. To cap it all off, again: Dynamic capping and recapping of coding and non-coding RNAs to control transcript fate and biological activity. Cell Cycle 2021, 20, 1347–1360. [Google Scholar] [CrossRef] [PubMed]
- Trotman, J.B.; Giltmier, A.J.; Mukherjee, C.; Schoenberg, D.R. RNA guanine-7 methyltransferase catalyzes the methylation of cytoplasmically recapped RNAs. Nucleic Acids Res. 2017, 45, 10726–10739. [Google Scholar] [CrossRef] [PubMed]
- Trotman, J.B.; Schoenberg, D.R. A recap of RNA recapping. Wiley Interdiscip. Rev. RNA 2019, 10, e1504. [Google Scholar] [CrossRef] [PubMed]
- Zhai, L.T.; Xiang, S. mRNA quality control at the 5′ end. J. Zhejiang Univ. Sci. B 2014, 15, 438–443. [Google Scholar] [CrossRef] [PubMed]
- Grasso, L.; Suska, O.; Davidson, L.; Gonatopoulos-Pournatzis, T.; Williamson, R.; Wasmus, L.; Wiedlich, S.; Peggie, M.; Stavridis, M.P.; Cowling, V.H. mRNA Cap Methylation in Pluripotency and Differentiation. Cell Rep. 2016, 16, 1352–1365. [Google Scholar] [CrossRef][Green Version]
- Culjkovic-Kraljacic, B.; Skrabanek, L.; Revuelta, M.V.; Gasiorek, J.; Cowling, V.H.; Cerchietti, L.; Borden, K.L.B. The eukaryotic translation initiation factor eIF4E elevates steady-state m(7)G capping of coding and noncoding transcripts. Proc. Natl. Acad. Sci. USA 2020, 117, 26773–26783. [Google Scholar] [CrossRef]
- Dunn, S.; Lombardi, O.; Lukoszek, R.; Cowling, V.H. Oncogenic PIK3CA mutations increase dependency on the mRNA cap methyltransferase, RNMT, in breast cancer cells. Open Biol. 2019, 9, 190052. [Google Scholar] [CrossRef]
- Shaheen, R.; Abdel-Salam, G.M.; Guy, M.P.; Alomar, R.; Abdel-Hamid, M.S.; Afifi, H.H.; Ismail, S.I.; Emam, B.A.; Phizicky, E.M.; Alkuraya, F.S. Mutation in WDR4 impairs tRNA m(7)G46 methylation and causes a distinct form of microcephalic primordial dwarfism. Genome Biol. 2015, 16, 210. [Google Scholar] [CrossRef]
- Zhang, L.S.; Liu, C.; Ma, H.; Dai, Q.; Sun, H.L.; Luo, G.; Zhang, Z.; Zhang, L.; Hu, L.; Dong, X.; et al. Transcriptome-wide Mapping of Internal N(7)-Methylguanosine Methylome in Mammalian mRNA. Mol. Cell 2019, 74, 1304–1316.e8. [Google Scholar] [CrossRef]
- Zorbas, C.; Nicolas, E.; Wacheul, L.; Huvelle, E.; Heurgue-Hamard, V.; Lafontaine, D.L. The human 18S rRNA base methyltransferases DIMT1L and WBSCR22-TRMT112 but not rRNA modification are required for ribosome biogenesis. Mol. Biol. Cell 2015, 26, 2080–2095. [Google Scholar] [CrossRef] [PubMed]
- Ounap, K.; Kasper, L.; Kurg, A.; Kurg, R. The human WBSCR22 protein is involved in the biogenesis of the 40S ribosomal subunits in mammalian cells. PLoS ONE 2013, 8, e75686. [Google Scholar] [CrossRef] [PubMed]
- Xie, M.; Li, M.; Vilborg, A.; Lee, N.; Shu, M.D.; Yartseva, V.; Sestan, N.; Steitz, J.A. Mammalian 5′-capped microRNA precursors that generate a single microRNA. Cell 2013, 155, 1568–1580. [Google Scholar] [CrossRef] [PubMed]
- Sheng, P.; Fields, C.; Aadland, K.; Wei, T.; Kolaczkowski, O.; Gu, T.; Kolaczkowski, B.; Xie, M. Dicer cleaves 5′-extended microRNA precursors originating from RNA polymerase II transcription start sites. Nucleic Acids Res. 2018, 46, 5737–5752. [Google Scholar] [CrossRef] [PubMed]
- Kaur, S.; Saldana, A.C.; Elkahloun, A.G.; Petersen, J.D.; Arakelyan, A.; Singh, S.P.; Jenkins, L.M.; Kuo, B.; Reginauld, B.; Jordan, D.G.; et al. CD47 interactions with exportin-1 limit the targeting of m(7)G-modified RNAs to extracellular vesicles. J. Cell Commun. Signal. 2021. [Google Scholar] [CrossRef] [PubMed]
- Martinez, I.; Hayes, K.E.; Barr, J.A.; Harold, A.D.; Xie, M.; Bukhari, S.I.A.; Vasudevan, S.; Steitz, J.A.; DiMaio, D. An Exportin-1-dependent microRNA biogenesis pathway during human cell quiescence. Proc. Natl. Acad. Sci. USA 2017, 114, E4961–E4970. [Google Scholar] [CrossRef]
- Kamel, W.; Akusjarvi, G. An Ago2-associated capped transcriptional start site small RNA suppresses adenovirus DNA replication. RNA 2017, 23, 1700–1711. [Google Scholar] [CrossRef]
- Pandolfini, L.; Barbieri, I.; Bannister, A.J.; Hendrick, A.; Andrews, B.; Webster, N.; Murat, P.; Mach, P.; Brandi, R.; Robson, S.C.; et al. METTL1 Promotes let-7 MicroRNA Processing via m7G Methylation. Mol. Cell 2019, 74, 1278–1290 e1279. [Google Scholar] [CrossRef]
- Vinther, J. No Evidence for N7-Methylation of Guanosine (m(7)G) in Human let-7e. Mol. Cell 2020, 79, 199–200. [Google Scholar] [CrossRef]
- Menezes, M.R.; Balzeau, J.; Hagan, J.P. 3′ RNA Uridylation in Epitranscriptomics, Gene Regulation, and Disease. Front. Mol. Biosci. 2018, 5, 61. [Google Scholar] [CrossRef]
- Lim, J.; Ha, M.; Chang, H.; Kwon, S.C.; Simanshu, D.K.; Patel, D.J.; Kim, V.N. Uridylation by TUT4 and TUT7 marks mRNA for degradation. Cell 2014, 159, 1365–1376. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.J.; West, S.; Norbury, C.J. The human cytoplasmic RNA terminal U-transferase ZCCHC11 targets histone mRNAs for degradation. RNA 2011, 17, 39–44. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Gonzales, M.L.; Mellman, D.L.; Anderson, R.A. CKIalpha is associated with and phosphorylates star-PAP and is also required for expression of select star-PAP target messenger RNAs. J. Biol. Chem. 2008, 283, 12665–12673. [Google Scholar] [CrossRef]
- Mroczek, S.; Krwawicz, J.; Kutner, J.; Lazniewski, M.; Kucinski, I.; Ginalski, K.; Dziembowski, A. C16orf57, a gene mutated in poikiloderma with neutropenia, encodes a putative phosphodiesterase responsible for the U6 snRNA 3′ end modification. Genes Dev. 2012, 26, 1911–1925. [Google Scholar] [CrossRef]
- Labno, A.; Warkocki, Z.; Kulinski, T.; Krawczyk, P.S.; Bijata, K.; Tomecki, R.; Dziembowski, A. Perlman syndrome nuclease DIS3L2 controls cytoplasmic non-coding RNAs and provides surveillance pathway for maturing snRNAs. Nucleic Acids Res. 2016, 44, 10437–10453. [Google Scholar] [CrossRef]
- Chang, H.; Lim, J.; Ha, M.; Kim, V.N. TAIL-seq: Genome-wide determination of poly(A) tail length and 3′ end modifications. Mol. Cell 2014, 53, 1044–1052. [Google Scholar] [CrossRef]
- Mullen, T.E.; Marzluff, W.F. Degradation of histone mRNA requires oligouridylation followed by decapping and simultaneous degradation of the mRNA both 5′ to 3′ and 3′ to 5′. Genes Dev. 2008, 22, 50–65. [Google Scholar] [CrossRef] [PubMed]
- Balzeau, J.; Menezes, M.R.; Cao, S.; Hagan, J.P. The LIN28/let-7 Pathway in Cancer. Front. Genet. 2017, 8, 31. [Google Scholar] [CrossRef]
- Kim, B.; Ha, M.; Loeff, L.; Chang, H.; Simanshu, D.K.; Li, S.; Fareh, M.; Patel, D.J.; Joo, C.; Kim, V.N. TUT7 controls the fate of precursor microRNAs by using three different uridylation mechanisms. EMBO J. 2015, 34, 1801–1815. [Google Scholar] [CrossRef]
- Hagan, J.P.; Piskounova, E.; Gregory, R.I. Lin28 recruits the TUTase Zcchc11 to inhibit let-7 maturation in mouse embryonic stem cells. Nat. Struct. Mol. Biol. 2009, 16, 1021–1025. [Google Scholar] [CrossRef]
- Wang, X.; Cao, L.; Wang, Y.; Wang, X.; Liu, N.; You, Y. Regulation of let-7 and its target oncogenes (Review). Oncol. Lett. 2012, 3, 955–960. [Google Scholar] [CrossRef]
- Jones, M.R.; Quinton, L.J.; Blahna, M.T.; Neilson, J.R.; Fu, S.; Ivanov, A.R.; Wolf, D.A.; Mizgerd, J.P. Zcchc11-dependent uridylation of microRNA directs cytokine expression. Nat. Cell Biol. 2009, 11, 1157–1163. [Google Scholar] [CrossRef] [PubMed]
- Bass, B.L.; Weintraub, H. An unwinding activity that covalently modifies its double-stranded RNA substrate. Cell 1988, 55, 1089–1098. [Google Scholar] [CrossRef]
- Wagner, R.W.; Smith, J.E.; Cooperman, B.S.; Nishikura, K. A double-stranded RNA unwinding activity introduces structural alterations by means of adenosine to inosine conversions in mammalian cells and Xenopus eggs. Proc. Natl. Acad. Sci. USA 1989, 86, 2647–2651. [Google Scholar] [CrossRef] [PubMed]
- Sommer, B.; Kohler, M.; Sprengel, R.; Seeburg, P.H. RNA editing in brain controls a determinant of ion flow in glutamate-gated channels. Cell 1991, 67, 11–19. [Google Scholar] [CrossRef]
- Melcher, T.; Maas, S.; Herb, A.; Sprengel, R.; Higuchi, M.; Seeburg, P.H. RED2, a brain-specific member of the RNA-specific adenosine deaminase family. J. Biol. Chem. 1996, 271, 31795–31798. [Google Scholar] [CrossRef]
- Romano, G.; Saviana, M.; Le, P.; Li, H.; Micalo, L.; Nigita, G.; Acunzo, M.; Nana-Sinkam, P. Non-Coding RNA Editing in Cancer Pathogenesis. Cancers 2020, 12, 1845. [Google Scholar] [CrossRef]
- Nishikura, K. A-to-I editing of coding and non-coding RNAs by ADARs. Nat. Rev. Mol. Cell Biol. 2016, 17, 83–96. [Google Scholar] [CrossRef]
- Rueter, S.M.; Dawson, T.R.; Emeson, R.B. Regulation of alternative splicing by RNA editing. Nature 1999, 399, 75–80. [Google Scholar] [CrossRef]
- Laurencikiene, J.; Kallman, A.M.; Fong, N.; Bentley, D.L.; Ohman, M. RNA editing and alternative splicing: The importance of co-transcriptional coordination. EMBO Rep. 2006, 7, 303–307. [Google Scholar] [CrossRef]
- Heraud-Farlow, J.E.; Walkley, C.R. What do editors do? Understanding the physiological functions of A-to-I RNA editing by adenosine deaminase acting on RNAs. Open Biol. 2020, 10, 200085. [Google Scholar] [CrossRef] [PubMed]
- Ohman, M. A-to-I editing challenger or ally to the microRNA process. Biochimie 2007, 89, 1171–1176. [Google Scholar] [CrossRef] [PubMed]
- Slotkin, W.; Nishikura, K. Adenosine-to-inosine RNA editing and human disease. Genome Med. 2013, 5, 105. [Google Scholar] [CrossRef]
- Burns, C.M.; Chu, H.; Rueter, S.M.; Hutchinson, L.K.; Canton, H.; Sanders-Bush, E.; Emeson, R.B. Regulation of serotonin-2C receptor G-protein coupling by RNA editing. Nature 1997, 387, 303–308. [Google Scholar] [CrossRef] [PubMed]
- Tariq, A.; Jantsch, M.F. Transcript diversification in the nervous system: A to I RNA editing in CNS function and disease development. Front. Neurosci. 2012, 6, 99. [Google Scholar] [CrossRef]
- Zhang, X.J.; He, P.P.; Li, M.; He, C.D.; Yan, K.L.; Cui, Y.; Yang, S.; Zhang, K.Y.; Gao, M.; Chen, J.J.; et al. Seven novel mutations of the ADAR gene in Chinese families and sporadic patients with dyschromatosis symmetrica hereditaria (DSH). Hum. Mutat. 2004, 23, 629–630. [Google Scholar] [CrossRef]
- Rice, G.I.; Kasher, P.R.; Forte, G.M.; Mannion, N.M.; Greenwood, S.M.; Szynkiewicz, M.; Dickerson, J.E.; Bhaskar, S.S.; Zampini, M.; Briggs, T.A.; et al. Mutations in ADAR1 cause Aicardi-Goutieres syndrome associated with a type I interferon signature. Nat. Genet. 2012, 44, 1243–1248. [Google Scholar] [CrossRef]
- Livingston, J.H.; Crow, Y.J. Neurologic Phenotypes Associated with Mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, ADAR1, and IFIH1: Aicardi-Goutieres Syndrome and Beyond. Neuropediatrics 2016, 47, 355–360. [Google Scholar] [CrossRef]
- Higuchi, M.; Maas, S.; Single, F.N.; Hartner, J.; Rozov, A.; Burnashev, N.; Feldmeyer, D.; Sprengel, R.; Seeburg, P.H. Point mutation in an AMPA receptor gene rescues lethality in mice deficient in the RNA-editing enzyme ADAR2. Nature 2000, 406, 78–81. [Google Scholar] [CrossRef]
- Maas, S.; Patt, S.; Schrey, M.; Rich, A. Underediting of glutamate receptor GluR-B mRNA in malignant gliomas. Proc. Natl. Acad. Sci. USA 2001, 98, 14687–14692. [Google Scholar] [CrossRef]
- Ishiuchi, S.; Yoshida, Y.; Sugawara, K.; Aihara, M.; Ohtani, T.; Watanabe, T.; Saito, N.; Tsuzuki, K.; Okado, H.; Miwa, A.; et al. Ca2+-permeable AMPA receptors regulate growth of human glioblastoma via Akt activation. J. Neurosci. 2007, 27, 7987–8001. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Li, Y.; Lin, C.H.; Chan, T.H.; Chow, R.K.; Song, Y.; Liu, M.; Yuan, Y.F.; Fu, L.; Kong, K.L.; et al. Recoding RNA editing of AZIN1 predisposes to hepatocellular carcinoma. Nat. Med. 2013, 19, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Bass, B.L. Double-stranded RNA as a template for gene silencing. Cell 2000, 101, 235–238. [Google Scholar] [CrossRef]
- Yang, W.; Chendrimada, T.P.; Wang, Q.; Higuchi, M.; Seeburg, P.H.; Shiekhattar, R.; Nishikura, K. Modulation of microRNA processing and expression through RNA editing by ADAR deaminases. Nat. Struct. Mol. Biol. 2006, 13, 13–21. [Google Scholar] [CrossRef]
- Heale, B.S.; Keegan, L.P.; McGurk, L.; Michlewski, G.; Brindle, J.; Stanton, C.M.; Caceres, J.F.; O’Connell, M.A. Editing independent effects of ADARs on the miRNA/siRNA pathways. EMBO J. 2009, 28, 3145–3156. [Google Scholar] [CrossRef]
- Kawahara, Y.; Zinshteyn, B.; Sethupathy, P.; Iizasa, H.; Hatzigeorgiou, A.G.; Nishikura, K. Redirection of silencing targets by adenosine-to-inosine editing of miRNAs. Science 2007, 315, 1137–1140. [Google Scholar] [CrossRef]
- Kume, H.; Hino, K.; Galipon, J.; Ui-Tei, K. A-to-I editing in the miRNA seed region regulates target mRNA selection and silencing efficiency. Nucleic Acids Res. 2014, 42, 10050–10060. [Google Scholar] [CrossRef]
- Cesarini, V.; Silvestris, D.A.; Tassinari, V.; Tomaselli, S.; Alon, S.; Eisenberg, E.; Locatelli, F.; Gallo, A. ADAR2/miR-589-3p axis controls glioblastoma cell migration/invasion. Nucleic Acids Res. 2018, 46, 2045–2059. [Google Scholar] [CrossRef]
- Velazquez-Torres, G.; Shoshan, E.; Ivan, C.; Huang, L.; Fuentes-Mattei, E.; Paret, H.; Kim, S.J.; Rodriguez-Aguayo, C.; Xie, V.; Brooks, D.; et al. A-to-I miR-378a-3p editing can prevent melanoma progression via regulation of PARVA expression. Nat. Commun. 2018, 9, 461. [Google Scholar] [CrossRef]
- Xu, X.; Wang, Y.; Mojumdar, K.; Zhou, Z.; Jeong, K.J.; Mangala, L.S.; Yu, S.; Tsang, Y.H.; Rodriguez-Aguayo, C.; Lu, Y.; et al. A-to-I-edited miRNA-379-5p inhibits cancer cell proliferation through CD97-induced apoptosis. J. Clin. Investig. 2019, 129, 5343–5356. [Google Scholar] [CrossRef]
- Nigita, G.; Acunzo, M.; Romano, G.; Veneziano, D.; Lagana, A.; Vitiello, M.; Wernicke, D.; Ferro, A.; Croce, C.M. microRNA editing in seed region aligns with cellular changes in hypoxic conditions. Nucleic Acids Res. 2016, 44, 6298–6308. [Google Scholar] [CrossRef] [PubMed]
- Lagana, A.; Paone, A.; Veneziano, D.; Cascione, L.; Gasparini, P.; Carasi, S.; Russo, F.; Nigita, G.; Macca, V.; Giugno, R.; et al. miR-EdiTar: A database of predicted A-to-I edited miRNA target sites. Bioinformatics 2012, 28, 3166–3168. [Google Scholar] [CrossRef] [PubMed]
- Magnaye, K.M.; Naughton, K.A.; Huffman, J.; Hogarth, D.K.; Naureckas, E.T.; White, S.R.; Ober, C. A-to-I editing of miR-200b-3p in airway cells is associated with moderate-to-severe asthma. Eur. Respir. J. 2021, 58, 2003862. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xu, X.; Yu, S.; Jeong, K.J.; Zhou, Z.; Han, L.; Tsang, Y.H.; Li, J.; Chen, H.; Mangala, L.S.; et al. Systematic characterization of A-to-I RNA editing hotspots in microRNAs across human cancers. Genome Res. 2017, 27, 1112–1125. [Google Scholar] [CrossRef]
- Pinto, Y.; Buchumenski, I.; Levanon, E.Y.; Eisenberg, E. Human cancer tissues exhibit reduced A-to-I editing of miRNAs coupled with elevated editing of their targets. Nucleic Acids Res. 2018, 46, 71–82. [Google Scholar] [CrossRef]
- Ramirez-Moya, J.; Baker, A.R.; Slack, F.J.; Santisteban, P. ADAR1-mediated RNA editing is a novel oncogenic process in thyroid cancer and regulates miR-200 activity. Oncogene 2020, 39, 3738–3753. [Google Scholar] [CrossRef]
- Maemura, K.; Watanabe, K.; Ando, T.; Hiyama, N.; Sakatani, T.; Amano, Y.; Kage, H.; Nakajima, J.; Yatomi, Y.; Nagase, T.; et al. Altered editing level of microRNAs is a potential biomarker in lung adenocarcinoma. Cancer Sci. 2018, 109, 3326–3335. [Google Scholar] [CrossRef]
- Nigita, G.; Distefano, R.; Veneziano, D.; Romano, G.; Rahman, M.; Wang, K.; Pass, H.; Croce, C.M.; Acunzo, M.; Nana-Sinkam, P. Tissue and exosomal miRNA editing in Non-Small Cell Lung Cancer. Sci. Rep. 2018, 8, 10222. [Google Scholar] [CrossRef]
- Li, Q.; Li, X.; Tang, H.; Jiang, B.; Dou, Y.; Gorospe, M.; Wang, W. NSUN2-Mediated m5C Methylation and METTL3/METTL14-Mediated m6A Methylation Cooperatively Enhance p21 Translation. J. Cell Biochem. 2017, 118, 2587–2598. [Google Scholar] [CrossRef]
- Tassinari, V.; Cesarini, V.; Tomaselli, S.; Ianniello, Z.; Silvestris, D.A.; Ginistrelli, L.C.; Martini, M.; De Angelis, B.; De Luca, G.; Vitiani, L.R.; et al. ADAR1 is a new target of METTL3 and plays a pro-oncogenic role in glioblastoma by an editing-independent mechanism. Genome Biol. 2021, 22, 51. [Google Scholar] [CrossRef]
- Xiang, J.F.; Yang, Q.; Liu, C.X.; Wu, M.; Chen, L.L.; Yang, L. N(6)-Methyladenosines Modulate A-to-I RNA Editing. Mol. Cell 2018, 69, 126–135 e126. [Google Scholar] [CrossRef] [PubMed]
- Terajima, H.; Lu, M.; Zhang, L.; Cui, Q.; Shi, Y.; Li, J.; He, C. N6-methyladenosine promotes induction of ADAR1-mediated A-to-I RNA editing to suppress aberrant antiviral innate immune responses. PLoS Biol. 2021, 19, e3001292. [Google Scholar] [CrossRef]
- Wei, C.; Gershowitz, A.; Moss, B. N6, O2′-dimethyladenosine a novel methylated ribonucleoside next to the 5′ terminal of animal cell and virus mRNAs. Nature 1975, 257, 251–253. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Zhang, M.; Li, K.; Bai, D.; Yi, C. Cap-specific, terminal N(6)-methylation by a mammalian m(6)Am methyltransferase. Cell Res. 2019, 29, 80–82. [Google Scholar] [CrossRef] [PubMed]
- Akichika, S.; Hirano, S.; Shichino, Y.; Suzuki, T.; Nishimasu, H.; Ishitani, R.; Sugita, A.; Hirose, Y.; Iwasaki, S.; Nureki, O.; et al. Cap-specific terminal N (6)-methylation of RNA by an RNA polymerase II-associated methyltransferase. Science 2019, 363, eaav0080. [Google Scholar] [CrossRef]
- Nariman-Saleh-Fam, Z.; Saadatian, Z.; Daraei, A.; Mansoori, Y.; Bastami, M.; Tavakkoli-Bazzaz, J. The intricate role of miR-155 in carcinogenesis: Potential implications for esophageal cancer research. Biomark. Med. 2019, 13, 147–159. [Google Scholar] [CrossRef]
- Li, Y.; Su, R.; Deng, X.; Chen, Y.; Chen, J. FTO in cancer: Functions, molecular mechanisms, and therapeutic implications. Trends Cancer 2022, 8, 598–614. [Google Scholar] [CrossRef]
- Courtney, D.G.; Chalem, A.; Bogerd, H.P.; Law, B.A.; Kennedy, E.M.; Holley, C.L.; Cullen, B.R. Extensive Epitranscriptomic Methylation of A and C Residues on Murine Leukemia Virus Transcripts Enhances Viral Gene Expression. mBio 2019, 10, e01209-19. [Google Scholar] [CrossRef]
- Cristinelli, S.; Angelino, P.; Janowczyk, A.; Delorenzi, M.; Ciuffi, A. HIV Modifies the m6A and m5C Epitranscriptomic Landscape of the Host Cell. Front. Virol. 2021, 1, 714475. [Google Scholar] [CrossRef]
- Chhabra, R. miRNA and methylation: A multifaceted liaison. ChemBioChem 2015, 16, 195–203. [Google Scholar] [CrossRef]
- Santiago, M.; Strobel, S. Thin layer chromatography. Methods Enzymol. 2013, 533, 303–324. [Google Scholar] [CrossRef] [PubMed]
- Bodi, Z.; Fray, R.G. Detection and Quantification of N (6)-Methyladenosine in Messenger RNA by TLC. Methods Mol. Biol. 2017, 1562, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Abner, J.J.; Franklin, J.L.; Clement, M.A.; Hinger, S.A.; Allen, R.M.; Liu, X.; Kellner, S.; Wu, J.; Karijolich, J.; Liu, Q.; et al. Depletion of METTL3 alters cellular and extracellular levels of miRNAs containing m(6)A consensus sequences. Heliyon 2021, 7, e08519. [Google Scholar] [CrossRef] [PubMed]
- Thuring, K.; Schmid, K.; Keller, P.; Helm, M. LC-MS Analysis of Methylated RNA. Methods Mol. Biol. 2017, 1562, 3–18. [Google Scholar] [CrossRef] [PubMed]
- Schmid, K.; Thuring, K.; Keller, P.; Ochel, A.; Kellner, S.; Helm, M. Variable presence of 5-methylcytosine in commercial RNA and DNA. RNA Biol. 2015, 12, 1152–1158. [Google Scholar] [CrossRef] [PubMed]
- Ross, R.; Cao, X.; Yu, N.; Limbach, P.A. Sequence mapping of transfer RNA chemical modifications by liquid chromatography tandem mass spectrometry. Methods 2016, 107, 73–78. [Google Scholar] [CrossRef]
- Zhang, N.; Shi, S.; Jia, T.Z.; Ziegler, A.; Yoo, B.; Yuan, X.; Li, W.; Zhang, S. A general LC-MS-based RNA sequencing method for direct analysis of multiple-base modifications in RNA mixtures. Nucleic Acids Res. 2019, 47, e125. [Google Scholar] [CrossRef]
- Dominissini, D.; Moshitch-Moshkovitz, S.; Schwartz, S.; Salmon-Divon, M.; Ungar, L.; Osenberg, S.; Cesarkas, K.; Jacob-Hirsch, J.; Amariglio, N.; Kupiec, M.; et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature 2012, 485, 201–206. [Google Scholar] [CrossRef]
- Edelheit, S.; Schwartz, S.; Mumbach, M.R.; Wurtzel, O.; Sorek, R. Transcriptome-wide mapping of 5-methylcytidine RNA modifications in bacteria, archaea, and yeast reveals m5C within archaeal mRNAs. PLoS Genet. 2013, 9, e1003602. [Google Scholar] [CrossRef]
- Linder, B.; Grozhik, A.V.; Olarerin-George, A.O.; Meydan, C.; Mason, C.E.; Jaffrey, S.R. Single-nucleotide-resolution mapping of m6A and m6Am throughout the transcriptome. Nat. Methods 2015, 12, 767–772. [Google Scholar] [CrossRef]
- Trixl, L.; Lusser, A. The dynamic RNA modification 5-methylcytosine and its emerging role as an epitranscriptomic mark. Wiley Interdiscip. Rev. RNA 2019, 10, e1510. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Lu, Z.; Wang, X.; Fu, Y.; Luo, G.Z.; Liu, N.; Han, D.; Dominissini, D.; Dai, Q.; Pan, T.; et al. High-resolution N(6)-methyladenosine (m(6) A) map using photo-crosslinking-assisted m(6) A sequencing. Angew. Chem. Int. Ed. Engl. 2015, 54, 1587–1590. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Xiong, X.; Yi, C. Epitranscriptome sequencing technologies: Decoding RNA modifications. Nat. Methods 2016, 14, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Baquero-Perez, B.; Geers, D.; Diez, J. From A to m(6)A: The Emerging Viral Epitranscriptome. Viruses 2021, 13, 1049. [Google Scholar] [CrossRef]
- Molinie, B.; Wang, J.; Lim, K.S.; Hillebrand, R.; Lu, Z.X.; Van Wittenberghe, N.; Howard, B.D.; Daneshvar, K.; Mullen, A.C.; Dedon, P.; et al. m(6)A-LAIC-seq reveals the census and complexity of the m(6)A epitranscriptome. Nat. Methods 2016, 13, 692–698. [Google Scholar] [CrossRef] [PubMed]
- Flatau, E.; Gonzales, F.A.; Michalowsky, L.A.; Jones, P.A. DNA methylation in 5-aza-2′-deoxycytidine-resistant variants of C3H 10T1/2 C18 cells. Mol. Cell Biol. 1984, 4, 2098–2102. [Google Scholar] [CrossRef]
- Juttermann, R.; Li, E.; Jaenisch, R. Toxicity of 5-aza-2′-deoxycytidine to mammalian cells is mediated primarily by covalent trapping of DNA methyltransferase rather than DNA demethylation. Proc. Natl. Acad. Sci. USA 1994, 91, 11797–11801. [Google Scholar] [CrossRef]
- Schaefer, M.; Pollex, T.; Hanna, K.; Lyko, F. RNA cytosine methylation analysis by bisulfite sequencing. Nucleic Acids Res. 2009, 37, e12. [Google Scholar] [CrossRef]
- Amort, T.; Rieder, D.; Wille, A.; Khokhlova-Cubberley, D.; Riml, C.; Trixl, L.; Jia, X.Y.; Micura, R.; Lusser, A. Distinct 5-methylcytosine profiles in poly(A) RNA from mouse embryonic stem cells and brain. Genome Biol. 2017, 18, 1. [Google Scholar] [CrossRef]
- Huang, T.; Chen, W.; Liu, J.; Gu, N.; Zhang, R. Genome-wide identification of mRNA 5-methylcytosine in mammals. Nat. Struct. Mol. Biol. 2019, 26, 380–388. [Google Scholar] [CrossRef]
- Incarnato, D.; Anselmi, F.; Morandi, E.; Neri, F.; Maldotti, M.; Rapelli, S.; Parlato, C.; Basile, G.; Oliviero, S. High-throughput single-base resolution mapping of RNA 2-O-methylated residues. Nucleic Acids Res. 2017, 45, 1433–1441. [Google Scholar] [CrossRef] [PubMed]
- Birkedal, U.; Christensen-Dalsgaard, M.; Krogh, N.; Sabarinathan, R.; Gorodkin, J.; Nielsen, H. Profiling of ribose methylations in RNA by high-throughput sequencing. Angew. Chem. Int. Ed. Engl. 2015, 54, 451–455. [Google Scholar] [CrossRef] [PubMed]
- Krogh, N.; Birkedal, U.; Nielsen, H. RiboMeth-seq: Profiling of 2′-O-Me in RNA. Methods Mol. Biol. 2017, 1562, 189–209. [Google Scholar] [CrossRef] [PubMed]
- Krogh, N.; Nielsen, H. Sequencing-based methods for detection and quantitation of ribose methylations in RNA. Methods 2019, 156, 5–15. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Holley, C.L.; Carmichael, G.G. Transcriptome-Wide Identification of 2′-O-Methylation Sites with RibOxi-Seq. Methods Mol. Biol. 2022, 2404, 393–407. [Google Scholar] [CrossRef]
- Sakurai, M.; Ueda, H.; Yano, T.; Okada, S.; Terajima, H.; Mitsuyama, T.; Toyoda, A.; Fujiyama, A.; Kawabata, H.; Suzuki, T. A biochemical landscape of A-to-I RNA editing in the human brain transcriptome. Genome Res. 2014, 24, 522–534. [Google Scholar] [CrossRef]
- Marceca, G.P.; Tomasello, L.; Distefano, R.; Acunzo, M.; Croce, C.M.; Nigita, G. Detecting and Characterizing A-To-I microRNA Editing in Cancer. Cancers 2021, 13, 1699. [Google Scholar] [CrossRef]
- Yuting, K.; Ding, D.; Iizasa, H. Adenosine-to-Inosine RNA Editing Enzyme ADAR and microRNAs. Methods Mol. Biol. 2021, 2181, 83–95. [Google Scholar] [CrossRef]
- de Hoon, M.J.; Taft, R.J.; Hashimoto, T.; Kanamori-Katayama, M.; Kawaji, H.; Kawano, M.; Kishima, M.; Lassmann, T.; Faulkner, G.J.; Mattick, J.S.; et al. Cross-mapping and the identification of editing sites in mature microRNAs in high-throughput sequencing libraries. Genome Res. 2010, 20, 257–264. [Google Scholar] [CrossRef]
- Alon, S.; Mor, E.; Vigneault, F.; Church, G.M.; Locatelli, F.; Galeano, F.; Gallo, A.; Shomron, N.; Eisenberg, E. Systematic identification of edited microRNAs in the human brain. Genome Res. 2012, 22, 1533–1540. [Google Scholar] [CrossRef]
- Alon, S.; Eisenberg, E. Identifying RNA editing sites in miRNAs by deep sequencing. Methods Mol. Biol. 2013, 1038, 159–170. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Ji, B.; Song, R.; Wang, S.; Li, T.; Zhang, X.; Chen, K.; Li, T.; Li, J. Accurate detection for a wide range of mutation and editing sites of microRNAs from small RNA high-throughput sequencing profiles. Nucleic Acids Res. 2016, 44, e123. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Baras, A.S.; Halushka, M.K. miRge 2.0 for comprehensive analysis of microRNA sequencing data. BMC Bioinform. 2018, 19, 275. [Google Scholar] [CrossRef]
- Garalde, D.R.; Snell, E.A.; Jachimowicz, D.; Sipos, B.; Lloyd, J.H.; Bruce, M.; Pantic, N.; Admassu, T.; James, P.; Warland, A.; et al. Highly parallel direct RNA sequencing on an array of nanopores. Nat. Methods 2018, 15, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Parker, M.T.; Knop, K.; Sherwood, A.V.; Schurch, N.J.; Mackinnon, K.; Gould, P.D.; Hall, A.J.; Barton, G.J.; Simpson, G.G. Nanopore direct RNA sequencing maps the complexity of Arabidopsis mRNA processing and m(6)A modification. eLife 2020, 9, e49658. [Google Scholar] [CrossRef] [PubMed]
- Campos, J.H.C.; Maricato, J.T.; Braconi, C.T.; Antoneli, F.; Janini, L.M.R.; Briones, M.R.S. Direct RNA Sequencing Reveals SARS-CoV-2 m6A Sites and Possible Differential DRACH Motif Methylation among Variants. Viruses 2021, 13, 2108. [Google Scholar] [CrossRef]
- Yang, L.; Perrera, V.; Saplaoura, E.; Apelt, F.; Bahin, M.; Kramdi, A.; Olas, J.; Mueller-Roeber, B.; Sokolowska, E.; Zhang, W.; et al. m(5)C Methylation Guides Systemic Transport of Messenger RNA over Graft Junctions in Plants. Curr. Biol. 2019, 29, 2465–2476 e2465. [Google Scholar] [CrossRef]
- Zhang, J.; Yan, S.; Chang, L.; Guo, W.; Wang, Y.; Wang, Y.; Zhang, P.; Chen, H.Y.; Huang, S. Direct microRNA Sequencing Using Nanopore-Induced Phase-Shift Sequencing. iScience 2020, 23, 100916. [Google Scholar] [CrossRef]
- Lee, F.C.Y.; Ule, J. Advances in CLIP Technologies for Studies of Protein-RNA Interactions. Mol. Cell 2018, 69, 354–369. [Google Scholar] [CrossRef]
- Bailey, A.D.; Talkish, J.; Ding, H.; Igel, H.; Duran, A.; Mantripragada, S.; Paten, B.; Ares, M. Concerted modification of nucleotides at functional centers of the ribosome revealed by single-molecule RNA modification profiling. eLife 2022, 11, e76562. [Google Scholar] [CrossRef]
- Nguyen, T.A.; Heng, J.W.J.; Kaewsapsak, P.; Kok, E.P.L.; Stanojevic, D.; Liu, H.; Cardilla, A.; Praditya, A.; Yi, Z.; Lin, M.; et al. Direct identification of A-to-I editing sites with nanopore native RNA sequencing. Nat. Methods 2022, 19, 833–844. [Google Scholar] [CrossRef] [PubMed]
- Leger, A.; Amaral, P.P.; Pandolfini, L.; Capitanchik, C.; Capraro, F.; Miano, V.; Migliori, V.; Toolan-Kerr, P.; Sideri, T.; Enright, A.J.; et al. RNA modifications detection by comparative Nanopore direct RNA sequencing. Nat. Commun. 2021, 12, 7198. [Google Scholar] [CrossRef] [PubMed]
- Vo, J.M.; Mulroney, L.; Quick-Cleveland, J.; Jain, M.; Akeson, M.; Ares, M., Jr. Synthesis of modified nucleotide polymers by the poly(U) polymerase Cid1: Application to direct RNA sequencing on nanopores. RNA 2021, 27, 1497–1511. [Google Scholar] [CrossRef] [PubMed]
- Rang, F.J.; Kloosterman, W.P.; de Ridder, J. From squiggle to basepair: Computational approaches for improving nanopore sequencing read accuracy. Genome Biol. 2018, 19, 90. [Google Scholar] [CrossRef] [PubMed]
- Yankova, E.; Blackaby, W.; Albertella, M.; Rak, J.; De Braekeleer, E.; Tsagkogeorga, G.; Pilka, E.S.; Aspris, D.; Leggate, D.; Hendrick, A.G.; et al. Small-molecule inhibition of METTL3 as a strategy against myeloid leukaemia. Nature 2021, 593, 597–601. [Google Scholar] [CrossRef]
- Bhate, A.; Sun, T.; Li, J.B. ADAR1: A New Target for Immuno-oncology Therapy. Mol. Cell 2019, 73, 866–868. [Google Scholar] [CrossRef]
- Thornton, J.E.; Chang, H.M.; Piskounova, E.; Gregory, R.I. Lin28-mediated control of let-7 microRNA expression by alternative TUTases Zcchc11 (TUT4) and Zcchc6 (TUT7). RNA 2012, 18, 1875–1885. [Google Scholar] [CrossRef]
- Lin, S.; Gregory, R.I. Identification of small molecule inhibitors of Zcchc11 TUTase activity. RNA Biol. 2015, 12, 792–800. [Google Scholar] [CrossRef]
- Zhu, J.; Djukovic, D.; Deng, L.; Gu, H.; Himmati, F.; Chiorean, E.G.; Raftery, D. Colorectal cancer detection using targeted serum metabolic profiling. J. Proteome Res. 2014, 13, 4120–4130. [Google Scholar] [CrossRef]
- Djukovic, D.; Baniasadi, H.R.; Kc, R.; Hammoud, Z.; Raftery, D. Targeted serum metabolite profiling of nucleosides in esophageal adenocarcinoma. Rapid Commun. Mass Spectrom. 2010, 24, 3057–3062. [Google Scholar] [CrossRef]
- Chen, F.; Xue, J.; Zhou, L.; Wu, S.; Chen, Z. Identification of serum biomarkers of hepatocarcinoma through liquid chromatography/mass spectrometry-based metabonomic method. Anal. Bioanal. Chem. 2011, 401, 1899–1904. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Hu, Q.; Hou, H.; Wang, S.; Zhang, Y.; Luo, Y.; Chen, H.; Deng, H.; Zhu, H.; Zhang, L.; et al. Metabolite analysis-aided diagnosis of papillary thyroid cancer. Endocr. Relat. Cancer 2019, 26, 829–841. [Google Scholar] [CrossRef] [PubMed]
- Distefano, R.; Nigita, G.; Le, P.; Romano, G.; Acunzo, M.; Nana-Sinkam, P. Disparities in Lung Cancer: miRNA Isoform Characterization in Lung Adenocarcinoma. Cancers 2022, 14, 773. [Google Scholar] [CrossRef] [PubMed]
- Relier, S.; Ripoll, J.; Guillorit, H.; Amalric, A.; Achour, C.; Boissiere, F.; Vialaret, J.; Attina, A.; Debart, F.; Choquet, A.; et al. FTO-mediated cytoplasmic m(6)Am demethylation adjusts stem-like properties in colorectal cancer cell. Nat. Commun. 2021, 12, 1716. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Method | Specificity | Description | Advantages and Limitations | Suitability for miRNAs | References |
|---|---|---|---|---|---|
| Thin-layer chromatography | All modifications | Separates compounds in a mixture by their chemical properties. Each component migrates differentially based on affinity for the stationary (adherent) phase vs. mobile (liquid) phase. In 2D-TLC, the RNA is digested to form a 5′ OH prior to labeling with radioactive ATP. The migration is compared to a synthetic RNA standard, allowing for identification of specific epitranscriptomic modifications. | Can identify RNA modifications and be utilized for studying enzymatic activity and kinetics but does not provide the exact sites of the modifications. | Yes | [81,182,183,184] |
| Liquid chromatography–mass spectrometry (LC-MS) | All modifications | RNA samples are digested into nucleosides, which are separated into nucleotides by liquid chromatography, and the corresponding mass is determined by mass spectrometry. Using a ladder with known fragmentation patterns, the RNA sequence can be determined. | Can detect and quantify epitranscriptomic modifications with high sensitivity. | Yes | [185,186,187,188] |
| Methylated RNA immunoprecipitation coupled with high-throughput sequencing (MeRIP-seq), m5C-RIP-seq, and m7G-MeRIP | m6A, m5C m7G | Purified mRNA is randomly fragmented (~100–150 nucleotides) prior to immunoprecipitation with an anti-m6A antibody (MeRIP-seq), an anti-m5C (m5C-RIP-seq) antibody, or an anti m7G antibody (m7G-MeRIP). A library is constructed and sequenced. | High specificity but does not have single-nucleotide resolution and cannot detect methylation of non-abundant RNAs. | Yes | [7,22,82,111,189,190] |
| m6A-individual nucleotide resolution crosslinking and immunoprecipitation (miCLIP-m6A) | m6A | Implements UV crosslinking at the anti-m6A-bound site, which induces a mutation that can be identified by sequencing | Can identify the exact sites of m6A. | Not tested | [191] |
| m5C-individual nucleotide resolution crosslinking and immunoprecipitation (miCLIP-m5C) | m5C | A mutant of NSUN2 (C271A) is overexpressed which forms a covalent bond with m5C. The bond can be detected with an anti-NSUN2 antibody. This complex induces a stop position during RT-PCR, interpreted as a truncation site. | Can identify the exact sites of m5C. | Yes | [83,85,192] |
| Photo-crosslinking-assisted m6A sequencing (PA-m6A-seq) | m6A | Incorporates 4-thiouridine into the RNA, which induces a T-to-C mutation at crosslinked anti-m6A-bound sites that can be identified by sequencing. | Can identify the exact sites of m6A. | Not tested | [193,194,195] |
| m6A-level and isoform-characterization sequencing (m6A-LAIC-seq) | m6A | An excess of anti-m6A antibody is utilized for pulling down methylated RNA. A spike-in internal standard is added to allow for relative quantification of m6A RNAs | Permits evaluation of methylation status. | Not tested | [194,196] |
| 5-azacytidine-mediated RNA immunoprecipitation (Aza-IP-seq) | m5C | 5-azaC, a cytidine analog, is randomly incorporated into RNA. RCMT will form an irreversible bond with its RNA targets, which can be detected using an anti-RCMT antibody. m5C sites are recognized as C-to-G conversions due to a ring-opening of 5-azaC. | Can identify the exact sites of m5C, but only a short treatment is conducted due to the high toxicity of 5-azaC, thereby reducing its incorporation into RNA. | Not tested | [192,194,197,198] |
| RNA bisulfite sequencing technology (RNA-BisSeq) | Methylated cytosines such as m5C | Sodium bisulfite is added, which deaminates unmethylated cytosines (at acidic pHs) or uracil (at basic pHs), leaving methylated cytosines intact. | Has single-nucleotide resolution and does not use high concentrations of RNA. However, it cannot react with base-paired cytosines and does not distinguish 5-methylcytosine from 5-hydroxymethylcytosine | Yes | [82,192,199,200,201] |
| 2′-O-methyl sequencing (2′-O-Me-Seq) | 2′-O-methyl | Reverse transcription halts once it reaches a 2′-O-methylated nucleotide, thereby truncating the cDNA. These sites can be detected by sequencing. | Can detect specific 2′-O-methyl sites. | Not tested | [202] |
| RiboMeth-seq | 2′-O-methyl | RNA is treated at an alkaline pH and high temperature to fragment the RNA into 20–40 nucleotides. The resulting fragments are sequenced. Sites that contain 2′-O-methyl sites are not fragmented and do not generate read ends. | Can detect omitted peak regions that corresponds to 2′-O-methyl sites. | Not tested | [203,204] |
| RibOxi-Seq | 2′-O-methyl | RNAs are fragmented with Benzonase and oxidized to remove 3′ phosphates. 3′ ends that contain 2′-O-methyl are resistant to oxidation and are enriched with linker ligation. | Can detect 2′-O-methyl in rRNAs but requires microgram amounts of input. | No | [205,206] |
| Nm-seq | 2′-O-methyl | Fragmented RNAs are treated with repeated cycles of OED, removing 3′ nucleotides that are not 2′-O-methylated. A final OE cycle is implemented to dephosphorylate non-2′-O-methylated 3′ ends, preventing adapter ligation. | Provides single nucleotide detection of 2′-O-methylation. Can be used for a wide range of RNAs. | Not tested | [71] |
| TAIL-seq | Poly-U | rRNA-depleted RNA samples are ligated in the 3′ end with a biotinylated adapter. RNA is fragmented with RNAse T1, and 3′ ends are recovered using streptavidin pulldown. | Provides information on poly-A tail length and the addition of poly-U at the 3′ end. | No | [127] |
| Borohydride Reduction (BoRed-seq) | m7G | RNA is decapped and treated with NaBH4 at a low pH. The abasic m7G site is treated with biotin-coupled aldehyde-reactive probe. The biotinylated RNA is recovered with streptavidin pulldown | Detects m7G site in RNAs without cleavage of the m7G sites. Suitable for small RNAs and low abundant RNAs | Yes | [119] |
| Inosine chemical erasing sequencing (ICE-seq) | A-to-I | Inosine is treated with acrylonitrile to form N1-cyanoethylinosinem, which halts retrotranscription and truncates the cDNA. These sites can be detected by sequencing. | Can identify A-to-I sites. | Yes | [207,208] |
| Bioinformatic detection of A-to-I editing from RNAseq | A-to-I | A-to-I editing is detected directly from RNAseq using bioinformatic tools to identify editing sites from SNPs | Can detect editing from RNAseq but requires high sequencing depth. | Yes | [169,194,208,209,210,211,212,213,214] |
| Nanopore sequencing | All modifications | Utilizes nanopore proteins that are inserted into the membrane. RNAs are translocated through these proteins, which leads to a perturbation of the nanopore current. | Has single-nucleotide resolution and does not require the processing of the amplified RNA. However, it has a high signal-to-noise ratio and may not distinguish similar nucleotides. | Yes | [83,215,216,217,218,219] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
del Valle-Morales, D.; Le, P.; Saviana, M.; Romano, G.; Nigita, G.; Nana-Sinkam, P.; Acunzo, M. The Epitranscriptome in miRNAs: Crosstalk, Detection, and Function in Cancer. Genes 2022, 13, 1289. https://doi.org/10.3390/genes13071289
del Valle-Morales D, Le P, Saviana M, Romano G, Nigita G, Nana-Sinkam P, Acunzo M. The Epitranscriptome in miRNAs: Crosstalk, Detection, and Function in Cancer. Genes. 2022; 13(7):1289. https://doi.org/10.3390/genes13071289
Chicago/Turabian Styledel Valle-Morales, Daniel, Patricia Le, Michela Saviana, Giulia Romano, Giovanni Nigita, Patrick Nana-Sinkam, and Mario Acunzo. 2022. "The Epitranscriptome in miRNAs: Crosstalk, Detection, and Function in Cancer" Genes 13, no. 7: 1289. https://doi.org/10.3390/genes13071289
APA Styledel Valle-Morales, D., Le, P., Saviana, M., Romano, G., Nigita, G., Nana-Sinkam, P., & Acunzo, M. (2022). The Epitranscriptome in miRNAs: Crosstalk, Detection, and Function in Cancer. Genes, 13(7), 1289. https://doi.org/10.3390/genes13071289