
Whole Transcriptome Sequencing Reveals Drought Resistance-Related Genes in Upland Cotton

 ,
,
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Plant Materials and Samples
2.2. RNA Extraction and Illumina Sequencing
2.3. Comparison with Reference Genome Sequences
2.4. Bioinformatics Analysis
2.5. Analysis of Differential Gene Expression
2.6. Screening of Differentially Expressed Genes
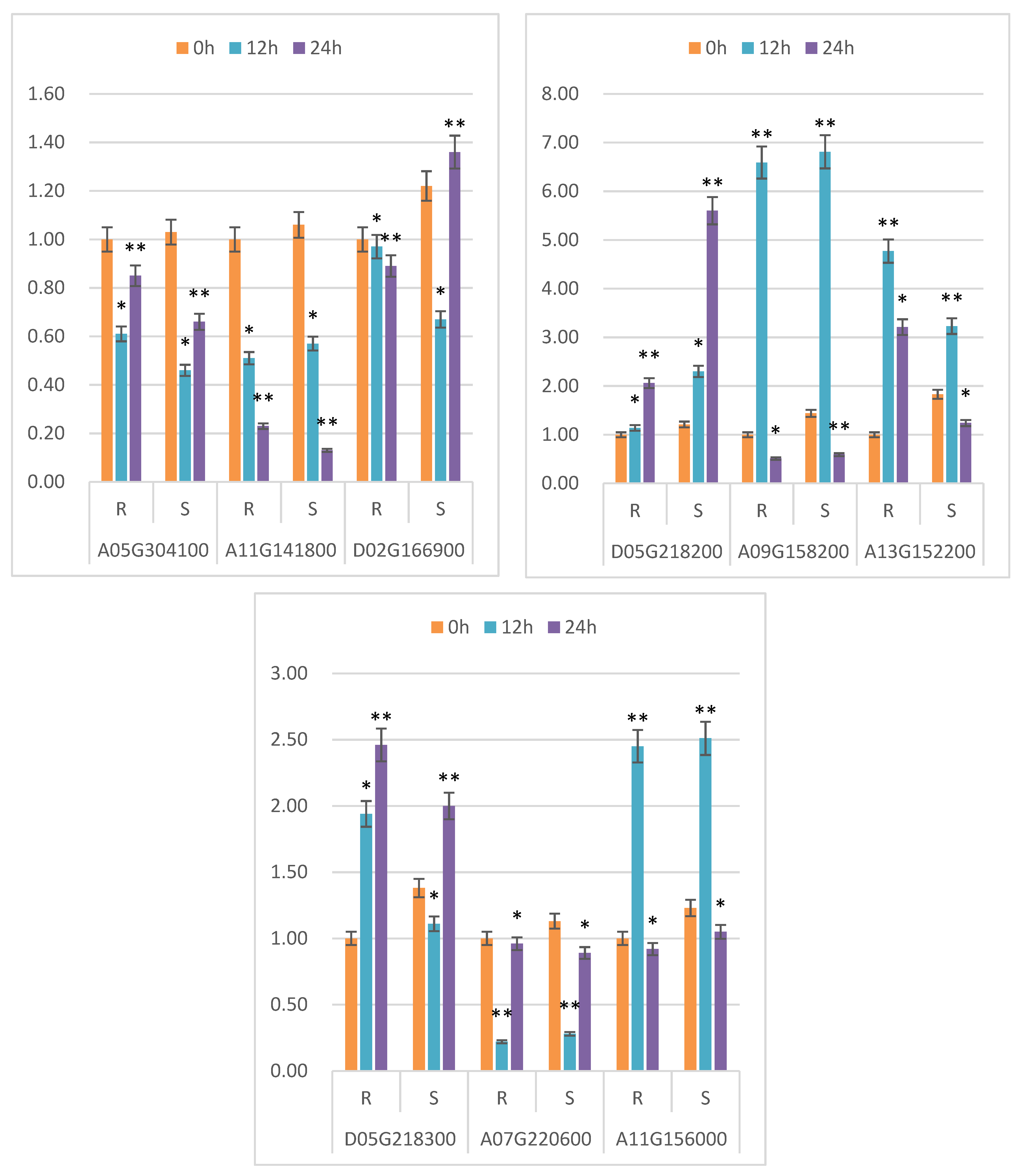
2.7. qRT-PCR Analysis
2.8. Statistical Analysis
3. Results
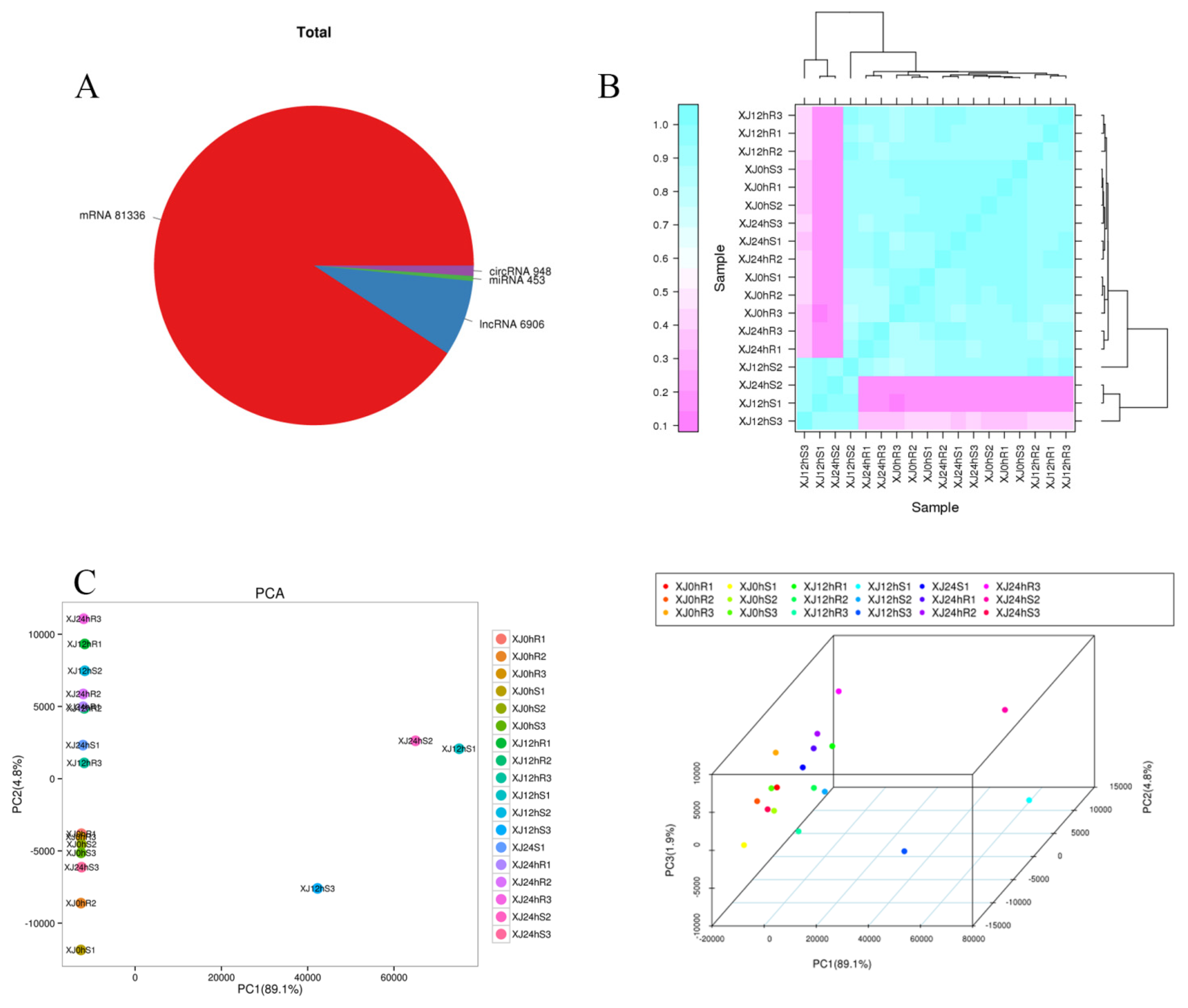
3.1. Quality Control Results for the Total RNA
3.2. Sequencing Results for the Cotton Transcriptome Were Analysed
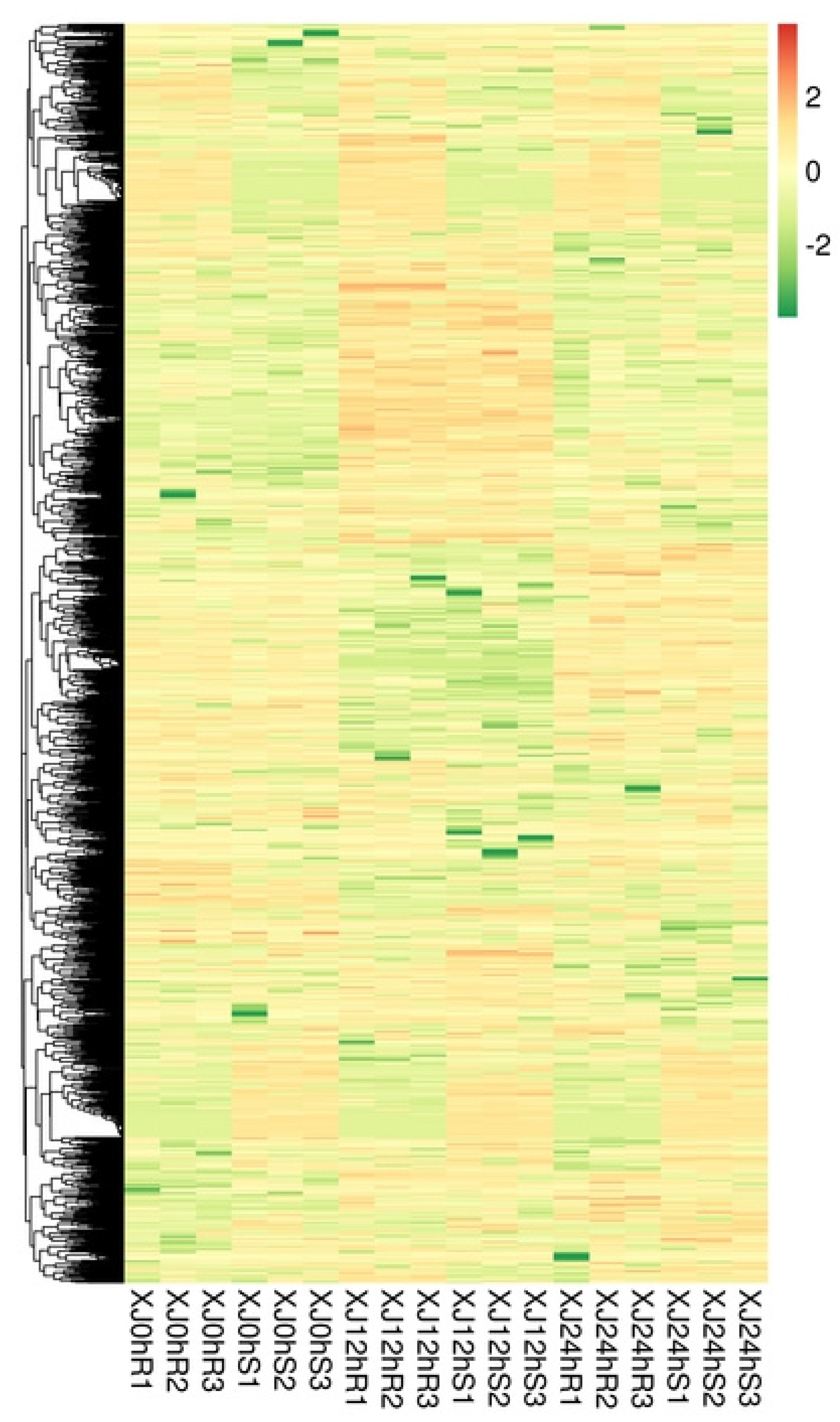
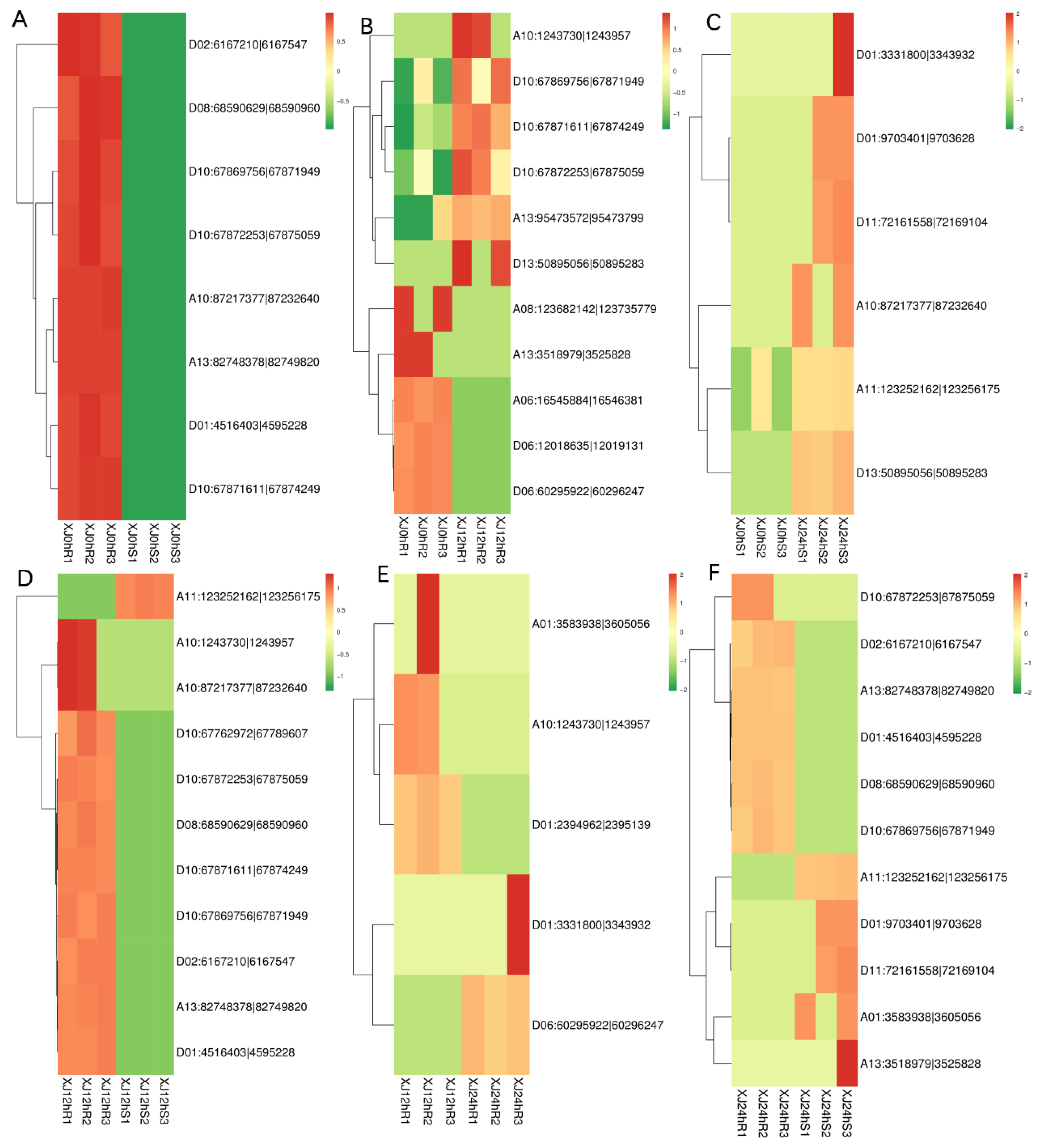
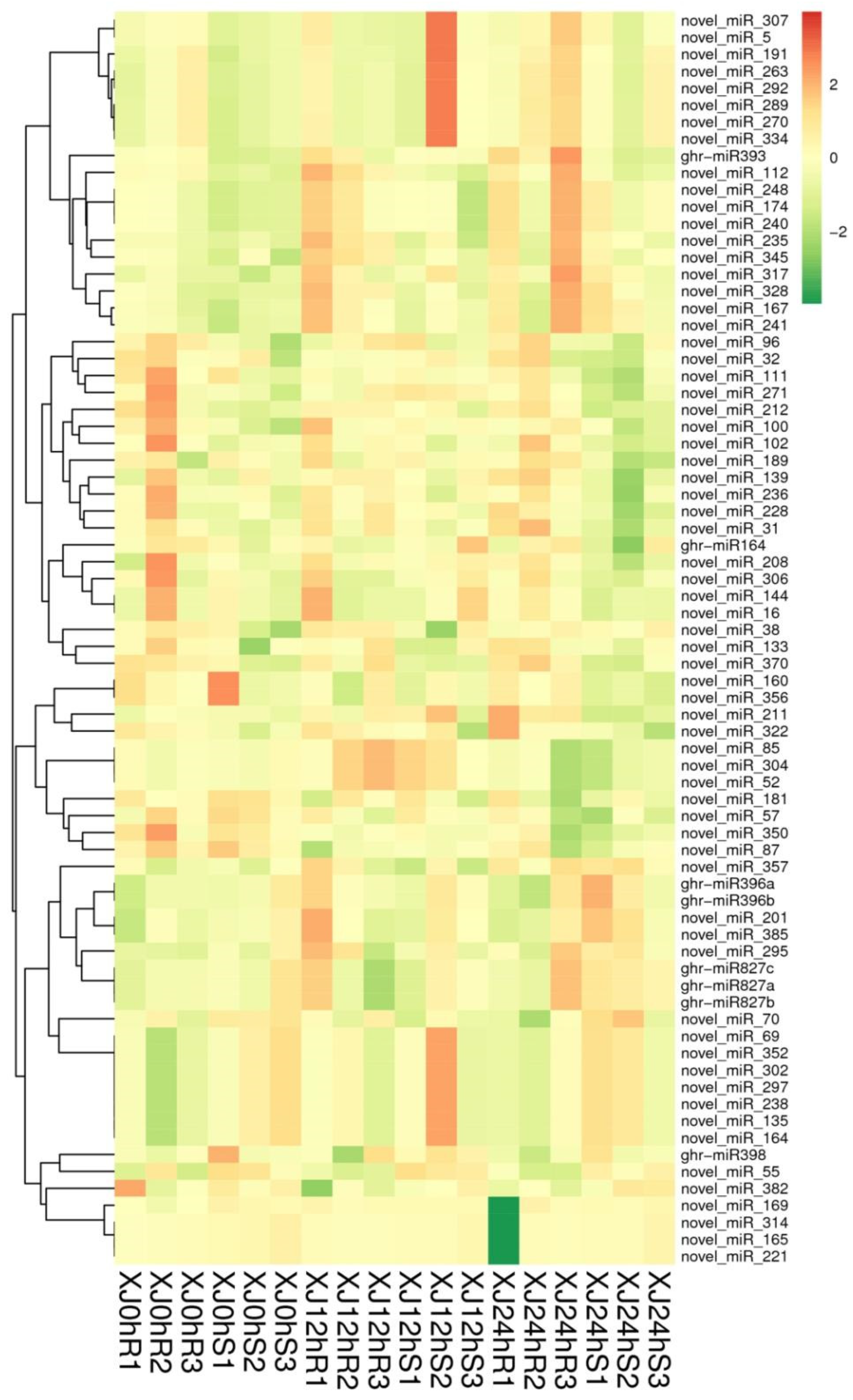
3.3. Analysis of Differential Gene Expression
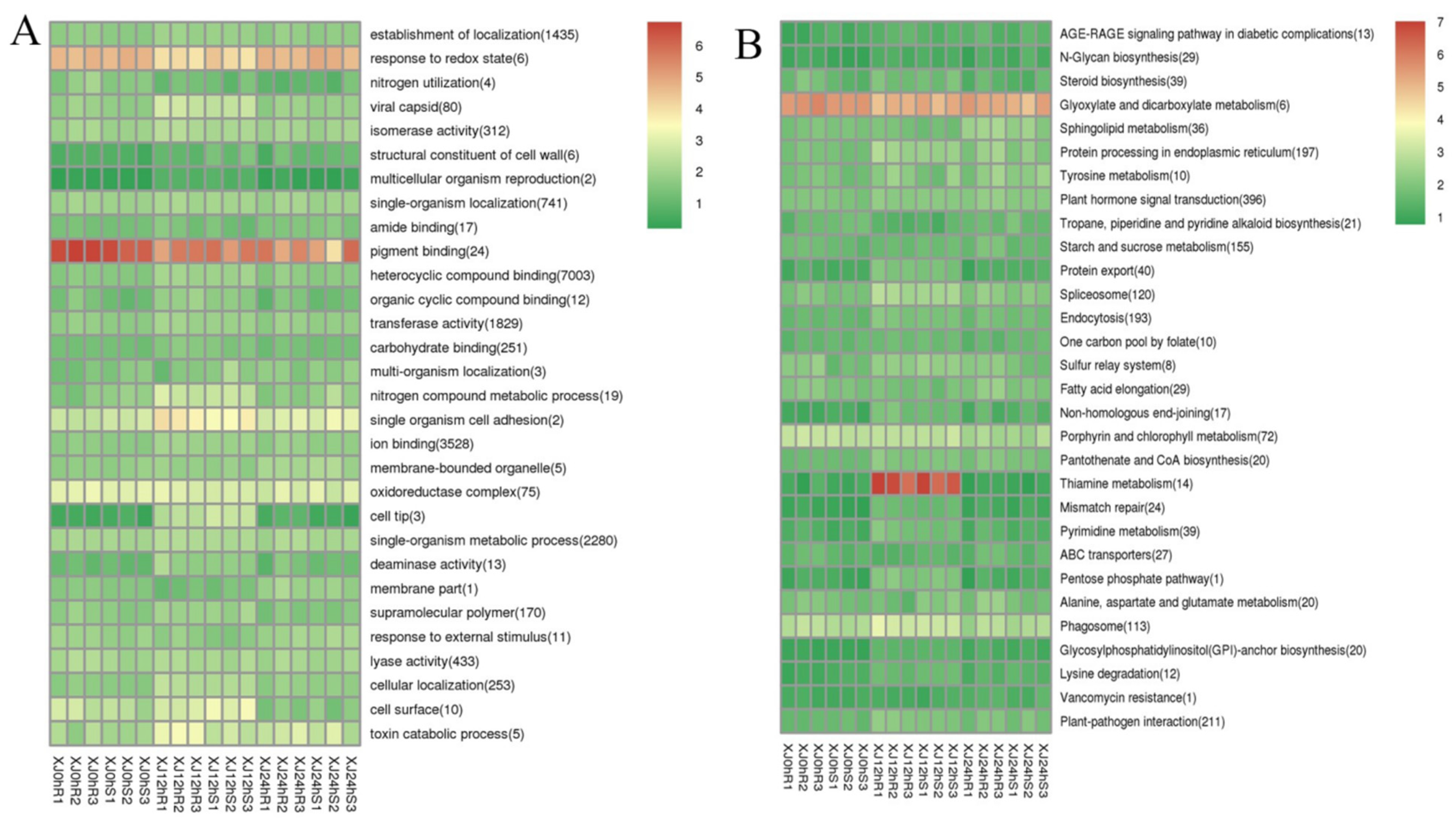
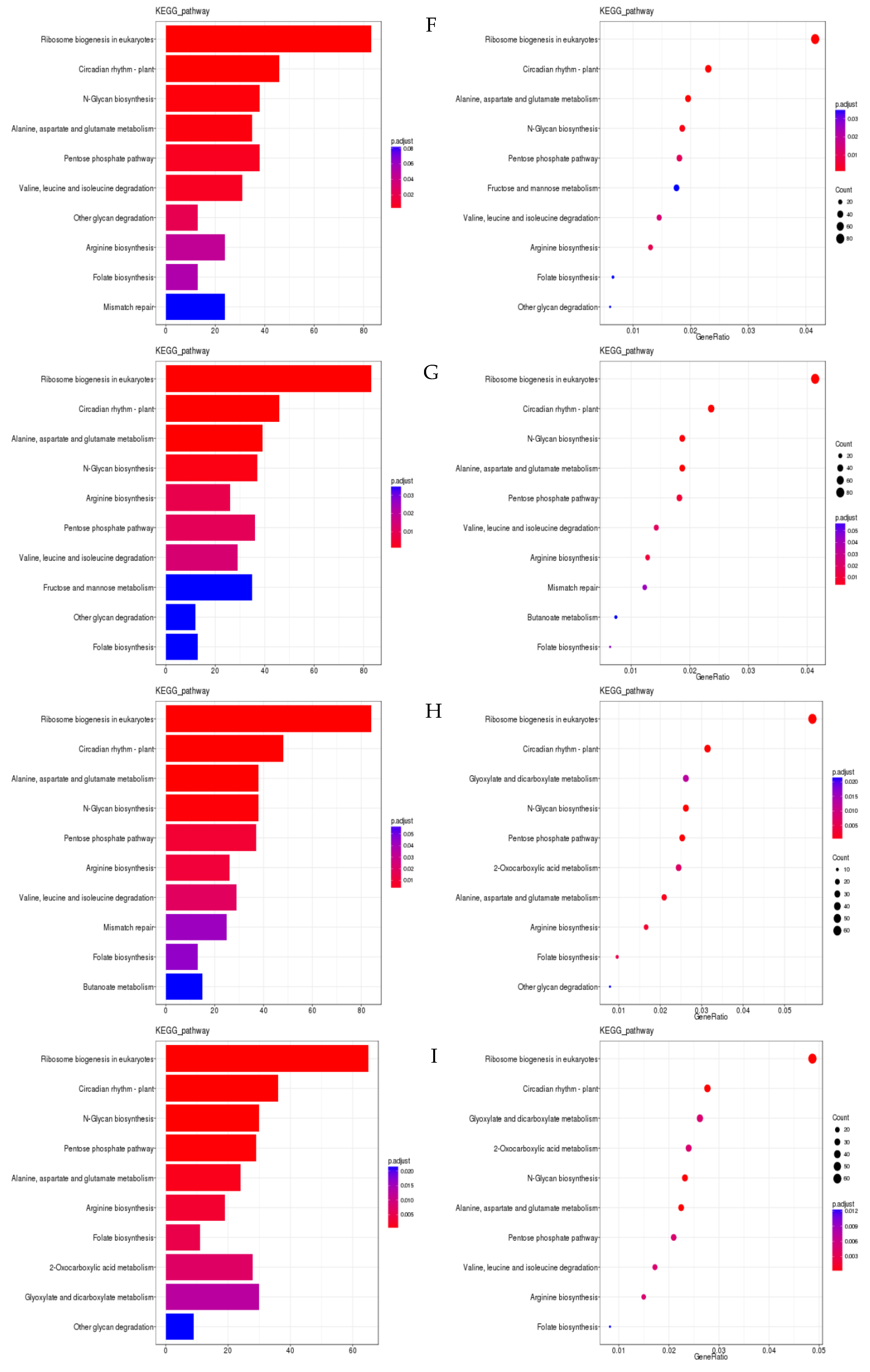
3.4. Analysis of Differentially Expressed Genes’ Functional Annotation and Enrichment
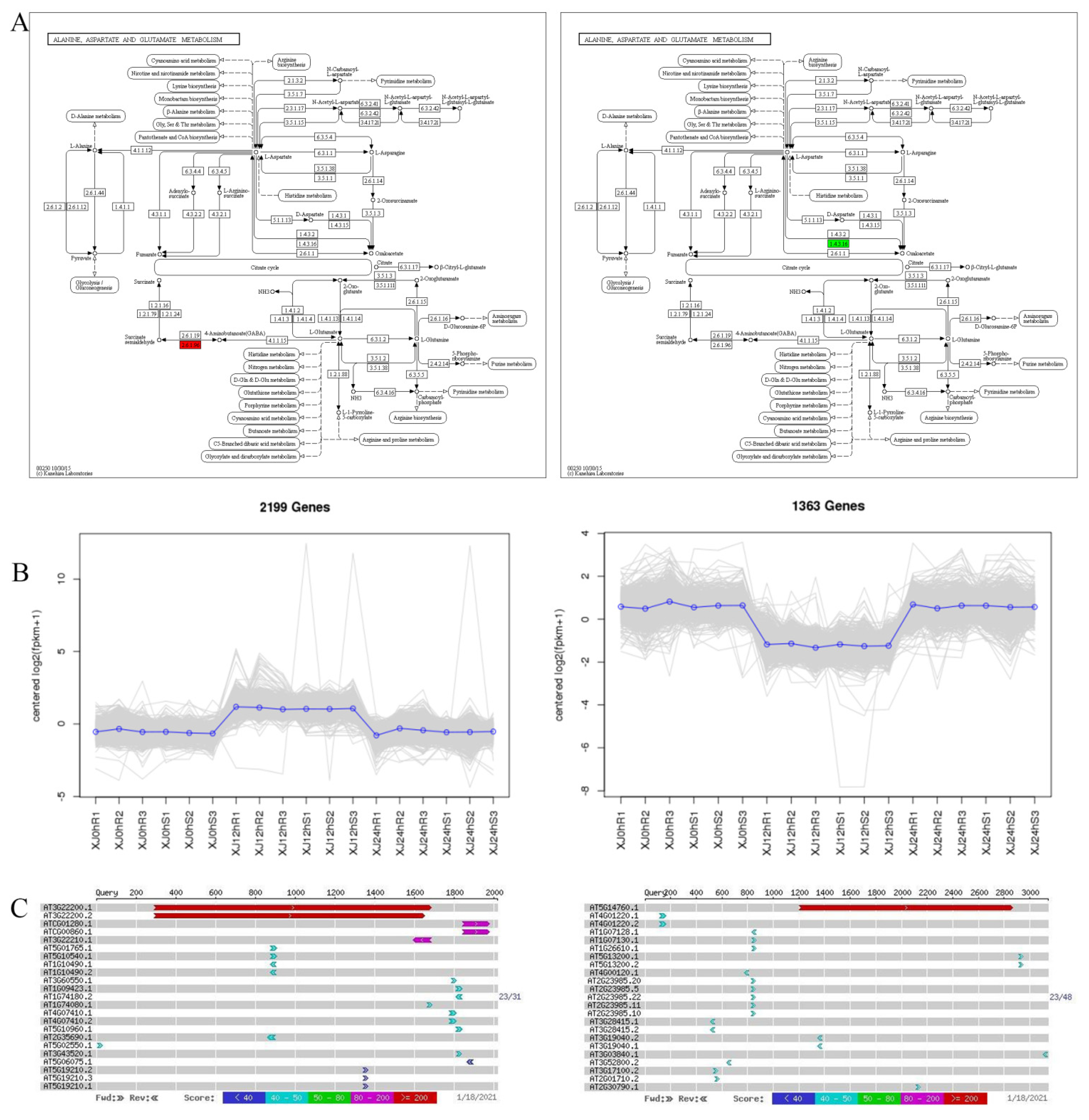
3.5. Construction of the ceRNA Regulatory Network and Integration Analysis of Key Pathways
3.6. Differential Gene Annotation and ceRNA Network Analysis Were Used to Screen Drought Resistance-Related Genes
4. Discussion
4.1. Rapid Screening of Drought Resistance-Related Genes by Differential Gene Annotation and ceRNA Regulatory Network Analysis
4.2. Study of GABA in Plant Stress Resistance
4.3. Mechanism of L-Aspartate Oxidase and NAD in Plant Stress Resistance
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| circRNAs | circular RNAs |
| lncRNAs | long non-coding RNAs |
| miRNAs | micro RNAs |
| mRNAs | messenger RNAs |
| ceRNAs | competing endogenous RNAs |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| DE | differentially expressed |
| GO | gene ontology |
| PCA | principal components analysis |
| GABA | γ-aminobutyric acid |
| NAD | nicotinamide adenine dinucleotide |
| PEG | polyethylene glycol |
| BP | biological process |
| CC | cell component |
| MF | molecular function |
| qRT-PCR | quantitative real-time PCR |
| TEM | transmission electron microscopy |
References
- Mehta, R.H.; Ponnuchamy, M.; Kumar, J. Exploring drought stress-regulated genes in senna (Cassiaangustifolia Vahl.): A transcriptomic approach. Funct. Integr. Genom. 2017, 17, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Do, P.T.; Degenkolbe, T.; Erban, A. Dissecting Rice Polyamine Metabolism under Controlled Long-Term Drought Stress. PLoS ONE 2013, 8, e60325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaves, M.M.; Maroco, J.P.; Pereira, J.S. Understanding Plant Responses to Drought-from Genes to the Whole Plant. Funct. Plant Biol. 2003, 30, 239. [Google Scholar] [CrossRef] [PubMed]
- Raney, J.A.; Reynolds, D.J.; Elzinga, D.B. Transcriptome analysis of drought induced stress in Chenopodium quinoa. Am. J. Plant Sci. 2014, 5, 338–357. [Google Scholar] [CrossRef]
- Bowman, M.J.; Wonkeun, P.; Bauer, P.J. RNA-Seq transcriptome profiling of upland cotton (G. hirsutum L) Root Tissue under water-deficit stress. PLoS ONE 2013, 8, e82634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thumma, B.R.; Sharma, N.; Southerton, S.G. Transcriptome sequencing of Eucalyptus camaldulensis seedlings subjected to water stressreveals functional single nucleotide polymorphisms and genes underselection. BMC Genom. 2012, 13, 364. [Google Scholar] [CrossRef] [Green Version]
- Luo, Z.B.; Janz, D.; Jiang, X. Upgrading root physiology for stress tolerance by ectomycorrhizas: Insights from metabolite and transcriptional profiling into reprogramming for stress anticipation. Plant Physiol. 2009, 151, 1902–1917. [Google Scholar] [CrossRef] [Green Version]
- Deyholos, M.K. Making the most of drought and salinity transcriptomics. Plant Cell Environ. 2010, 33, 648. [Google Scholar] [CrossRef]
- Chen, H. Gossypium darwinii cotton drought stress transcriptional group sequencing, EST-SSR development, and high-density genetic mapping construction. Chin. Acad. Agric. Sci. 2013. [Google Scholar]
- Zhang, C.; Sang, X.; Qu, M. De novo sequencing and analysis of root transcriptome to reveal regulation of gene expression by moderate drought stress in Glycyrrhiza uralensis. Chin. Mater. Med. 2015, 40, 4817–4823. [Google Scholar]
- Liu, C.; Zhang, X.; Zhang, K. Comparative analysis of the brassica napus root and leaf transcript profiling in response to drought stress. Int. J. Mol. Sci. 2015, 16, 18752–18777. [Google Scholar] [CrossRef] [Green Version]
- Wei, C.; Yao, Q.; Patil, G.B. Identification and comparative analysis of differential gene expression in soybean leaf tissue under drought and flooding dtress revealed by RNA-Seq. Front. Plant Sci. 2016, 7, 1044. [Google Scholar]
- Garg, R.; Shankar, R.; Thakkar, B. Transcriptome analyses reveal genotype and developmental stage-specific molecular responses to drought and salinity stresses in chickpea. Sci. Rep. 2016, 6, 19228. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Ma, W.; Zeng, P. LncTar: A tool for predicting the RNA targets of long noncoding RNAs. Brief. Bioinform. 2014, 16, 806–812. [Google Scholar] [CrossRef]
- Trapnell, C. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeck, W.R.; Sorrentino, J.A.; Wang, K. Circular RNAs are abundant, conserved, and associated with ALU repeats. RNA 2012, 19, 141–157. [Google Scholar] [CrossRef] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, Y.; Li, J.; Wu, S. Integrated nr Database in Protein Annotation System and Its Localization. Comput. Eng. 2006, 32, 71–74. [Google Scholar]
- Apweiler, R.; Bairoch, A.; Wu, C.H. UniProt: The universal protein knowledgebase. Nucleic Acids Res. 2004, 32 (suppl. 1), D115–D119. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, C29. [Google Scholar] [CrossRef] [Green Version]
- Tatusov, R.L.; Galperin, M.Y.; Natale, D.A. The COG database: A tool for genome scale analysis of protein functions and evolution. Nucleic Acids Res. 2000, 28, 33–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanehisa, M.; Goto, S.; Kawashima, S.; Okuno, Y.; Hattori, M. The KEGG resource for deciphering the genome. Nucleic Acids Res. 2004, 32, D277–D280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koonin, E.V.; Fedorova, N.D.; Jackson, J.D. A comprehensive evolutionary classification of proteins encoded in complete eukaryotic genomes. Genome Biol. Ital. 2004, 5, R7. [Google Scholar] [CrossRef] [Green Version]
- Yu, G.; Wang, L.G.; Han, Y. clusterProfiler: An R package for comparing biological themes among gene clusters. Omics A J. Integr. Biol. 2004, 16, 284–287. [Google Scholar] [CrossRef]
- Price, A.L.; Patterson, N.J. Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet. 2006, 38, 904–909. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Society. Ser. B Methodol. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, X.O.; Chen, T.; Xiang, J.F.; Yin, Q.F.; Xing, Y.H.; Zhu, S.; Yang, L.; Chen, L.L. Circular intronic long noncoding RNAs. Mol. Cell 2013, 51, 792–806. [Google Scholar] [CrossRef] [Green Version]
- Hansen, T.B.; Jensen, T.I.; Clausen, B.H.; Bramsen, J.B.; Finsen, B.; Damgaard, C.K.; Kjems, J. Natural RNA circles function as efficient microRNA sponges. Nature 2013, 495, 384–388. [Google Scholar] [CrossRef]
- Salmena, L.; Poliseno, L.; Tay, Y.; Kats, L.; Pandolfi, P.P. A ceRNA hypothesis: The Rosetta Stone of a hidden RNA language. Cell 2011, 146, 353–358. [Google Scholar] [CrossRef] [Green Version]
- Franco-Zorrilla, J.M.; Valli, A.; Todesco, M. Target mimicry provides a new mechanism forregulation of microRNA activity. Nat. Genet. 2007, 39, 1033–1037. [Google Scholar] [CrossRef]
- Deng, F.; Zhang, X.; Wang, W.; Yuan, R.; Shen, F. Identification of Gossypium hirsutum long non-coding RNAs (lncRNAs) under salt stress. BMC Plant Biol. 2018, 18, 23. [Google Scholar] [CrossRef] [PubMed]
- Breitkreuz, K.E.; Shelp, B.J. Subcellular compartmentation of the 4-aminobutyrate shunt in protoplasts from developing soybean cotyledons. Plant Physiol. 1995, 108, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Bouche, N.; Fait, A.; Bouchez, D. Mitochondrial succinie-semial-dehyde dehydrogenase of the-γ-aminobutyrate shunt is required to restrict levels of reactive oxygen intermediates in plants. Proc. Natl. Acad. Sci. USA 2003, 100, 6843–6848. [Google Scholar] [CrossRef] [Green Version]
- Palanivelu, R.; Brass, L.; Edlund, A.F.; Preuss, D. Pollen tube growth and guidance is regulated by POP2, an Arabidopsis gene that controls GABA levels. Cell 2003, 114, 47–59. [Google Scholar] [CrossRef] [Green Version]
- Renault, H.; El Amrani, A.; Palanivelu, R.; Updegraff, E.P.; Yu, A.; Renou, J.P. GABA accumulation causes cell elongation defects and a decrease in expression of genes encoding secreted and cell wall-related proteins in A. thaliana. Plant Cell Physiol. 2011, 52, 894–908. [Google Scholar] [CrossRef] [Green Version]
- Farooq, M.; Wahita, A.; Kobayash, N. Plant drought stress: Effects, mecharisms and management. Agron. Sustain. Dev. 2009, 29, 185–212. [Google Scholar] [CrossRef] [Green Version]
- Rongvaux, A.; Andris, F.; Van Gool, F.; Leo, O. Reconstructing eukaryotic NAD metabolism. Bioessays 2003, 25, 683–690. [Google Scholar] [CrossRef]
- Katoh, A.; Uenohara, K.; Akita, M.; Hashimoto, T. Early Steps in the Biosynthesis of NAD in Arabidopsis Start with Aspartate and Occur in the Plastid. Plant Physiol. 2006, 141, 851–857. [Google Scholar] [CrossRef] [Green Version]
- Katoh, A.; Hashimoto, T. Molecular biology of pyridine nucleotide and nicotine biosynthesis. Front. Biosci. 2004, 9, 1577–1586. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SampleID | ReadSum | BaseSum | GC (%) | N (%) | Q20 (%) | Q30 (%) | Total Reads | Mapped Reads |
|---|---|---|---|---|---|---|---|---|
| XJ0hR1 | 71,742,474 | 21,224,569,062 | 42.85 | 0 | 97.92 | 93.80 | 143,484,948 | 138,314,357 (96.40%) |
| XJ0hR2 | 53,952,826 | 16,058,270,844 | 42.78 | 0 | 97.93 | 93.88 | 107,905,652 | 104,104,314 (96.48%) |
| XJ0hR3 | 67,100,443 | 19,944,130,312 | 43.07 | 0 | 97.90 | 93.73 | 134,200,886 | 129,284,996 (96.34%) |
| XJ0hS1 | 53,931,117 | 16,041,131,562 | 42.77 | 0 | 98.34 | 94.70 | 107,862,234 | 103,981,706 (96.40%) |
| XJ0hS2 | 57,233,313 | 17,014,361,012 | 42.72 | 0 | 98.32 | 94.65 | 114,466,626 | 110,557,335 (96.58%) |
| XJ0hS3 | 56,289,536 | 16,740,674,196 | 42.84 | 0 | 98.21 | 94.50 | 112,579,072 | 108,590,412 (96.46%) |
| XJ12hR1 | 60,305,370 | 17,970,064,928 | 43.00 | 0 | 98.27 | 94.52 | 120,610,740 | 116,216,676 (96.36%) |
| XJ12hR2 | 53,171,614 | 15,800,217,442 | 42.67 | 0 | 98.28 | 94.59 | 106,343,228 | 102,417,327 (96.31%) |
| XJ12hR3 | 54,393,796 | 16,198,260,692 | 42.06 | 0 | 98.29 | 94.60 | 108,787,592 | 104,988,226 (96.51%) |
| XJ12hS1 | 55,632,640 | 16,553,339,614 | 42.5 | 0 | 98.27 | 94.51 | 111,265,280 | 107,405,318 (96.53%) |
| XJ12hS2 | 55,334,885 | 16,503,777,878 | 42.61 | 0 | 98.19 | 94.39 | 110,669,770 | 105,879,284 (95.67%) |
| XJ12hS3 | 56,241,086 | 16,733,164,858 | 42.56 | 0 | 98.20 | 94.39 | 112,482,172 | 107,971,513 (95.99%) |
| XJ24hR1 | 55,174,614 | 16,453,704,450 | 42.76 | 0 | 98.21 | 94.48 | 110,349,228 | 106,142,505 (96.19%) |
| XJ24hR2 | 55,843,015 | 16,660,958,134 | 42.43 | 0 | 98.34 | 94.76 | 111,686,030 | 107,264,860 (96.04%) |
| XJ24hR3 | 58,592,920 | 17,464,204,712 | 42.98 | 0 | 98.22 | 94.44 | 117,185,840 | 113,571,833 (96.92%) |
| XJ24hS1 | 55,363,695 | 16,530,287,984 | 42.26 | 0 | 98.47 | 94.98 | 110,727,390 | 106,999,856 (96.63%) |
| XJ24hS2 | 55,231,921 | 16,459,828,202 | 42.49 | 0 | 98.19 | 94.32 | 110,463,842 | 106,313,930 (96.24%) |
| XJ24hS3 | 56,920,057 | 16,913,326,528 | 42.8 | 0 | 98.45 | 94.91 | 113,840,114 | 109,889,372 (96.53%) |
| DEG Set | Number of DEGs | Upregulated | Downregulated |
|---|---|---|---|
| XJ0hR1_XJ0hR2_XJ0hR3_vs_XJ0hS1_XJ0hS2_XJ0hS3 | 2161 | 1065 | 1096 |
| XJ0hR1_XJ0hR2_XJ0hR3_vs_XJ12hR1_XJ12hR2_XJ12hR3 | 16,882 | 7808 | 9074 |
| XJ0hR1_XJ0hR2_XJ0hR3_vs_XJ24hR1_XJ24hR2_XJ24hR3 | 7168 | 2997 | 4171 |
| XJ0hS1_XJ0hS2_XJ0hS3_vs_XJ12hS1_XJ12hS2_XJ12hS3 | 15,626 | 7498 | 8128 |
| XJ0hS1_XJ0hS2_XJ0hS3_vs_XJ24hS1_XJ24hS2_XJ24hS3 | 3070 | 1224 | 1846 |
| XJ12hR1_XJ12hR2_XJ12hR3_vs_XJ12hS1_XJ12hS2_XJ12hS3 | 1614 | 615 | 999 |
| XJ12hR1_XJ12hR2_XJ12hR3_vs_XJ24hR1_XJ24hR2_XJ24hR3 | 15,240 | 7572 | 7668 |
| XJ12hS1_XJ12hS2_XJ12hS3_vs_XJ24hS1_XJ24hS2_XJ24hS3 | 14,396 | 7159 | 7237 |
| XJ24hR1_XJ24hR2_XJ24hR3_vs_XJ24hS1_XJ24hS2_XJ24hS3 | 1563 | 805 | 758 |
| DEG Set | Number of DEGs | Upregulated | Downregulated |
|---|---|---|---|
| XJ0hR1_XJ0hR2_XJ0hR3_vs_XJ0hS1_XJ0hS2_XJ0hS3 | 294 | 147 | 147 |
| XJ0hR1_XJ0hR2_XJ0hR3_vs_XJ12hR1_XJ12hR2_XJ12hR3 | 462 | 246 | 216 |
| XJ0hR1_XJ0hR2_XJ0hR3_vs_XJ24hR1_XJ24hR2_XJ24hR3 | 182 | 108 | 74 |
| XJ0hS1_XJ0hS2_XJ0hS3_vs_XJ12hS1_XJ12hS2_XJ12hS3 | 439 | 220 | 219 |
| XJ0hS1_XJ0hS2_XJ0hS3_vs_XJ24hS1_XJ24hS2_XJ24hS3 | 126 | 73 | 53 |
| XJ12hR1_XJ12hR2_XJ12hR3_vs_XJ12hS1_XJ12hS2_XJ12hS3 | 302 | 148 | 154 |
| XJ12hR1_XJ12hR2_XJ12hR3_vs_XJ24hR1_XJ24hR2_XJ24hR3 | 437 | 217 | 220 |
| XJ12hS1_XJ12hS2_XJ12hS3_vs_XJ24hS1_XJ24hS2_XJ24hS3 | 423 | 223 | 200 |
| XJ24hR1_XJ24hR2_XJ24hR3_vs_XJ24hS1_XJ24hS2_XJ24hS3 | 342 | 162 | 180 |
| DEG Set | Number of DEGs | Upregulated | Downregulated |
|---|---|---|---|
| XJ0hR1_XJ0hR2_XJ0hR3_vs_XJ0hS1_XJ0hS2_XJ0hS3 | 8 | 0 | 8 |
| XJ0hR1_XJ0hR2_XJ0hR3_vs_XJ12hR1_XJ12hR2_XJ12hR3 | 11 | 6 | 5 |
| XJ0hR1_XJ0hR2_XJ0hR3_vs_XJ24hR1_XJ24hR2_XJ24hR3 | 1 | 0 | 1 |
| XJ0hS1_XJ0hS2_XJ0hS3_vs_XJ12hS1_XJ12hS2_XJ12hS3 | 2 | 2 | 0 |
| XJ0hS1_XJ0hS2_XJ0hS3_vs_XJ24hS1_XJ24hS2_XJ24hS3 | 6 | 6 | 0 |
| XJ12hR1_XJ12hR2_XJ12hR3_vs_XJ12hS1_XJ12hS2_XJ12hS3 | 11 | 1 | 10 |
| XJ12hR1_XJ12hR2_XJ12hR3_vs_XJ24hR1_XJ24hR2_XJ24hR3 | 5 | 2 | 3 |
| XJ12hS1_XJ12hS2_XJ12hS3_vs_XJ24hS1_XJ24hS2_XJ24hS3 | 0 | 0 | 0 |
| XJ24hR1_XJ24hR2_XJ24hR3_vs_XJ24hS1_XJ24hS2_XJ24hS3 | 11 | 5 | 6 |
| DEG Set | Number of DEGs | Upregulated | Downregulated |
|---|---|---|---|
| XJ0hR1_XJ0hR2_XJ0hR3_vs_XJ0hS1_XJ0hS2_XJ0hS3 | 22 | 8 | 14 |
| XJ0hR1_XJ0hR2_XJ0hR3_vs_XJ12hR1_XJ12hR2_XJ12hR3 | 9 | 3 | 6 |
| XJ0hR1_XJ0hR2_XJ0hR3_vs_XJ24hR1_XJ24hR2_XJ24hR3 | 5 | 3 | 2 |
| XJ0hS1_XJ0hS2_XJ0hS3_vs_XJ12hS1_XJ12hS2_XJ12hS3 | 6 | 3 | 3 |
| XJ0hS1_XJ0hS2_XJ0hS3_vs_XJ24hS1_XJ24hS2_XJ24hS3 | 19 | 13 | 6 |
| XJ12hR1_XJ12hR2_XJ12hR3_vs_XJ12hS1_XJ12hS2_XJ12hS3 | 14 | 7 | 7 |
| XJ12hR1_XJ12hR2_XJ12hR3_vs_XJ24hR1_XJ24hR2_XJ24hR3 | 10 | 7 | 3 |
| XJ12hS1_XJ12hS2_XJ12hS3_vs_XJ24hS1_XJ24hS2_XJ24hS3 | 17 | 7 | 10 |
| XJ24hR1_XJ24hR2_XJ24hR3_vs_XJ24hS1_XJ24hS2_XJ24hS3 | 30 | 15 | 15 |
| DEG Set | Total | COG | GO | KEGG | KOG | NR | Pfam | Swiss-Prot | eggNOG |
|---|---|---|---|---|---|---|---|---|---|
| XJ0hR1_XJ0hR2_XJ0hR3_vs_XJ0hS1_XJ0hS2_XJ0hS3 | 2153 | 941 | 1745 | 782 | 1101 | 2153 | 1832 | 1776 | 2056 |
| XJ0hR1_XJ0hR2_XJ0hR3_vs_XJ12hR1_XJ12hR2_XJ12hR3 | 16,826 | 7311 | 13,680 | 6637 | 9786 | 16,822 | 14,425 | 13,455 | 16,415 |
| XJ0hR1_XJ0hR2_XJ0hR3_vs_XJ24hR1_XJ24hR2_XJ24hR3 | 7143 | 3241 | 5971 | 2867 | 3686 | 7143 | 6196 | 5830 | 6950 |
| XJ0hS1_XJ0hS2_XJ0hS3_vs_XJ12hS1_XJ12hS2_XJ12hS3 | 15,583 | 6888 | 12,632 | 6259 | 9292 | 15,579 | 13,394 | 12,432 | 15,198 |
| XJ0hS1_XJ0hS2_XJ0hS3_vs_XJ24hS1_XJ24hS2_XJ24hS3 | 3054 | 1394 | 2527 | 1280 | 1518 | 3054 | 2638 | 2541 | 2968 |
| XJ12hR1_XJ12hR2_XJ12hR3_vs_XJ12hS1_XJ12hS2_XJ12hS3 | 1602 | 667 | 1293 | 582 | 910 | 1602 | 1358 | 1296 | 1509 |
| XJ12hR1_XJ12hR2_XJ12hR3_vs_XJ24hR1_XJ24hR2_XJ24hR3 | 15,190 | 6738 | 12,328 | 6120 | 9132 | 15,188 | 13,139 | 12,083 | 14,848 |
| XJ12hS1_XJ12hS2_XJ12hS3_vs_XJ24hS1_XJ24hS2_XJ24hS3 | 14,362 | 6424 | 11,620 | 5739 | 8699 | 14,357 | 12,421 | 11,437 | 14,033 |
| XJ24hR1_XJ24hR2_XJ24hR3_vs_XJ24hS1_XJ24hS2_XJ24hS3 | 1557 | 700 | 1236 | 611 | 889 | 1557 | 1328 | 1255 | 1487 |
| DEG Set | Total | COG | GO | KEGG | KOG | NR | Pfam | Swiss-Prot | eggNOG |
|---|---|---|---|---|---|---|---|---|---|
| XJ0hR1_XJ0hR2_XJ0hR3_vs_XJ0hS1_XJ0hS2_XJ0hS3 | 1 | 1 | 1 | 0 | 1 | 1 | 1 | 1 | 1 |
| XJ0hR1_XJ0hR2_XJ0hR3_vs_XJ12hR1_XJ12hR2_XJ12hR3 | 7 | 5 | 7 | 3 | 7 | 7 | 7 | 7 | 7 |
| XJ0hR1_XJ0hR2_XJ0hR3_vs_XJ24hR1_XJ24hR2_XJ24hR3 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| XJ0hS1_XJ0hS2_XJ0hS3_vs_XJ12hS1_XJ12hS2_XJ12hS3 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
| XJ0hS1_XJ0hS2_XJ0hS3_vs_XJ24hS1_XJ24hS2_XJ24hS3 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 |
| XJ12hR1_XJ12hR2_XJ12hR3_vs_XJ12hS1_XJ12hS2_XJ12hS3 | 2 | 2 | 2 | 1 | 2 | 2 | 2 | 2 | 2 |
| XJ12hR1_XJ12hR2_XJ12hR3_vs_XJ24hR1_XJ24hR2_XJ24hR3 | 3 | 2 | 3 | 1 | 3 | 3 | 3 | 3 | 3 |
| XJ12hS1_XJ12hS2_XJ12hS3_vs_XJ24hS1_XJ24hS2_XJ24hS3 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| XJ24hR1_XJ24hR2_XJ24hR3_vs_XJ24hS1_XJ24hS2_XJ24hS3 | 2 | 2 | 2 | 1 | 2 | 2 | 2 | 2 | 2 |
| DEG Set | Total | COG | GO | KEGG | KOG | NR | Pfam | Swiss-Prot | eggNOG |
|---|---|---|---|---|---|---|---|---|---|
| XJ0hR1_XJ0hR2_XJ0hR3_vs_XJ0hS1_XJ0hS2_XJ0hS3 | 400 | 156 | 299 | 139 | 223 | 400 | 322 | 304 | 377 |
| XJ0hR1_XJ0hR2_XJ0hR3_vs_XJ12hR1_XJ12hR2_XJ12hR3 | 1009 | 385 | 763 | 377 | 575 | 1009 | 849 | 793 | 949 |
| XJ0hR1_XJ0hR2_XJ0hR3_vs_XJ24hR1_XJ24hR2_XJ24hR3 | 737 | 285 | 559 | 256 | 419 | 737 | 618 | 574 | 690 |
| XJ0hS1_XJ0hS2_XJ0hS3_vs_XJ12hS1_XJ12hS2_XJ12hS3 | 376 | 127 | 270 | 119 | 229 | 376 | 310 | 309 | 365 |
| XJ0hS1_XJ0hS2_XJ0hS3_vs_XJ24hS1_XJ24hS2_XJ24hS3 | 754 | 269 | 566 | 261 | 439 | 754 | 633 | 589 | 710 |
| XJ12hR1_XJ12hR2_XJ12hR3_vs_XJ12hS1_XJ12hS2_XJ12hS3 | 297 | 109 | 210 | 104 | 181 | 297 | 247 | 226 | 288 |
| XJ12hR1_XJ12hR2_XJ12hR3_vs_XJ24hR1_XJ24hR2_XJ24hR3 | 137 | 46 | 101 | 49 | 75 | 137 | 123 | 118 | 132 |
| XJ12hS1_XJ12hS2_XJ12hS3_vs_XJ24hS1_XJ24hS2_XJ24hS3 | 493 | 185 | 387 | 175 | 321 | 493 | 398 | 404 | 470 |
| XJ24hR1_XJ24hR2_XJ24hR3_vs_XJ24hS1_XJ24hS2_XJ24hS3 | 1999 | 716 | 1530 | 702 | 1175 | 1999 | 1645 | 1547 | 1900 |
| Key Gene | Homologous Genes in Arabidopsis | Homology Index % |
|---|---|---|
| Gohir.A11G156000 | AT3G22200 | 77 |
| Gohir.A11G227533 | AT3G27740 | 79 |
| Gohir.D01G114300 | AT5G10920 | 76 |
| Gohir.D04G022000 | AT3G24090 | 77 |
| Gohir.D05G088300 | AT3G47340 | 77 |
| Gohir.A07G220600 | AT5G14760 | 76 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zheng, J.; Zhang, Z.; Liang, Y.; Gong, Z.; Zhang, N.; Ditta, A.; Sang, Z.; Wang, J.; Li, X. Whole Transcriptome Sequencing Reveals Drought Resistance-Related Genes in Upland Cotton. Genes 2022, 13, 1159. https://doi.org/10.3390/genes13071159
Zheng J, Zhang Z, Liang Y, Gong Z, Zhang N, Ditta A, Sang Z, Wang J, Li X. Whole Transcriptome Sequencing Reveals Drought Resistance-Related Genes in Upland Cotton. Genes. 2022; 13(7):1159. https://doi.org/10.3390/genes13071159
Chicago/Turabian StyleZheng, Juyun, Zeliang Zhang, Yajun Liang, Zhaolong Gong, Nala Zhang, Allah Ditta, Zhiwei Sang, Junduo Wang, and Xueyuan Li. 2022. "Whole Transcriptome Sequencing Reveals Drought Resistance-Related Genes in Upland Cotton" Genes 13, no. 7: 1159. https://doi.org/10.3390/genes13071159
APA StyleZheng, J., Zhang, Z., Liang, Y., Gong, Z., Zhang, N., Ditta, A., Sang, Z., Wang, J., & Li, X. (2022). Whole Transcriptome Sequencing Reveals Drought Resistance-Related Genes in Upland Cotton. Genes, 13(7), 1159. https://doi.org/10.3390/genes13071159