
Germline Testing in a Cohort of Patients at High Risk of Hereditary Cancer Predisposition Syndromes: First Two-Year Results from South Italy

 ,
,  , ,
, , 
Abstract
1. Introduction
2. Materials and Methods
2.1. Patients and Enrolling Criteria
2.2. Genetic Testing: DNA Extraction and NGS
2.3. Sanger Sequencing
2.4. Variant Analysis (Classification): Germline Calling Variants and Filtering
2.5. Statistics
3. Results
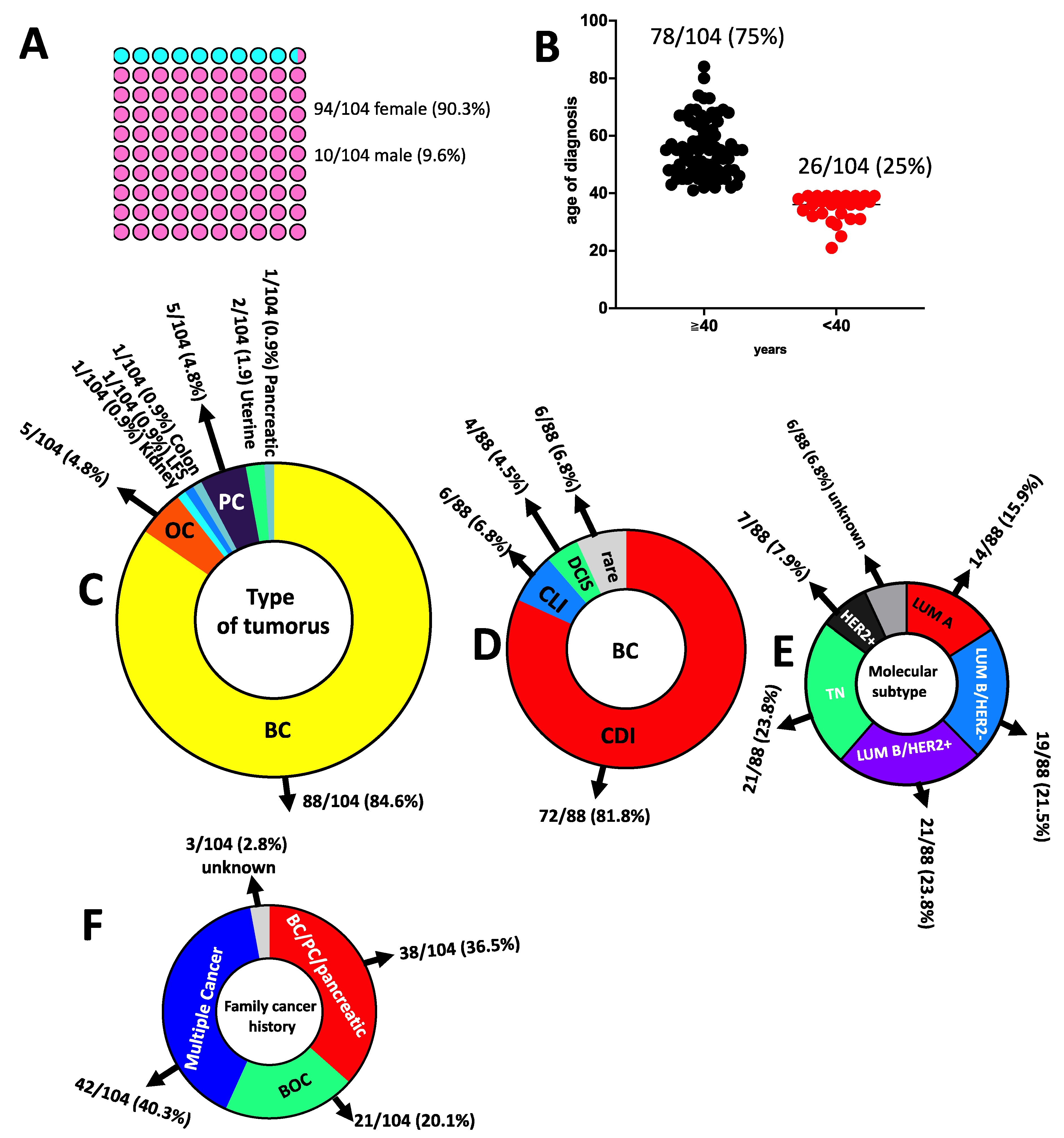
3.1. Genetic Counselling and Clinical Features of Cancer Patients
3.2. Likelihood of Carrying PVs in the Moderate-to-High-Risk Genes Calculated by the Cancer Risk Model BOADICEA in HBOC and Prostate Cancer Patients
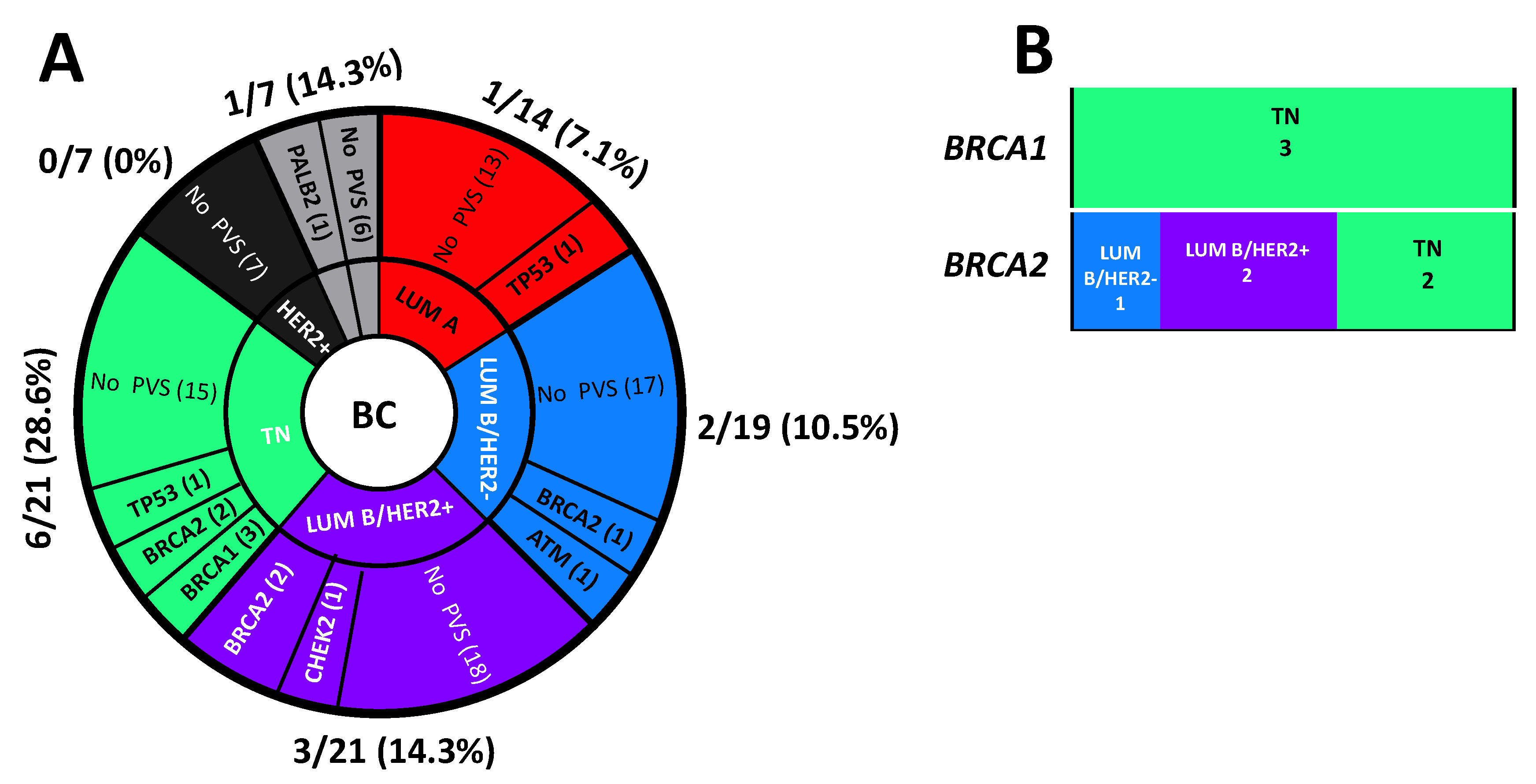
3.3. Genetic Testing and Variants Distribution

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| n. | Patient ID | Variant (HGVS) GRCh37 | Gene with Variant | dpSNP (Varsome Link) | Type of Variant | MAF gnomAD% | Clinvar Classification | Ref | Type of Cancer |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 558/19 | chr17:g.41258504A>C c.181T>G (p.Cys61Gly) | BRCA1 | rs28897672 | missense | 0.0031 | Pathogenic | [20] | Ovarian cancer |
| 2 | 673/19 | chr11:g.108186742C>T c.6100C>T (p.Arg2034Ter) | ATM | rs532480170 | nonsense | 0.0004 | Pathogenic | [21,22] | Breast cancer |
| 3 | 764/19 | chr17:g.7578204del c.645delT (p.Ser215ArgfsTer32) | TP53 | NR | frameshift | NR | Pathogenic | [11] | Li–Fraumeni |
| 4 | 775/19 | chr11:g.108236087G>A c.9023G>A (p.Arg3008His) | ATM | rs587781894 | missense | NR | Likely pathogenic | [23,24,25,26,27] | Prostate cancer |
| 5 | 99/21 | chr22:g.29105993C>A c.846+1G>C | CHEK2 | rs864622149 | splice-site | NR | Likely pathogenic | [28,29] | Breast cancer |
| 6 | 164/21 | chr17:g.41267741A>G c.134+2T>C | BRCA1 | rs80358131 | splice-site | NR | Pathogenic | [20,30] | Breast cancer |
| 7 | 223/21 | chr13:g.32944695G>A c.8487+1G>A | BRCA2 | rs81002798 | splice-site | NR | Pathogenic | [31,32,33] | Breast cancer |
| 8 | 279/21 | chr13:g.32921033G>A c.7007G>A (p.Arg2336His) | BRCA2 | rs28897743 | splice-site (*) | NR | Pathogenic | [34,35,36] | Breast cancer |
| 9 | 365/21 | chr13:g.32907285T>G c.1670T>G (p.Leu557Ter) | BRCA2 | rs80358452 | nonsense | NR | Pathogenic | [37,38,39] | Breast cancer |
| 10 | 432/21 | chr16:g.23646416A>C c.1451T>G (p.Leu484Ter) | PALB2 | rs786203714 | nonsense | NR | Pathogenic | [40,41,42,43] | Breast cancer |
| 11 | 488/21 | chr17:g.7578479G>C c.451C>G (p.Pro151Ala) | TP53 | NR | missense | NR | Likely pathogenic | [44,45] | Breast cancer |
| 12 | 713/21 | chr13:g.32907526T>A c.1909+2T>A | BRCA2 | rs876658577 | splice-site | NR | Likely pathogenic | NR | Breast cancer |
| 13 | 812/21 | Chr2:g.47429869del c.1204del (p.Gln402LysfsTer10) | MSH2 | rs63751413 | frameshift | NR | Pathogenic | [46] | Colon cancer |
| 14 | 930/21 | chr13:g.32333148T>G c.1670T>G (p.Leu557Ter) | BRCA2 | rs80358452 | nonsense | NR | Pathogenic | [37] | Breast cancer |
| 15 | 943/21 | chr17:g.41223012_41223030del c.4964_4982del p.(Ser1655TyrfsTer16) | BRCA1 | rs1555580678 | frameshift | NR | Pathogenic | NR | Breast cancer |
| 16 | 22/22 | chr17:g.7578555C>T c.376-1G>A | TP53 | rs868137297 | splice-site | 0.00000657 | Pathogenic | [47] | Breast cancer |
| 17 | 69/22 | chr17:g.431157724A>C c.134+2T>G | BRCA1 | rs80358131 | splice-site | NR | Pathogenic | [48] | Breast cancer |
3.4. VUS, Variants Classification by ACMG Guidelines and Reclassification by SHERLOC Framework
| n. | Patient ID | Variant (HGVS) GRCh37 | Gene with Variant | dpSNP | Type of Variant | MAF gnomAD% | Clinvar Classification | Ref | Type of Cancer |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 674/19 | chr11:g.108201108T>G c.7475T>G (p.Leu2492Arg) | ATM | rs56399857 | missense | 0.0099 | VUS | [51,52] | Breast cancer |
| 2 | 348/20 | chr11:g.108150289C>T c.3356C>T (p.Ala1119Val) | ATM | rs778882461 | missense | 0.0039 | VUS | NR | Breast cancer |
| 3 | 704/20 | chr11:g.108200949T>C c.7316T>C (p.Val2439Ala) | ATM | rs776266049 | missense | 0.0004 | VUS | [53,54,55] | Prostate cancer |
| 4 | 87/21 | chr16: g.23652442C>T c.37G>A (p.Glu13Lys) | PALB2 | rs373287455 | missense | 0.0004 | VUS | [41,52,56,57,58,59] | Kidney cancer |
| 5 | 133/21 | chr22:g.29095923A>G c.911T>C (p.Met304Thr) | CHEK2 | rs587782033 | missense | NR | VUS | [60,61,62,63,64] | Breast cancer |
| 6 | 150/21 | chr17:g.41246204G>C c.1344C>G (p.His448Gln) | BRCA1 | NR | missense | NR | VUS | NR | Breast cancer |
| 7 | 182/21 | chr17:g.41203100G>T c.5312C>A (p.Pro1771His) | BRCA1 | NR | missense | NR | VUS | [20] | Breast cancer |
| 8 | 262/21 | chr2:g.48028063A>G c.2941A>G (p.Ile981Val) | MSH6 | rs730881799 | missense | NR | VUS | [65] | Breast cancer |
| 9 | 282/21 | chr22: g.29091178C>A c.1312G>T (p.Asp438Tyr) chr3:g.37055919A>G c.678-4A>G chr2:g.48026120C>T c.998C>T (p.Thr333Ile) | CHEK2 MLH1 MSH6 | rs2000508 83 rs766711342 rs587781983 | missense splice-site missense | 0.039 0.0012 0.0032 | VUS VUS VUS | [66,67,68,69,70,71,72,73] | Pancreatic cancer |
| 10 | 310/21 | chr16:g.23647304G>C c.563C>G (p.Ala188Gly) | PALB2 | rs587781975 | missense | 0.0011 | VUS | NR | Breast cancer |
| 11 | 344/21 | chr22:g.29091178C>A c.1312G>T (p.Asp438Tyr) | CHEK2 | rs200050883 | missense | 0.039 | VUS | [66,67,68] | Breast cancer |
| 12 | 465/21 | chr11:g.108142010A>G c.2954A>G (p.Asp985Gly) | ATM | rs864622159 | missense | 0.0004 | VUS | NR | Breast cancer |
| 13 | 489/21 | chr2:g.48026433-48026434delinsGC c.1311_ 1312delinsGC (p.437_438delinsGlnLeu) | MSH6 | NR | missense | VUS | [74] | Ovarian cancer | |
| 14 | 620/21 | chr22:g.28711914C>G c.787G>C (p.Glu263Gln) | CHEK2 | rs730881686 | missense | 0.00000796 | VUS | [60,75] | Breast cancer |
| 15 | 665/21 | chr22:g.29091797G>A c.1160C>T (p.Thr387Ile) | CHEK2 | rs587780168 | missense | 0.00000398 | VUS | [76,77] | Breast cancer |
| 16 | 760/21 | chr7:g.6043346C>A c.328G>T (p.Ala110Ser) | PMS2 | rs767775907 | missense | 0.0000169 | VUS | [78] | Breast cancer |
| 17 | 761/21 | chr7:g.6043346C>A c.328G>T (p.Ala110Ser) | PMS2 | rs767775907 | missense | 0.0000169 | VUS | [78] | Breast cancer |
| 18 | 979/21 | chr22:g.29130713G>A c.-4C>T chr3:g.37092003C>G c.2130C>G (p.Asn710Lys) | CHEK2 MLH1 | rs3749381 48 rs7491000 96 | 5′-UTR variant missense | 0.0000573 0.00000398 | VUS VUS | [79] NR | Breast cancer |
| 19 | 1006/21 | chr11:g.108300949T>C c.7316T>C (p.Val2439Ala) | ATM | rs776266049 | missense | 0.00000398 | VUS | [53] | Breast cancer |
| 20 | 68/22 | chr11:g.108224555 c.8734A>G (p.Arg2912Gly) | ATM | rs376676328 | missense | 0.000219 | VUS | [80] | Prostate cancer |
| 21 | 156/22 | chr17:g.41246298T>C c.1250A>G (p.Asn417Ser) | BRCA1 | rs80357113 | missense | NR | VUS | [81] | Breast Cancer |
3.5. Likelihood of Carrying PVs in LP/P Variant-Positive HBOC and Prostate Cancer Patients versus Negative or Patients Carrying VUS
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tsaousis, G.N.; Papadopoulou, E.; Apessos, A.; Agiannitopoulos, K.; Pepe, G.; Kampouri, S.; Diamantopoulos, N.; Floros, T.; Iosifidou, R.; Katopodi, O. Analysis of hereditary cancer syndromes by using a panel of genes: Novel and multiple pathogenic mutations. BMC Cancer 2019, 19, 535. [Google Scholar] [CrossRef] [PubMed]
- Ramírez-Calvo, M.; García-Casado, Z.; Fernández-Serra, A.; de Juan, I.; Palanca, S.; Oltra, S.; Soto, J.L.; Castillejo, A.; Barbera, V.M.; Juan-Fita, M.J. Implementation of massive sequencing in the genetic diagnosis of hereditary cancer syndromes: Diagnostic performance in the Hereditary Cancer Programme of the Valencia Community (FamCan-NGS). Hered. Cancer Clin. Pract. 2019, 17, 1–7. [Google Scholar] [CrossRef]
- Nagy, R.; Sweet, K.; Eng, C. Highly penetrant hereditary cancer syndromes. Oncogene 2004, 23, 6445–6470. [Google Scholar] [CrossRef] [PubMed]
- Bhai, P.; Levy, M.A.; Rooney, K.; Carere, D.A.; Reilly, J.; Kerkhof, J.; Volodarsky, M.; Stuart, A.; Kadour, M.; Panabaker, K. Page Title: Analysis of sequence and copy number variants in Canadian patient cohort with familial cancer syndromes using a unique next generation sequencing based approach. Front. Genet. 2021, 12, 1145. [Google Scholar] [CrossRef]
- Hampel, H.; Bennett, R.L.; Buchanan, A.; Pearlman, R.; Wiesner, G.L. A practice guideline from the American College of Medical Genetics and Genomics and the National Society of Genetic Counselors: Referral indications for cancer predisposition assessment. Genet. Med. 2015, 17, 70–87. [Google Scholar] [CrossRef] [PubMed]
- Angeli, D.; Salvi, S.; Tedaldi, G. Genetic predisposition to breast and ovarian cancers: How many and which genes to test? Int. J. Mol. Sci. 2020, 21, 1128. [Google Scholar] [CrossRef]
- Daly, M.B.; Pilarski, R.; Yurgelun, M.B.; Berry, M.P.; Buys, S.S.; Dickson, P.; Domchek, S.M.; Elkhanany, A.; Friedman, S.; Garber, J.E. NCCN guidelines insights: Genetic/familial high-risk assessment: Breast, ovarian, and pancreatic, version 1.2020: Featured updates to the NCCN guidelines. J. Natl. Compr. Cancer Netw. 2020, 18, 380–391. [Google Scholar] [CrossRef]
- AIOM Guidelines. Available online: https://www.aiom.it/raccomandazioni-per-limplementazione-del-test-brca-predittivo-e-preventivo-nei-tumori-della-mammella-dellovaio-del-pancreas-e-della-prostata/ (accessed on 22 June 2022).
- Goldhirsch, A.; Winer, E.P.; Coates, A.; Gelber, R.; Piccart-Gebhart, M.; Thürlimann, B.; Senn, H.-J.; Albain, K.S.; André, F.; Bergh, J. Personalizing the treatment of women with early breast cancer: Highlights of the St Gallen International Expert Consensus on the Primary Therapy of Early Breast Cancer 2013. Ann. Oncol. 2013, 24, 2206–2223. [Google Scholar] [CrossRef]
- BODICEA. Available online: https://www.canrisk.org/canrisk_tool (accessed on 22 June 2022).
- Paduano, F.; Fabiani, F.; Colao, E.; Trapasso, F.; Perrotti, N.; Barbieri, V.; Baudi, F.; Iuliano, R. Case Report: Identification of a Novel Pathogenic Germline TP53 Variant in a Family With Li-Fraumeni Syndrome. Front. Genet. 2021, 12, 734809. [Google Scholar] [CrossRef]
- Paduano, F.; Colao, E.; Grillone, T.; Vismara, M.F.M.; Amato, R.; Nisticò, S.; Mignogna, C.; Dastoli, S.; Fabiani, F.; Zucco, R. A Familial Form of Epidermolysis Bullosa Simplex Associated with a Pathogenic Variant in KRT5. Genes 2021, 12, 1503. [Google Scholar] [CrossRef]
- Paduano, F.; Colao, E.; Loddo, S.; Orlando, V.; Trapasso, F.; Novelli, A.; Perrotti, N.; Iuliano, R. 7q35 Microdeletion and 15q13.3 and Xp22.33 Microduplications in a Patient with Severe Myoclonic Epilepsy, Microcephaly, Dysmorphisms, Severe Psychomotor Delay and Intellectual Disability. Genes 2020, 11, 525. [Google Scholar] [CrossRef] [PubMed]
- Clinvar. Available online: http://clinvar.com/ (accessed on 22 June 2022).
- LOVD. Available online: https://www.lovd.nl/ (accessed on 22 June 2022).
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–423. [Google Scholar] [CrossRef]
- Nykamp, K.; Anderson, M.; Powers, M.; Garcia, J.; Herrera, B.; Ho, Y.-Y.; Kobayashi, Y.; Patil, N.; Thusberg, J.; Westbrook, M. Sherloc: A comprehensive refinement of the ACMG–AMP variant classification criteria. Genet. Med. 2017, 19, 1105–1117. [Google Scholar] [CrossRef] [PubMed]
- Hung, F.-H.; Wang, Y.A.; Jian, J.-W.; Peng, H.-P.; Hsieh, L.-L.; Hung, C.-F.; Yang, M.M.; Yang, A.-S. Evaluating BRCA mutation risk predictive models in a Chinese cohort in Taiwan. Sci. Rep. 2019, 9, 1–10. [Google Scholar] [CrossRef]
- Lee, A.; Mavaddat, N.; Cunningham, A.P.; Carver, T.; Archer, S.; Walter, F.M.; Tischkowitz, M.; Roberts, J.; Usher-Smith, J.; Simard, J. Enhancing the BOADICEA cancer risk prediction model to incorporate new data on RAD51C, RAD51D, BARD1, updates to tumour pathology and cancer incidences. medRxiv 2022. [Google Scholar] [CrossRef]
- Findlay, G.M.; Daza, R.M.; Martin, B.; Zhang, M.D.; Leith, A.P.; Gasperini, M.; Janizek, J.D.; Huang, X.; Starita, L.M.; Shendure, J. Accurate classification of BRCA1 variants with saturation genome editing. Nature 2018, 562, 217–222. [Google Scholar] [CrossRef]
- Yang, Z.; Ouyang, T.; Li, J.; Wang, T.; Fan, Z.; Fan, T.; Lin, B.; Zhang, J.; Xie, Y. Prevalence and characterization of ATM germline mutations in Chinese BRCA1/2-negative breast cancer patients. Breast Cancer Res. Treat. 2019, 174, 639–647. [Google Scholar] [CrossRef]
- Perkins, B.A.; Caskey, C.T.; Brar, P.; Dec, E.; Karow, D.S.; Kahn, A.M.; Hou, Y.C.; Shah, N.; Boeldt, D.; Coughlin, E.; et al. Precision medicine screening using whole-genome sequencing and advanced imaging to identify disease risk in adults. Proc. Natl. Acad. Sci. USA 2018, 115, 3686–3691. [Google Scholar] [CrossRef]
- Feliubadaló, L.; Moles-Fernández, A.; Santamariña-Pena, M.; Sánchez, A.T.; López-Novo, A.; Porras, L.M.; Blanco, A.; Capellá, G.; de la Hoya, M.; Molina, I.J.; et al. A Collaborative Effort to Define Classification Criteria for ATM Variants in Hereditary Cancer Patients. Clin. Chem. 2021, 67, 518–533. [Google Scholar] [CrossRef]
- Milanovic, M.; Houghton, L.M.; Menolfi, D.; Lee, J.H.; Yamamoto, K.; Li, Y.; Lee, B.J.; Xu, J.; Estes, V.M.; Wang, D.; et al. The Cancer-Associated ATM R3008H Mutation Reveals the Link between ATM Activation and Its Exchange. Cancer Res. 2021, 81, 426–437. [Google Scholar] [CrossRef]
- Ida, K.; Miyamoto, T.; Higuchi, S.; Takeuchi, H.; Yamada, S.; Ono, M.; Nishihara, H.; Shiozawa, T. Effectiveness of a genetic test panel designed for gynecological cancer: An exploratory study. Med. Oncol. 2019, 36, 62. [Google Scholar] [CrossRef] [PubMed]
- Gong, R.; He, Y.; Liu, X.Y.; Wang, H.Y.; Sun, L.Y.; Yang, X.H.; Li, B.; Cao, X.K.; Ye, Z.L.; Kong, L.H.; et al. Mutation spectrum of germline cancer susceptibility genes among unselected Chinese colorectal cancer patients. Cancer Manag. Res. 2019, 11, 3721–3739. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, S.M.; De Simone, L.M.; Olopade, O.I. Cancer Susceptibility Genetic Testing in a High-Risk Cohort of Urban Ashkenazi Jewish Individuals. J. Genet. Couns. 2018, 27, 1405–1410. [Google Scholar] [CrossRef] [PubMed]
- Bernstein-Molho, R.; Singer, A.; Laitman, Y.; Netzer, I.; Zalmanoviz, S.; Friedman, E. Multigene panel testing in unselected Israeli breast cancer cases: Mutational spectrum and use of BRCA1/2 mutation prediction algorithms. Breast Cancer Res. Treat. 2019, 176, 165–170. [Google Scholar] [CrossRef] [PubMed]
- Bąk, A.; Janiszewska, H.; Junkiert-Czarnecka, A.; Heise, M.; Pilarska-Deltow, M.; Laskowski, R.; Pasińska, M.; Haus, O. A risk of breast cancer in women-carriers of constitutional CHEK2 gene mutations, originating from the North-Central Poland. Hered. Cancer Clin. Pract. 2014, 12, 10. [Google Scholar] [CrossRef]
- Baralle, D.; Baralle, M. Splicing in action: Assessing disease causing sequence changes. J. Med. Genet. 2005, 42, 737–748. [Google Scholar] [CrossRef]
- Chen, X.; Truong, T.T.; Weaver, J.; Bove, B.A.; Cattie, K.; Armstrong, B.A.; Daly, M.B.; Godwin, A.K. Intronic alterations in BRCA1 and BRCA2: Effect on mRNA splicing fidelity and expression. Hum. Mutat. 2006, 27, 427–435. [Google Scholar] [CrossRef]
- Tedaldi, G.; Tebaldi, M.; Zampiga, V.; Danesi, R.; Arcangeli, V.; Ravegnani, M.; Cangini, I.; Pirini, F.; Petracci, E.; Rocca, A.; et al. Multiple-gene panel analysis in a case series of 255 women with hereditary breast and ovarian cancer. Oncotarget 2017, 8, 47064–47075. [Google Scholar] [CrossRef]
- Lang, G.T.; Shi, J.X.; Hu, X.; Zhang, C.H.; Shan, L.; Song, C.G.; Zhuang, Z.G.; Cao, A.Y.; Ling, H.; Yu, K.D.; et al. The spectrum of BRCA mutations and characteristics of BRCA-associated breast cancers in China: Screening of 2,991 patients and 1,043 controls by next-generation sequencing. Int. J. Cancer 2017, 141, 129–142. [Google Scholar] [CrossRef]
- Machackova, E.; Foretova, L.; Lukesova, M.; Vasickova, P.; Navratilova, M.; Coene, I.; Pavlu, H.; Kosinova, V.; Kuklova, J.; Claes, K. Spectrum and characterisation of BRCA1 and BRCA2 deleterious mutations in high-risk Czech patients with breast and/or ovarian cancer. BMC Cancer 2008, 8, 140. [Google Scholar] [CrossRef]
- Coppa, A.; Buffone, A.; Capalbo, C.; Nicolussi, A.; D’Inzeo, S.; Belardinilli, F.; Colicchia, V.; Petroni, M.; Granato, T.; Midulla, C.; et al. Novel and recurrent BRCA2 mutations in Italian breast/ovarian cancer families widen the ovarian cancer cluster region boundaries to exons 13 and 14. Breast Cancer Res. Treat. 2014, 148, 629–635. [Google Scholar] [CrossRef] [PubMed]
- Parsons, M.T.; Tudini, E.; Li, H.; Hahnen, E.; Wappenschmidt, B.; Feliubadaló, L.; Aalfs, C.M.; Agata, S.; Aittomäki, K.; Alducci, E.; et al. Large scale multifactorial likelihood quantitative analysis of BRCA1 and BRCA2 variants: An ENIGMA resource to support clinical variant classification. Hum. Mutat. 2019, 40, 1557–1578. [Google Scholar] [CrossRef] [PubMed]
- Susswein, L.R.; Marshall, M.L.; Nusbaum, R.; Vogel Postula, K.J.; Weissman, S.M.; Yackowski, L.; Vaccari, E.M.; Bissonnette, J.; Booker, J.K.; Cremona, M.L.; et al. Pathogenic and likely pathogenic variant prevalence among the first 10,000 patients referred for next-generation cancer panel testing. Genet. Med. Off. J. Am. Coll. Med. Genet. 2016, 18, 823–832. [Google Scholar] [CrossRef] [PubMed]
- Borg, A.; Haile, R.W.; Malone, K.E.; Capanu, M.; Diep, A.; Törngren, T.; Teraoka, S.; Begg, C.B.; Thomas, D.C.; Concannon, P.; et al. Characterization of BRCA1 and BRCA2 deleterious mutations and variants of unknown clinical significance in unilateral and bilateral breast cancer: The WECARE study. Hum. Mutat. 2010, 31, E1200–E1240. [Google Scholar] [CrossRef]
- De Brakeleer, S.; Bogdani, M.; De Grève, J.; Decock, J.; Sermijn, E.; Bonduelle, M.; Goelen, G.; Teugels, E. Loss of nuclear BRCA1 protein staining in normal tissue cells derived from BRCA1 and BRCA2 mutation carriers. Mutat. Res. 2007, 619, 104–112. [Google Scholar] [CrossRef]
- Kaneyasu, T.; Mori, S.; Yamauchi, H.; Ohsumi, S.; Ohno, S.; Aoki, D.; Baba, S.; Kawano, J.; Miki, Y.; Matsumoto, N.; et al. Prevalence of disease-causing genes in Japanese patients with BRCA1/2-wildtype hereditary breast and ovarian cancer syndrome. NPJ Breast Cancer 2020, 6, 25. [Google Scholar] [CrossRef]
- Decker, B.; Allen, J.; Luccarini, C.; Pooley, K.A.; Shah, M.; Bolla, M.K.; Wang, Q.; Ahmed, S.; Baynes, C.; Conroy, D.M.; et al. Rare, protein-truncating variants in ATM, CHEK2 and PALB2, but not XRCC2, are associated with increased breast cancer risks. J. Med. Genet. 2017, 54, 732–741. [Google Scholar] [CrossRef]
- Yadav, S.; Reeves, A.; Campian, S.; Paine, A.; Zakalik, D. Outcomes of retesting BRCA negative patients using multigene panels. Fam. Cancer 2017, 16, 319–328. [Google Scholar] [CrossRef]
- Liu, X.; Li, H.; Shao, B.; Wu, J.; Kong, W.; Song, G.; Jiang, H.; Wang, J.; Wan, F. Identification of recurrent BRCA1 mutation and its clinical relevance in Chinese Triple-negative breast cancer cohort. Cancer Med. 2017, 6, 547–554. [Google Scholar] [CrossRef]
- Giacomelli, A.O.; Yang, X.; Lintner, R.E.; McFarland, J.M.; Duby, M.; Kim, J.; Howard, T.P.; Takeda, D.Y.; Ly, S.H.; Kim, E.; et al. Mutational processes shape the landscape of TP53 mutations in human cancer. Nat. Genet. 2018, 50, 1381–1387. [Google Scholar] [CrossRef]
- Kotler, E.; Shani, O.; Goldfeld, G.; Lotan-Pompan, M.; Tarcic, O.; Gershoni, A.; Hopf, T.A.; Marks, D.S.; Oren, M.; Segal, E. A Systematic p53 Mutation Library Links Differential Functional Impact to Cancer Mutation Pattern and Evolutionary Conservation. Mol. Cell 2018, 71, 178–190.e178. [Google Scholar] [CrossRef] [PubMed]
- Lagerstedt-Robinson, K.; Rohlin, A.; Aravidis, C.; Melin, B.; Nordling, M.; Stenmark-Askmalm, M.; Lindblom, A.; Nilbert, M. Mismatch repair gene mutation spectrum in the Swedish Lynch syndrome population. Oncol. Rep. 2016, 36, 2823–2835. [Google Scholar] [CrossRef]
- Wang, Y.; Wu, J.; Li, W.; Li, J.; Liu, R.; Yang, B.; Li, C.; Jiang, T. Retrospective investigation of hereditary syndromes in patients with medulloblastoma in a single institution. Child’s Nerv. Syst. ChNS Off. J. Int. Soc. Pediatric Neurosurg. 2021, 37, 411–417. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Zhang, J.; Ouyang, T.; Li, J.; Wang, T.; Fan, Z.; Fan, T.; Lin, B.; Xie, Y. Incidence of BRCA1 somatic mutations and response to neoadjuvant chemotherapy in Chinese women with triple-negative breast cancer. Gene 2016, 584, 26–30. [Google Scholar] [CrossRef] [PubMed]
- HGVS. Available online: http://hgvs.org (accessed on 22 June 2022).
- Biswas, K.; Das, R.; Alter, B.P.; Kuznetsov, S.G.; Stauffer, S.; North, S.L.; Burkett, S.; Brody, L.C.; Meyer, S.; Byrd, R.A.; et al. A comprehensive functional characterization of BRCA2 variants associated with Fanconi anemia using mouse ES cell-based assay. Blood 2011, 118, 2430–2442. [Google Scholar] [CrossRef] [PubMed]
- Tiao, G.; Improgo, M.R.; Kasar, S.; Poh, W.; Kamburov, A.; Landau, D.A.; Tausch, E.; Taylor-Weiner, A.; Cibulskis, C.; Bahl, S.; et al. Rare germline variants in ATM are associated with chronic lymphocytic leukemia. Leukemia 2017, 31, 2244–2247. [Google Scholar] [CrossRef]
- Yurgelun, M.B.; Kulke, M.H.; Fuchs, C.S.; Allen, B.A.; Uno, H.; Hornick, J.L.; Ukaegbu, C.I.; Brais, L.K.; McNamara, P.G.; Mayer, R.J.; et al. Cancer Susceptibility Gene Mutations in Individuals With Colorectal Cancer. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2017, 35, 1086–1095. [Google Scholar] [CrossRef]
- Bandeira, G.; Rocha, K.; Lazar, M.; Ezquina, S.; Yamamoto, G.; Varela, M.; Takahashi, V.; Aguena, M.; Gollop, T.; Zatz, M.; et al. Germline variants of Brazilian women with breast cancer and detection of a novel pathogenic ATM deletion in early-onset breast cancer. Breast Cancer 2021, 28, 346–354. [Google Scholar] [CrossRef]
- Akcay, I.M.; Celik, E.; Agaoglu, N.B.; Alkurt, G.; Kizilboga Akgun, T.; Yildiz, J.; Enc, F.; Kir, G.; Canbek, S.; Kilic, A.; et al. Germline pathogenic variant spectrum in 25 cancer susceptibility genes in Turkish breast and colorectal cancer patients and elderly controls. Int. J. Cancer 2021, 148, 285–295. [Google Scholar] [CrossRef]
- Kelsen, J.R.; Dawany, N.; Moran, C.J.; Petersen, B.S.; Sarmady, M.; Sasson, A.; Pauly-Hubbard, H.; Martinez, A.; Maurer, K.; Soong, J.; et al. Exome sequencing analysis reveals variants in primary immunodeficiency genes in patients with very early onset inflammatory bowel disease. Gastroenterology 2015, 149, 1415–1424. [Google Scholar] [CrossRef]
- Hauke, J.; Horvath, J.; Groß, E.; Gehrig, A.; Honisch, E.; Hackmann, K.; Schmidt, G.; Arnold, N.; Faust, U.; Sutter, C.; et al. Gene panel testing of 5589 BRCA1/2-negative index patients with breast cancer in a routine diagnostic setting: Results of the German Consortium for Hereditary Breast and Ovarian Cancer. Cancer Med. 2018, 7, 1349–1358. [Google Scholar] [CrossRef]
- Shindo, K.; Yu, J.; Suenaga, M.; Fesharakizadeh, S.; Cho, C.; Macgregor-Das, A.; Siddiqui, A.; Witmer, P.D.; Tamura, K.; Song, T.J.; et al. Deleterious Germline Mutations in Patients With Apparently Sporadic Pancreatic Adenocarcinoma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2017, 35, 3382–3390. [Google Scholar] [CrossRef] [PubMed]
- Kraus, C.; Hoyer, J.; Vasileiou, G.; Wunderle, M.; Lux, M.P.; Fasching, P.A.; Krumbiegel, M.; Uebe, S.; Reuter, M.; Beckmann, M.W.; et al. Gene panel sequencing in familial breast/ovarian cancer patients identifies multiple novel mutations also in genes others than BRCA1/2. Int. J. Cancer 2017, 140, 95–102. [Google Scholar] [CrossRef]
- Akbari, M.R.; Malekzadeh, R.; Lepage, P.; Roquis, D.; Sadjadi, A.R.; Aghcheli, K.; Yazdanbod, A.; Shakeri, R.; Bashiri, J.; Sotoudeh, M.; et al. Mutations in Fanconi anemia genes and the risk of esophageal cancer. Hum. Genet. 2011, 129, 573–582. [Google Scholar] [CrossRef] [PubMed]
- Delimitsou, A.; Fostira, F.; Kalfakakou, D.; Apostolou, P.; Konstantopoulou, I.; Kroupis, C.; Papavassiliou, A.G.; Kleibl, Z.; Stratikos, E.; Voutsinas, G.E.; et al. Functional characterization of CHEK2 variants in a Saccharomyces cerevisiae system. Hum. Mutat. 2019, 40, 631–648. [Google Scholar] [CrossRef] [PubMed]
- Ciceri, S.; Gamba, B.; Corbetta, P.; Mondini, P.; Terenziani, M.; Catania, S.; Nantron, M.; Bianchi, M.; D’Angelo, P.; Torri, F.; et al. Genetic and epigenetic analyses guided by high resolution whole-genome SNP array reveals a possible role of CHEK2 in Wilms tumour susceptibility. Oncotarget 2018, 9, 34079–34089. [Google Scholar] [CrossRef]
- Isaacsson Velho, P.; Silberstein, J.L.; Markowski, M.C.; Luo, J.; Lotan, T.L.; Isaacs, W.B.; Antonarakis, E.S. Intraductal/ductal histology and lymphovascular invasion are associated with germline DNA-repair gene mutations in prostate cancer. Prostate 2018, 78, 401–407. [Google Scholar] [CrossRef]
- Young, E.L.; Feng, B.J.; Stark, A.W.; Damiola, F.; Durand, G.; Forey, N.; Francy, T.C.; Gammon, A.; Kohlmann, W.K.; Kaphingst, K.A.; et al. Multigene testing of moderate-risk genes: Be mindful of the missense. J. Med. Genet. 2016, 53, 366–376. [Google Scholar] [CrossRef]
- Le Calvez-Kelm, F.; Lesueur, F.; Damiola, F.; Vallée, M.; Voegele, C.; Babikyan, D.; Durand, G.; Forey, N.; McKay-Chopin, S.; Robinot, N.; et al. Rare, evolutionarily unlikely missense substitutions in CHEK2 contribute to breast cancer susceptibility: Results from a breast cancer family registry case-control mutation-screening study. Breast Cancer Res. BCR 2011, 13, R6. [Google Scholar] [CrossRef]
- Liccardo, R.; De Rosa, M.; Rossi, G.B.; Carlomagno, N.; Izzo, P.; Duraturo, F. Incomplete Segregation of MSH6 Frameshift Variants with Phenotype of Lynch Syndrome. Int. J. Mol. Sci. 2017, 18, 999. [Google Scholar] [CrossRef]
- Baloch, A.H.; Daud, S.; Raheem, N.; Luqman, M.; Ahmad, A.; Rehman, A.; Shuja, J.; Rasheed, S.; Ali, A.; Kakar, N.; et al. Missense mutations (p.H371Y, p.D438Y) in gene CHEK2 are associated with breast cancer risk in women of Balochistan origin. Mol. Biol. Rep. 2014, 41, 1103–1107. [Google Scholar] [CrossRef] [PubMed]
- Tischkowitz, M.D.; Yilmaz, A.; Chen, L.Q.; Karyadi, D.M.; Novak, D.; Kirchhoff, T.; Hamel, N.; Tavtigian, S.V.; Kolb, S.; Bismar, T.A.; et al. Identification and characterization of novel SNPs in CHEK2 in Ashkenazi Jewish men with prostate cancer. Cancer Lett. 2008, 270, 173–180. [Google Scholar] [CrossRef] [PubMed]
- Moradian, M.M.; Babikyan, D.T.; Markarian, S.; Petrosyan, J.G.; Avanesian, N.; Arutunyan, T.; Sarkisian, T.F. Germline mutational spectrum in Armenian breast cancer patients suspected of hereditary breast and ovarian cancer. Hum. Genome Var. 2021, 8, 9. [Google Scholar] [CrossRef] [PubMed]
- Dominguez-Valentin, M.; Nakken, S.; Tubeuf, H.; Vodak, D.; Ekstrøm, P.O.; Nissen, A.M.; Morak, M.; Holinski-Feder, E.; Holth, A.; Capella, G.; et al. Results of multigene panel testing in familial cancer cases without genetic cause demonstrated by single gene testing. Sci. Rep. 2019, 9, 18555. [Google Scholar] [CrossRef] [PubMed]
- Scarpitta, R.; Zanna, I.; Aretini, P.; Gambino, G.; Scatena, C.; Mei, B.; Ghilli, M.; Rossetti, E.; Roncella, M.; Congregati, C.; et al. Germline investigation in male breast cancer of DNA repair genes by next-generation sequencing. Breast Cancer Res. Treat. 2019, 178, 557–564. [Google Scholar] [CrossRef]
- Ansari, N.; Shahrabi, S.; Khosravi, A.; Shirzad, R.; Rezaeean, H. Prognostic Significance of CHEK2 Mutation in Progression of Breast Cancer. Lab. Med. 2019, 50, e36–e41. [Google Scholar] [CrossRef]
- Grandval, P.; Fabre, A.J.; Gaildrat, P.; Baert-Desurmont, S.; Buisine, M.P.; Ferrari, A.; Wang, Q.; Béroud, C.; Olschwang, S. UMD-MLH1/MSH2/MSH6 databases: Description and analysis of genetic variations in French Lynch syndrome families. Database J. Biol. Databases Curation 2013, 2013, bat036. [Google Scholar] [CrossRef]
- Simbolo, M.; Mafficini, A.; Agostini, M.; Pedrazzani, C.; Bedin, C.; Urso, E.D.; Nitti, D.; Turri, G.; Scardoni, M.; Fassan, M.; et al. Next-generation sequencing for genetic testing of familial colorectal cancer syndromes. Hered. Cancer Clin. Pract. 2015, 13, 18. [Google Scholar] [CrossRef][Green Version]
- Baudi, F.; Quaresima, B.; Grandinetti, C.; Cuda, G.; Faniello, C.; Tassone, P.; Barbieri, V.; Bisegna, R.; Ricevuto, E.; Conforti, S.; et al. Evidence of a founder mutation of BRCA1 in a highly homogeneous population from southern Italy with breast/ovarian cancer. Hum. Mutat. 2001, 18, 163–164. [Google Scholar] [CrossRef]
- Tung, N.; Lin, N.U.; Kidd, J.; Allen, B.A.; Singh, N.; Wenstrup, R.J.; Hartman, A.R.; Winer, E.P.; Garber, J.E. Frequency of Germline Mutations in 25 Cancer Susceptibility Genes in a Sequential Series of Patients With Breast Cancer. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2016, 34, 1460–1468. [Google Scholar] [CrossRef]
- Lee, C.H.; Chung, J.H. The hCds1 (Chk2)-FHA domain is essential for a chain of phosphorylation events on hCds1 that is induced by ionizing radiation. J. Biol. Chem. 2001, 276, 30537–30541. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, J.K.; Lovly, C.M.; Piwnica-Worms, H. Regulation of the Chk2 protein kinase by oligomerization-mediated cis- and trans-phosphorylation. Mol. Cancer Res. MCR 2003, 1, 598–609. [Google Scholar] [PubMed]
- Yehia, L.; Ni, Y.; Sesock, K.; Niazi, F.; Fletcher, B.; Chen, H.J.L.; LaFramboise, T.; Eng, C. Unexpected cancer-predisposition gene variants in Cowden syndrome and Bannayan-Riley-Ruvalcaba syndrome patients without underlying germline PTEN mutations. PLoS Genet. 2018, 14, e1007352. [Google Scholar] [CrossRef] [PubMed]
- Bono, M.; Fanale, D.; Incorvaia, L.; Cancelliere, D.; Fiorino, A.; Calò, V.; Dimino, A.; Filorizzo, C.; Corsini, L.R.; Brando, C.; et al. Impact of deleterious variants in other genes beyond BRCA1/2 detected in breast/ovarian and pancreatic cancer patients by NGS-based multi-gene panel testing: Looking over the hedge. ESMO Open 2021, 6, 100235. [Google Scholar] [CrossRef] [PubMed]
- Pylkäs, K.; Tommiska, J.; Syrjäkoski, K.; Kere, J.; Gatei, M.; Waddell, N.; Allinen, M.; Karppinen, S.M.; Rapakko, K.; Kääriäinen, H.; et al. Evaluation of the role of Finnish ataxia-telangiectasia mutations in hereditary predisposition to breast cancer. Carcinogenesis 2007, 28, 1040–1045. [Google Scholar] [CrossRef]
- Tram, E.; Savas, S.; Ozcelik, H. Missense variants of uncertain significance (VUS) altering the phosphorylation patterns of BRCA1 and BRCA2. PLoS ONE 2013, 8, e62468. [Google Scholar] [CrossRef]
- Couch, F.J.; DeShano, M.L.; Blackwood, M.A.; Calzone, K.; Stopfer, J.; Campeau, L.; Ganguly, A.; Rebbeck, T.; Weber, B.L. BRCA1 mutations in women attending clinics that evaluate the risk of breast cancer. New Engl. J. Med. 1997, 336, 1409–1415. [Google Scholar] [CrossRef]
- Sermijn, E.; Goelen, G.; Teugels, E.; Kaufman, L.; Bonduelle, M.; Neyns, B.; Poppe, B.; De Paepe, A.; De Grève, J. The impact of proband mediated information dissemination in families with a BRCA1/2 gene mutation. J. Med. Genet. 2004, 41, e23. [Google Scholar] [CrossRef]
- Figlioli, G.; De Nicolo, A.; Catucci, I.; Manoukian, S.; Peissel, B.; Azzollini, J.; Beltrami, B.; Bonanni, B.; Calvello, M.; Bondavalli, D. Analysis of Italian BRCA1/2 pathogenic variants identifies a private spectrum in the population from the Bergamo province in Northern Italy. Cancers 2021, 13, 532. [Google Scholar] [CrossRef]
- Li, N.; Zethoven, M.; McInerny, S.; Healey, E.; DeSilva, D.; Devereux, L.; Scott, R.J.; James, P.A.; Campbell, I.G. Contribution of large genomic rearrangements in PALB2 to familial breast cancer: Implications for genetic testing. J. Med. Genet. 2022. [Google Scholar] [CrossRef]
- Li, J.Y.; Jing, R.; Wei, H.; Wang, M.; Xiaowei, Q.; Liu, H.; Jian, L.; Ou, J.H.; Jiang, W.H.; Tian, F.G. Germline mutations in 40 cancer susceptibility genes among C hinese patients with high hereditary risk breast cancer. Int. J. Cancer 2019, 144, 281–289. [Google Scholar] [CrossRef] [PubMed]
- Tommiska, J.; Jansen, L.; Kilpivaara, O.; Edvardsen, H.; Kristensen, V.; Tamminen, A.; Aittomäki, K.; Blomqvist, C.; Børresen-Dale, A.L.; Nevanlinna, H. ATM variants and cancer risk in breast cancer patients from Southern Finland. BMC Cancer 2006, 6, 209. [Google Scholar] [CrossRef] [PubMed]
- Goldgar, D.E.; Healey, S.; Dowty, J.G.; Da Silva, L.; Chen, X.; Spurdle, A.B.; Terry, M.B.; Daly, M.J.; Buys, S.M.; Southey, M.C.; et al. Rare variants in the ATM gene and risk of breast cancer. Breast Cancer Res. BCR 2011, 13, R73. [Google Scholar] [CrossRef] [PubMed]
- Kleiblova, P.; Stolarova, L.; Krizova, K.; Lhota, F.; Hojny, J.; Zemankova, P.; Havranek, O.; Vocka, M.; Cerna, M.; Lhotova, K. Identification of deleterious germline CHEK2 mutations and their association with breast and ovarian cancer. Int. J. Cancer 2019, 145, 1782–1797. [Google Scholar] [CrossRef]
- Terkelsen, T.; Christensen, L.-L.; Fenton, D.C.; Jensen, U.B.; Sunde, L.; Thomassen, M.; Skytte, A.-B. Population frequencies of pathogenic alleles of BRCA1 and BRCA2: Analysis of 173 Danish breast cancer pedigrees using the BOADICEA model. Fam. Cancer 2019, 18, 381–388. [Google Scholar] [CrossRef]
- Antoniou, A.C.; Hardy, R.; Walker, L.; Evans, D.G.; Shenton, A.; Eeles, R.; Shanley, S.; Pichert, G.; Izatt, L.; Rose, S. Predicting the likelihood of carrying a BRCA1 or BRCA2 mutation: Validation of BOADICEA, BRCAPRO, IBIS, Myriad and the Manchester scoring system using data from UK genetics clinics. J. Med. Genet. 2008, 45, 425–431. [Google Scholar] [CrossRef]
- O’leary, E.; Iacoboni, D.; Holle, J.; Michalski, S.T.; Esplin, E.D.; Yang, S.; Ouyang, K. Expanded gene panel use for women with breast cancer: Identification and intervention beyond breast cancer risk. Ann. Surg. Oncol. 2017, 24, 3060–3066. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paduano, F.; Colao, E.; Fabiani, F.; Rocca, V.; Dinatolo, F.; Dattola, A.; D’Antona, L.; Amato, R.; Trapasso, F.; Baudi, F.; et al. Germline Testing in a Cohort of Patients at High Risk of Hereditary Cancer Predisposition Syndromes: First Two-Year Results from South Italy. Genes 2022, 13, 1286. https://doi.org/10.3390/genes13071286
Paduano F, Colao E, Fabiani F, Rocca V, Dinatolo F, Dattola A, D’Antona L, Amato R, Trapasso F, Baudi F, et al. Germline Testing in a Cohort of Patients at High Risk of Hereditary Cancer Predisposition Syndromes: First Two-Year Results from South Italy. Genes. 2022; 13(7):1286. https://doi.org/10.3390/genes13071286
Chicago/Turabian StylePaduano, Francesco, Emma Colao, Fernanda Fabiani, Valentina Rocca, Francesca Dinatolo, Adele Dattola, Lucia D’Antona, Rosario Amato, Francesco Trapasso, Francesco Baudi, and et al. 2022. "Germline Testing in a Cohort of Patients at High Risk of Hereditary Cancer Predisposition Syndromes: First Two-Year Results from South Italy" Genes 13, no. 7: 1286. https://doi.org/10.3390/genes13071286
APA StylePaduano, F., Colao, E., Fabiani, F., Rocca, V., Dinatolo, F., Dattola, A., D’Antona, L., Amato, R., Trapasso, F., Baudi, F., Perrotti, N., & Iuliano, R. (2022). Germline Testing in a Cohort of Patients at High Risk of Hereditary Cancer Predisposition Syndromes: First Two-Year Results from South Italy. Genes, 13(7), 1286. https://doi.org/10.3390/genes13071286