
Pathogenesis of Choledochal Cyst: Insights from Genomics and Transcriptomics


Abstract
1. Introduction
1.1. Chromosomal Anomalies
1.2. Transcriptomics Analysis
1.3. Genetic Variants
1.4. Genetics in Type V CC
1.5. Animal Model
1.6. Malignancy Transformation
1.7. Potential Future Research Directions
2. Conclusions
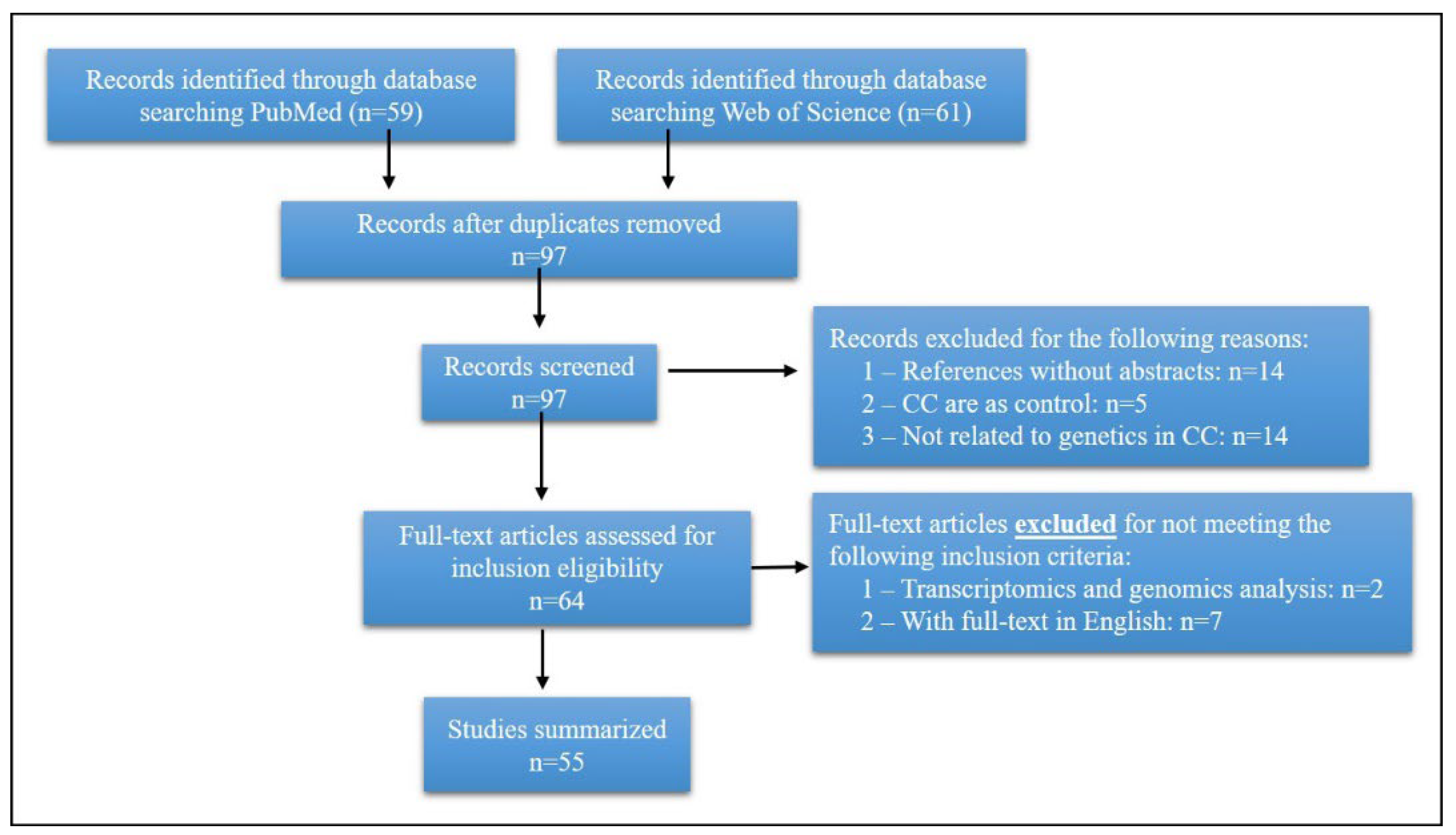
3. Search Strategy and Selection Criteria
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Todani, T.; Watanabe, Y.; Narusue, M.; Tabuchi, K.; Okajima, K. Congenital bile duct cysts: Classification, operative procedures, and review of thirty-seven cases including cancer arising from choledochal cyst. Am. J. Surg. 1977, 134, 263–269. [Google Scholar] [CrossRef]
- Makin, E.; Davenport, M. Understanding choledochal malformation. Arch. Dis. Child 2012, 97, 69–72. [Google Scholar] [CrossRef] [PubMed]
- Kemmotsu, H.; Mouri, T.; Muraji, T. Congenital stenosis of the hepatic duct at the porta hepatis in children with choledochal cyst. J. Pediatr. Surg. 2009, 44, 512–516. [Google Scholar] [CrossRef] [PubMed]
- Kato, T. Etiological relationships between choledochal cyst and anomalous junction of the pancreaticobiliary ductal system. Keio J. Med. 1989, 38, 136–151. [Google Scholar] [CrossRef][Green Version]
- Cao, J.; Wang, L.; Zou, X. Coexistence of Complete Pancreas Divisum and Anomalous Pancreaticobiliary Junction in a Patient with Type IA Choledochal Cyst. Clin. Gastroenterol. Hepatol. 2019, 17, A27–A28. [Google Scholar] [CrossRef]
- Singham, J.; Yoshida, E.M.; Scudamore, C.H. Choledochal cysts: Part 1 of 3: Classification and pathogenesis. Can. J. Surg. 2009, 52, 434–440. [Google Scholar]
- Shimotake, T.; Iwai, N.; Yanagihara, J.; Inoue, K.; Fushiki, S. Innervation patterns in congenital biliary dilatation. Eur. J. Pediatr. Surg. 1995, 5, 265–270. [Google Scholar] [CrossRef]
- Lewis, V.A.; Adam, S.Z.; Nikolaidis, P.; Wood, C.; Wu, J.G.; Yaghmai, V.; Miller, F.H. Imaging of choledochal cysts. Abdom. Imaging 2015, 40, 1567–1580. [Google Scholar] [CrossRef]
- Kusunoki, M.; Yamamura, T.; Takahashi, T.; Kantoh, M.; Ishikawa, Y.; Utsunomiya, J. Choledochal cyst. Its possible autonomic involvement in the bile duct. Arch. Surg. 1987, 122, 997–1000. [Google Scholar] [CrossRef]
- Davenport, M.; Basu, R. Under pressure: Choledochal malformation manometry. J. Pediatr. Surg. 2005, 40, 331–335. [Google Scholar] [CrossRef]
- Iwata, F.; Uchida, A.; Miyaki, T.; Aoki, S.; Fujioka, T.; Yamada, J.; Joh, T.; Itoh, M. Familial occurrence of congenital bile duct cysts. J. Gastroenterol. Hepatol. 1998, 13, 316–319. [Google Scholar] [CrossRef]
- Iwama, T. Familial case of choledochocele. J. Gastroenterol. Hepatol. 1998, 13, 237. [Google Scholar] [CrossRef]
- Clifton, M.S.; Goldstein, R.B.; Slavotinek, A.; Norton, M.E.; Lee, H.; Farrell, J.; Nobuhara, K.K. Prenatal diagnosis of familial type I choledochal cyst. Pediatrics 2006, 117, e596–e600. [Google Scholar] [CrossRef]
- Hodgson, S.V.; Coonar, A.S.; Hanson, P.J.; Cottrell, S.; Scriven, P.N.; Jones, T.; Hawley, P.R.; Wilkinson, M.L. Two cases of 5q deletions in patients with familial adenomatous polyposis: Possible link with Caroli’s disease. J. Med. Genet. 1993, 30, 369–375. [Google Scholar] [CrossRef]
- Behrns, K.E.; Shaheen, N.J.; Grimm, I.S. Type I choledochal cyst in association with familial adenomatous polyposis. Am. J. Gastroenterol. 1998, 93, 1377–1379. [Google Scholar] [CrossRef]
- Yao, X.; Ao, W.; Fang, J.; Mao, G.; Chen, C.; Yu, L.; Cai, H.; Xu, C. Imaging manifestations of Caroli disease with autosomal recessive polycystic kidney disease: A case report and literature review. BMC Pregnancy Childbirth 2021, 21, 294. [Google Scholar] [CrossRef]
- Torra, R.; Badenas, C.; Darnell, A.; Brú, C.; Escorsell, A.; Estivill, X. Autosomal dominant polycystic kidney disease with anticipation and Caroli’s disease associated with a PKD1 mutation. Rapid communication. Kidney Int. 1997, 52, 33–38. [Google Scholar] [CrossRef][Green Version]
- Lee, J.M.; Ahn, Y.H.; Kang, H.G.; Ha, I.I.; Lee, K.; Moon, K.C.; Lee, J.H.; Park, Y.S.; Cho, Y.M.; Bae, J.S.; et al. Nephronophthisis 13: Implications of its association with Caroli disease and altered intracellular localization of WDR19 in the kidney. Pediatr. Nephrol. 2015, 30, 1451–1458. [Google Scholar] [CrossRef]
- Hasegawa, E.; Sawa, N.; Hoshino, J.; Suwabe, T.; Hayami, N.; Yamanouchi, M.; Sekine, A.; Hiramatsu, R.; Imafuku, A.; Kawada, M.; et al. Recurrent Cholangitis in a Patient with Autosomal Dominant Polycystic Kidney Disease (ADPKD) and Caroli’s Disease. Intern. Med. 2016, 55, 3009–3012. [Google Scholar] [CrossRef][Green Version]
- Chung, E.M.; Conran, R.M.; Schroeder, J.W.; Rohena-Quinquilla, I.R.; Rooks, V.J. From the radiologic pathology archives: Pediatric polycystic kidney disease and other ciliopathies: Radiologic-pathologic correlation. Radiographics 2014, 34, 155–178. [Google Scholar] [CrossRef]
- Kotalova, R.; Dusatkova, P.; Drabova, J.; Elblova, L.; Dedic, T.; Cinek, O.; Lebl, J.; Pruhova, S. Choledochal Cyst with 17q12 Chromosomal Duplication. Ann. Hum. Genet. 2018, 82, 48–51. [Google Scholar] [CrossRef]
- Rogdaki, M.; Jauhar, S.; McCutcheon, R.; Howes, O. Treatment-Resistant Schizophrenia in a Patient With 17q12 Duplication. Biol. Psychiatry 2016, 80, e19–e20. [Google Scholar] [CrossRef]
- Kamath, A.; Linden, S.C.; Evans, F.M.; Hall, J.; Jose, S.F.; Spillane, S.A.; Hardie, A.D.R.; Morgan, S.M.; Pilz, D.T. Chromosome 17q12 duplications: Further delineation of the range of psychiatric and clinical phenotypes. Am. J. Med. Genet. B Neuropsychiatry Genet. 2018, 177, 520–528. [Google Scholar] [CrossRef]
- Hardies, K.; Weckhuysen, S.; Peeters, E.; Holmgren, P.; Van Esch, H.; De Jonghe, P.; Van Paesschen, W.; Suls, A. Duplications of 17q12 can cause familial fever-related epilepsy syndromes. Neurology 2013, 81, 1434–1440. [Google Scholar] [CrossRef]
- Clissold, R.L.; Shaw-Smith, C.; Turnpenny, P.; Bunce, B.; Bockenhauer, D.; Kerecuk, L.; Waller, S.; Bowman, P.; Ford, T.; Ellard, S.; et al. Chromosome 17q12 microdeletions but not intragenic HNF1B mutations link developmental kidney disease and psychiatric disorder. Kidney Int. 2016, 90, 203–211. [Google Scholar] [CrossRef]
- Clissold, R.L.; Hamilton, A.J.; Hattersley, A.T.; Ellard, S.; Bingham, C. HNF1B-associated renal and extra-renal disease-an expanding clinical spectrum. Nat. Rev. Nephrol. 2015, 11, 102–112. [Google Scholar] [CrossRef]
- Calle Sánchez, X.; Helenius, D.; Bybjerg-Grauholm, J.; Pedersen, C.; Hougaard, D.M.; Børglum, A.D.; Nordentoft, M.; Mors, O.; Mortensen, P.B.; Geschwind, D.H.; et al. Comparing Copy Number Variations in a Danish Case Cohort of Individuals with Psychiatric Disorders. JAMA Psychiatry 2022, 79, 59–69. [Google Scholar] [CrossRef]
- Strazzabosco, M.; Fabris, L. Development of the bile ducts: Essentials for the clinical hepatologist. J. Hepatol. 2012, 56, 1159–1170. [Google Scholar] [CrossRef] [PubMed]
- Kotalova, R.; Dusatkova, P.; Cinek, O.; Dusatkova, L.; Dedic, T.; Seeman, T.; Lebl, J.; Pruhova, S. Hepatic phenotypes of HNF1B gene mutations: A case of neonatal cholestasis requiring portoenterostomy and literature review. World J. Gastroenterol. 2015, 21, 2550–2557. [Google Scholar] [CrossRef] [PubMed]
- Kettunen, J.L.T.; Parviainen, H.; Miettinen, P.J.; Färkkilä, M.; Tamminen, M.; Salonen, P.; Lantto, E.; Tuomi, T. Biliary Anomalies in Patients with HNF1B Diabetes. J. Clin. Endocrinol. Metab. 2017, 102, 2075–2082. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Piccione, M.; Piro, E.; Serraino, F.; Cavani, S.; Ciccone, R.; Malacarne, M.; Pierluigi, M.; Vitaloni, M.; Zuffardi, O.; Corsello, G. Interstitial deletion of chromosome 2p15-16.1: Report of two patients and critical review of current genotype-phenotype correlation. Eur. J. Med. Genet. 2012, 55, 238–244. [Google Scholar] [CrossRef]
- Lévy, J.; Coussement, A.; Dupont, C.; Guimiot, F.; Baumann, C.; Viot, G.; Passemard, S.; Capri, Y.; Drunat, S.; Verloes, A.; et al. Molecular and clinical delineation of 2p15p16.1 microdeletion syndrome. Am. J. Med. Genet. A 2017, 173, 2081–2087. [Google Scholar] [CrossRef]
- Jorgez, C.J.; Rosenfeld, J.A.; Wilken, N.R.; Vangapandu, H.V.; Sahin, A.; Pham, D.; Carvalho, C.M.; Bandholz, A.; Miller, A.; Weaver, D.D.; et al. Genitourinary defects associated with genomic deletions in 2p15 encompassing OTX1. PLoS ONE 2014, 9, e107028. [Google Scholar] [CrossRef]
- Hucthagowder, V.; Liu, T.C.; Paciorkowski, A.R.; Thio, L.L.; Keller, M.S.; Anderson, C.D.; Herman, T.; Dehner, L.P.; Grange, D.K.; Kulkarni, S. Chromosome 2p15p16.1 microdeletion syndrome: 2.5 Mb deletion in a patient with renal anomalies, intractable seizures and a choledochal cyst. Eur. J. Med. Genet. 2012, 55, 485–489. [Google Scholar] [CrossRef]
- Florisson, J.M.; Mathijssen, I.M.; Dumee, B.; Hoogeboom, J.A.; Poddighe, P.J.; Oostra, B.A.; Frijns, J.P.; Koster, L.; de Klein, A.; Eussen, B.; et al. Complex craniosynostosis is associated with the 2p15p16.1 microdeletion syndrome. Am. J. Med. Genet. A 2013, 161, 244–253. [Google Scholar] [CrossRef]
- de Leeuw, N.; Pfundt, R.; Koolen, D.A.; Neefs, I.; Scheltinga, I.; Mieloo, H.; Sistermans, E.A.; Nillesen, W.; Smeets, D.F.; de Vries, B.B.; et al. A newly recognised microdeletion syndrome involving 2p15p16.1: Narrowing down the critical region by adding another patient detected by genome wide tiling path array comparative genomic hybridisation analysis. J. Med. Genet. 2008, 45, 122–124. [Google Scholar] [CrossRef]
- Bagheri, H.; Badduke, C.; Qiao, Y.; Colnaghi, R.; Abramowicz, I.; Alcantara, D.; Dunham, C.; Wen, J.; Wildin, R.S.; Nowaczyk, M.J.; et al. Identifying candidate genes for 2p15p16.1 microdeletion syndrome using clinical, genomic, and functional analysis. JCI Insight 2016, 1, e85461. [Google Scholar] [CrossRef]
- Vieira, J.; Pinto, C.; Afonso, M.; do Bom Sucesso, M.; Lopes, P.; Pinheiro, M.; Veiga, I.; Henrique, R.; Teixeira, M.R. Identification of previously unrecognized FAP in children with Gardner fibroma. Eur. J. Hum. Genet. 2015, 23, 715–718. [Google Scholar] [CrossRef]
- Li, J.; Woods, S.L.; Healey, S.; Beesley, J.; Chen, X.; Lee, J.S.; Sivakumaran, H.; Wayte, N.; Nones, K.; Waterfall, J.J.; et al. Point Mutations in Exon 1B of APC Reveal Gastric Adenocarcinoma and Proximal Polyposis of the Stomach as a Familial Adenomatous Polyposis Variant. Am. J. Hum. Genet. 2016, 98, 830–842. [Google Scholar] [CrossRef]
- Parada, L.A.; Hallén, M.; Hägerstrand, I.; Tranberg, K.G.; Johansson, B. Clonal chromosomal abnormalities in congenital bile duct dilatation (Caroli’s disease). Gut 1999, 45, 780–782. [Google Scholar] [CrossRef]
- Meier, T.; Timm, M.; Montani, M.; Wilkens, L. Gene networks and transcriptional regulators associated with liver cancer development and progression. BMC Med. Genom. 2021, 14, 41. [Google Scholar] [CrossRef]
- Cao, P.; Yang, A.; Li, P.; Xia, X.; Han, Y.; Zhou, G.; Wang, R.; Yang, F.; Li, Y.; Zhang, Y.; et al. Genomic gain of RRS1 promotes hepatocellular carcinoma through reducing the RPL11-MDM2-p53 signaling. Sci Adv. 2021, 7, 4304. [Google Scholar] [CrossRef]
- Soares, K.C.; Goldstein, S.D.; Ghaseb, M.A.; Kamel, I.; Hackam, D.J.; Pawlik, T.M. Pediatric choledochal cysts: Diagnosis and current management. Pediatr. Surg. Int. 2017, 33, 637–650. [Google Scholar] [CrossRef]
- Moslim, M.A.; Takahashi, H.; Seifarth, F.G.; Walsh, R.M.; Morris-Stiff, G. Choledochal Cyst Disease in a Western Center: A 30-Year Experience. J. Gastrointest. Surg. 2016, 20, 1453–1463. [Google Scholar] [CrossRef]
- Madadi-Sanjani, O.; Wirth, T.C.; Kuebler, J.F.; Petersen, C.; Ure, B.M. Choledochal Cyst and Malignancy: A Plea for Lifelong Follow-Up. Eur. J. Pediatr. Surg. 2019, 29, 143–149. [Google Scholar] [CrossRef]
- Lv, Y.; Xie, X.; Pu, L.; Wang, Q.; Pu, S.; Ai, C.; Liu, Y.; Chen, J.; Xiang, B. Molecular Characteristics of Choledochal Cysts in Children: Transcriptome Sequencing. Front. Genet. 2021, 12, 709340. [Google Scholar] [CrossRef]
- Muthucumaru, M.; Ljuhar, D.; Panabokke, G.; Paul, E.; Nataraja, R.; Ferguson, P.; Dagia, C.; Clarnette, T.; King, S. Acute pancreatitis complicating choledochal cysts in children. J. Paediatr. Child. Health 2017, 53, 291–294. [Google Scholar] [CrossRef]
- Fujishiro, J.; Masumoto, K.; Urita, Y.; Shinkai, T.; Gotoh, C. Pancreatic complications in pediatric choledochal cysts. J. Pediatr. Surg. 2013, 48, 1897–1902. [Google Scholar] [CrossRef]
- Xue, Z.; Huang, K.; Cai, C.; Cai, L.; Jiang, C.Y.; Feng, Y.; Liu, Z.; Zeng, Q.; Cheng, L.; Sun, Y.E.; et al. Genetic programs in human and mouse early embryos revealed by single-cell RNA sequencing. Nature 2013, 500, 593–597. [Google Scholar] [CrossRef]
- Wang, Y.; Li, X.; Ruiz, R. Weighted General Group Lasso for Gene Selection in Cancer Classification. IEEE Trans. Cybern 2019, 49, 2860–2873. [Google Scholar] [CrossRef]
- Scavo, M.P.; Depalo, N.; Rizzi, F.; Ingrosso, C.; Fanizza, E.; Chieti, A.; Messa, C.; Denora, N.; Laquintana, V.; Striccoli, M.; et al. FZD10 Carried by Exosomes Sustains Cancer Cell Proliferation. Cells 2019, 8, 777. [Google Scholar] [CrossRef] [PubMed]
- Noll, A.T.; Cramer, T.; Olde Damink, S.W.; Schaap, F.G. Cholangiocarcinoma, gone without the Wnt? World J. Hepatol. 2016, 8, 1093–1096. [Google Scholar] [CrossRef] [PubMed]
- Rizvi, S.; Gores, G.J. Pathogenesis, diagnosis, and management of cholangiocarcinoma. Gastroenterology 2013, 145, 1215–1229. [Google Scholar] [CrossRef] [PubMed]
- Montal, R.; Sia, D.; Montironi, C.; Leow, W.Q.; Esteban-Fabró, R.; Pinyol, R.; Torres-Martin, M.; Bassaganyas, L.; Moeini, A.; Peix, J.; et al. Molecular classification and therapeutic targets in extrahepatic cholangiocarcinoma. J. Hepatol. 2020, 73, 315–327. [Google Scholar] [CrossRef]
- Jusakul, A.; Cutcutache, I.; Yong, C.H.; Lim, J.Q.; Huang, M.N.; Padmanabhan, N.; Nellore, V.; Kongpetch, S.; Ng, A.W.T.; Ng, L.M.; et al. Whole-Genome and Epigenomic Landscapes of Etiologically Distinct Subtypes of Cholangiocarcinoma. Cancer Discov. 2017, 7, 1116–1135. [Google Scholar] [CrossRef]
- Wong, J.K.; Campbell, D.; Ngo, N.D.; Yeung, F.; Cheng, G.; Tang, C.S.; Chung, P.H.; Tran, N.S.; So, M.T.; Cherny, S.S.; et al. Genetic study of congenital bile-duct dilatation identifies de novo and inherited variants in functionally related genes. BMC Med. Genom. 2016, 9, 75. [Google Scholar] [CrossRef]
- Ziv, A.; Werner, L.; Konnikova, L.; Awad, A.; Jeske, T.; Hastreiter, M.; Mitsialis, V.; Stauber, T.; Wall, S.; Kotlarz, D.; et al. An RTEL1 Mutation Links to Infantile-Onset Ulcerative Colitis and Severe Immunodeficiency. J. Clin. Immunol. 2020, 40, 1010–1019. [Google Scholar] [CrossRef]
- Wargo, J.J.; Carr, D.R.; Plaza, J.A.; Verschraegen, C.F. Metastatic Spiradenocarcinoma Managed with PD-1 Inhibition. J. Natl. Compr. Cancer Netw. 2022, 20, 1–3. [Google Scholar] [CrossRef]
- Walne, A.J.; Vulliamy, T.; Kirwan, M.; Plagnol, V.; Dokal, I. Constitutional mutations in RTEL1 cause severe dyskeratosis congenita. Am. J. Hum. Genet. 2013, 92, 448–453. [Google Scholar] [CrossRef]
- Vannier, J.B.; Sarek, G.; Boulton, S.J. RTEL1: Functions of a disease-associated helicase. Trends Cell Biol. 2014, 24, 416–425. [Google Scholar] [CrossRef]
- Tompson, S.W.; Merriman, B.; Funari, V.A.; Fresquet, M.; Lachman, R.S.; Rimoin, D.L.; Nelson, S.F.; Briggs, M.D.; Cohn, D.H.; Krakow, D. A recessive skeletal dysplasia, SEMD aggrecan type, results from a missense mutation affecting the C-type lectin domain of aggrecan. Am. J. Hum. Genet. 2009, 84, 72–79. [Google Scholar] [CrossRef]
- Hu, X.; Gui, B.; Su, J.; Li, H.; Li, N.; Yu, T.; Zhang, Q.; Xu, Y.; Li, G.; Chen, Y.; et al. Novel pathogenic ACAN variants in non-syndromic short stature patients. Clin. Chim. Acta 2017, 469, 126–129. [Google Scholar] [CrossRef]
- Dubois, A.; Alonso-Sanchez, A.; Bajaj, V.; Husain, A.; Rajan, N. Multiple Facial Trichoepitheliomas and Vulval Cysts: Extending the Phenotypic Spectrum in CYLD Cutaneous Syndrome. JAMA Dermatol. 2017, 153, 826–828. [Google Scholar] [CrossRef]
- Choi, A.; Lao, R.; Ling-Fung Tang, P.; Wan, E.; Mayer, W.; Bardakjian, T.; Shaw, G.M.; Kwok, P.Y.; Schneider, A.; Slavotinek, A. Novel mutations in PXDN cause microphthalmia and anterior segment dysgenesis. Eur. J. Hum. Genet. 2015, 23, 337–341. [Google Scholar] [CrossRef]
- Cao, Y.; Guan, X.; Li, S.; Wu, N.; Chen, X.; Yang, T.; Yang, B.; Zhao, X. Identification of variants in ACAN and PAPSS2 leading to spondyloepi(meta)physeal dysplasias in four Chinese families. Mol. Genet. Genom. Med. 2022, 10, e1916. [Google Scholar] [CrossRef]
- Meng, D.; Li, Z.; Ma, X.; Wu, L.; Fu, L.; Qin, G. ETV5 overexpression contributes to tumor growth and progression of thyroid cancer through PIK3CA. Life Sci. 2020, 253, 117693. [Google Scholar] [CrossRef]
- Mei, Z.B.; Duan, C.Y.; Li, C.B.; Cui, L.; Ogino, S. Prognostic role of tumor PIK3CA mutation in colorectal cancer: A systematic review and meta-analysis. Ann. Oncol. 2016, 27, 1836–1848. [Google Scholar] [CrossRef]
- Martínez-Sáez, O.; Chic, N.; Pascual, T.; Adamo, B.; Vidal, M.; González-Farré, B.; Sanfeliu, E.; Schettini, F.; Conte, B.; Brasó-Maristany, F.; et al. Frequency and spectrum of PIK3CA somatic mutations in breast cancer. Breast Cancer Res. 2020, 22, 45. [Google Scholar] [CrossRef]
- Madsen, R.R.; Vanhaesebroeck, B.; Semple, R.K. Cancer-Associated PIK3CA Mutations in Overgrowth Disorders. Trends Mol. Med. 2018, 24, 856–870. [Google Scholar] [CrossRef]
- Herberts, C.; Murtha, A.J.; Fu, S.; Wang, G.; Schönlau, E.; Xue, H.; Lin, D.; Gleave, A.; Yip, S.; Angeles, A.; et al. Activating AKT1 and PIK3CA Mutations in Metastatic Castration-Resistant Prostate Cancer. Eur. Urol. 2020, 78, 834–844. [Google Scholar] [CrossRef]
- Quénet, D.; Gasser, V.; Fouillen, L.; Cammas, F.; Sanglier-Cianferani, S.; Losson, R.; Dantzer, F. The histone subcode: Poly(ADP-ribose) polymerase-1 (Parp-1) and Parp-2 control cell differentiation by regulating the transcriptional intermediary factor TIF1beta and the heterochromatin protein HP1alpha. FASEB J. 2008, 22, 3853–3865. [Google Scholar] [CrossRef]
- Park, H.H.; Kim, H.R.; Park, S.Y.; Hwang, S.M.; Hong, S.M.; Park, S.; Kang, H.C.; Morgan, M.J.; Cha, J.H.; Lee, D.; et al. RIPK3 activation induces TRIM28 derepression in cancer cells and enhances the anti-tumor microenvironment. Mol. Cancer 2021, 20, 107. [Google Scholar] [CrossRef]
- Czerwińska, P.; Mazurek, S.; Wiznerowicz, M. The complexity of TRIM28 contribution to cancer. J. Biomed. Sci. 2017, 24, 63. [Google Scholar] [CrossRef]
- Cammas, F.; Herzog, M.; Lerouge, T.; Chambon, P.; Losson, R. Association of the transcriptional corepressor TIF1beta with heterochromatin protein 1 (HP1): An essential role for progression through differentiation. Genes Dev. 2004, 18, 2147–2160. [Google Scholar] [CrossRef]
- Fahrner, R.; Dennler, S.G.; Inderbitzin, D. Risk of malignancy in Caroli disease and syndrome: A systematic review. World J. Gastroenterol. 2020, 26, 4718–4728. [Google Scholar] [CrossRef]
- Harjai, M.M.; Bal, R.K. Caroli syndrome. Pediatr. Surg. Int. 2000, 16, 431–432. [Google Scholar] [CrossRef]
- Yüce, A.; Koçak, N.; Akhan, O.; Gürakan, F.; Ozen, H. Caroli’s syndrome in two siblings. Am. J. Gastroenterol. 2002, 97, 1855–1856. [Google Scholar] [CrossRef]
- Yoshizawa, K.; Kiyosawa, K.; Yabu, K.; Usuda, S.; Shimizu, S.; Fujimori, Y.; Mukawa, K.; Tanaka, E.; Sodeyama, T.; Furuta, S. Caroli’s disease in three siblings. Gastroenterol. Jpn. 1992, 27, 780–784. [Google Scholar] [CrossRef]
- Shenoy, P.; Zaki, S.A.; Shanbag, P.; Bhongade, S. Caroli’s syndrome with autosomal recessive polycystic kidney disease. Saudi J. Kidney Dis. Transpl. 2014, 25, 840–843. [Google Scholar] [CrossRef]
- Ribeiro, A.; Reddy, R.K.; Bernstein, D.; Jeffers, L.; Schiff, E.R. Caroli’s syndrome in twin sisters. Am. J. Gastroenterol. 1996, 91, 1024–1026. [Google Scholar]
- Ananthakrishnan, A.N.; Saeian, K. Caroli’s disease: Identification and treatment strategy. Curr. Gastroenterol. Rep. 2007, 9, 151–155. [Google Scholar] [CrossRef] [PubMed]
- Nakanuma, Y.; Harada, K.; Sato, Y.; Ikeda, H. Recent progress in the etiopathogenesis of pediatric biliary disease, particularly Caroli’s disease with congenital hepatic fibrosis and biliary atresia. Histol. Histopathol. 2010, 25, 223–235. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Sato, Y.; Yamamura, M.; Nakada, S.; Tamano, Y.; Sasaki, M.; Harada, K. Notch-Hes1 signaling activation in Caroli disease and polycystic liver disease. Pathol. Int. 2021, 71, 521–529. [Google Scholar] [CrossRef] [PubMed]
- Sgro, M.; Rossetti, S.; Barozzino, T.; Toi, A.; Langer, J.; Harris, P.C.; Harvey, E.; Chitayat, D. Caroli’s disease: Prenatal diagnosis, postnatal outcome and genetic analysis. Ultrasound Obstet. Gynecol. 2004, 23, 73–76. [Google Scholar] [CrossRef]
- Mavlikeev, M.; Titova, A.; Saitburkhanova, R.; Abyzova, M.; Sayfutdinov, I.; Gizzatullina, N.; Kotov, I.; Plaksa, I.; Isaev, A.; Abdulkhakov, S.; et al. Caroli syndrome: A clinical case with detailed histopathological analysis. Clin. J. Gastroenterol. 2019, 12, 106–111. [Google Scholar] [CrossRef]
- Jang, D.G.; Chae, H.; Shin, J.C.; Park, I.Y.; Kim, M.; Kim, Y. Prenatal diagnosis of autosomal recessive polycystic kidney disease by molecular genetic analysis. J. Obstet. Gynaecol. Res. 2011, 37, 1744–1747. [Google Scholar] [CrossRef]
- Mi, X.X.; Li, X.G.; Wang, Z.R.; Lin, L.; Xu, C.H.; Shi, J.P. Abernethy malformation associated with Caroli’s syndrome in a patient with a PKHD1 mutation: A case report. Diagn. Pathol. 2017, 12, 61. [Google Scholar] [CrossRef][Green Version]
- Tsuchida, Y.; Sato, T.; Sanjo, K.; Etoh, T.; Hata, K.; Terawaki, K.; Suzuki, I.; Kawarasaki, H.; Idezuki, Y.; Nakagome, Y.; et al. Evaluation of long-term results of Caroli’s disease: 21 years’ observation of a family with autosomal “dominant” inheritance, and review of the literature. Hepatogastroenterology 1995, 42, 175–181. [Google Scholar]
- Shedda, S.; Robertson, A. Caroli’s syndrome and adult polycystic kidney disease. ANZ J. Surg. 2007, 77, 292–294. [Google Scholar] [CrossRef]
- Onori, P.; Mancinelli, R.; Franchitto, A.; Carpino, G.; Renzi, A.; Brozzetti, S.; Venter, J.; Francis, H.; Glaser, S.; Jefferson, D.M.; et al. Role of follicle-stimulating hormone on biliary cyst growth in autosomal dominant polycystic kidney disease. Liver Int. 2013, 33, 914–925. [Google Scholar] [CrossRef]
- Mousson, C.; Rabec, M.; Cercueil, J.P.; Virot, J.S.; Hillon, P.; Rifle, G. Caroli’s disease and autosomal dominant polycystic kidney disease: A rare association? Nephrol. Dial. Transpl. 1997, 12, 1481–1483. [Google Scholar] [CrossRef]
- Kumar, A.; Akselrod, D.; Prikis, M. Caroli Disease Revisited: A Case of a Kidney Transplant Patient with Autosomal Polycystic Kidney Disease and Recurrent Episodes of Cholangitis. Transplant. Proc. 2019, 51, 541–544. [Google Scholar] [CrossRef]
- Wang, Z.; Ng, C.; Liu, X.; Wang, Y.; Li, B.; Kashyap, P.; Chaudhry, H.A.; Castro, A.; Kalontar, E.M.; Ilyayev, L.; et al. The ion channel function of polycystin-1 in the polycystin-1/polycystin-2 complex. EMBO Rep. 2019, 20, e48336. [Google Scholar] [CrossRef]
- Newby, L.J.; Streets, A.J.; Zhao, Y.; Harris, P.C.; Ward, C.J.; Ong, A.C. Identification, characterization, and localization of a novel kidney polycystin-1-polycystin-2 complex. J. Biol. Chem. 2002, 277, 20763–20773. [Google Scholar] [CrossRef]
- Halbritter, J.; Porath, J.D.; Diaz, K.A.; Braun, D.A.; Kohl, S.; Chaki, M.; Allen, S.J.; Soliman, N.A.; Hildebrandt, F.; Otto, E.A. Identification of 99 novel mutations in a worldwide cohort of 1,056 patients with a nephronophthisis-related ciliopathy. Hum. Genet. 2013, 132, 865–884. [Google Scholar] [CrossRef]
- Calinescu-Tuleasca, A.M.; Bottani, A.; Rougemont, A.L.; Birraux, J.; Gubler, M.C.; Le Coultre, C.; Majno, P.; Mentha, G.; Girardin, E.; Belli, D.; et al. Caroli disease, bilateral diffuse cystic renal dysplasia, situs inversus, postaxial polydactyly, and preauricular fistulas: A ciliopathy caused by a homozygous NPHP3 mutation. Eur. J. Pediatr. 2013, 172, 877–881. [Google Scholar] [CrossRef]
- Katsuyama, M.; Masuyama, T.; Komura, I.; Hibino, T.; Takahashi, H. Characterization of a novel polycystic kidney rat model with accompanying polycystic liver. Exp. Anim. 2000, 49, 51–55. [Google Scholar] [CrossRef]
- Sanzen, T.; Harada, K.; Yasoshima, M.; Kawamura, Y.; Ishibashi, M.; Nakanuma, Y. Polycystic kidney rat is a novel animal model of Caroli’s disease associated with congenital hepatic fibrosis. Am. J. Pathol. 2001, 158, 1605–1612. [Google Scholar] [CrossRef]
- Yasoshima, M.; Sato, Y.; Furubo, S.; Kizawa, K.; Sanzen, T.; Ozaki, S.; Harada, K.; Nakanuma, Y. Matrix proteins of basement membrane of intrahepatic bile ducts are degraded in congenital hepatic fibrosis and Caroli’s disease. J. Pathol. 2009, 217, 442–451. [Google Scholar] [CrossRef]
- Sato, Y.; Harada, K.; Kizawa, K.; Sanzen, T.; Furubo, S.; Yasoshima, M.; Ozaki, S.; Ishibashi, M.; Nakanuma, Y. Activation of the MEK5/ERK5 cascade is responsible for biliary dysgenesis in a rat model of Caroli’s disease. Am. J. Pathol. 2005, 166, 49–60. [Google Scholar] [CrossRef]
- Nakanuma, Y.; Sato, Y.; Harada, K. Tissue culture correlational study of genetic cholangiopathy of autosomal recessive polycystic kidney disease. Methods Mol. Biol. 2013, 945, 303–318. [Google Scholar] [CrossRef]
- Ng, D.W.; Chiow, A.K.; Poh, W.T.; Tan, S.S. Metachronous cholangiocarcinoma 13 years post resection of choledochal cyst-is long-term follow-up useful?: A case study and review of the literature. Surg. Case Rep. 2016, 2, 60. [Google Scholar] [CrossRef]
- Søreide, K.; Søreide, J.A. Bile duct cyst as precursor to biliary tract cancer. Ann. Surg. Oncol 2007, 14, 1200–1211. [Google Scholar] [CrossRef]
- Ronnekleiv-Kelly, S.M.; Soares, K.C.; Ejaz, A.; Pawlik, T.M. Management of choledochal cysts. Curr. Opin. Gastroenterol. 2016, 32, 225–231. [Google Scholar] [CrossRef]
- Sastry, A.V.; Abbadessa, B.; Wayne, M.G.; Steele, J.G.; Cooperman, A.M. What is the incidence of biliary carcinoma in choledochal cysts, when do they develop, and how should it affect management? World J. Surg. 2015, 39, 487–492. [Google Scholar] [CrossRef]
- Schwab, M.E.; Song, H.; Mattis, A.; Phelps, A.; Vu, L.T.; Huang, F.W.; Nijagal, A. De novo somatic mutations and KRAS amplification are associated with cholangiocarcinoma in a patient with a history of choledochal cyst. J. Pediatr. Surg. 2020, 55, 2657–2661. [Google Scholar] [CrossRef]
- Zou, S.; Li, J.; Zhou, H.; Frech, C.; Jiang, X.; Chu, J.S.; Zhao, X.; Li, Y.; Li, Q.; Wang, H.; et al. Mutational landscape of intrahepatic cholangiocarcinoma. Nat. Commun. 2014, 5, 5696. [Google Scholar] [CrossRef]
- Tian, W.; Hu, W.; Shi, X.; Liu, P.; Ma, X.; Zhao, W.; Qu, L.; Zhang, S.; Shi, W.; Liu, A.; et al. Comprehensive genomic profile of cholangiocarcinomas in China. Oncol. Lett. 2020, 19, 3101–3110. [Google Scholar] [CrossRef]
- Farshidfar, F.; Zheng, S.; Gingras, M.C.; Newton, Y.; Shih, J.; Robertson, A.G.; Hinoue, T.; Hoadley, K.A.; Gibb, E.A.; Roszik, J.; et al. Integrative Genomic Analysis of Cholangiocarcinoma Identifies Distinct IDH-Mutant Molecular Profiles. Cell Rep. 2017, 19, 2878–2880. [Google Scholar] [CrossRef]
- Böger, C.; Haag, J.; Egberts, J.H.; Röcken, C. Complex APC germline mutation associated metaplasia and intraepithelial neoplasia (CAM-IEN) of the gallbladder. Pathol. Res. Pract. 2016, 212, 54–58. [Google Scholar] [CrossRef]
- Mori, H.; Masahata, K.; Umeda, S.; Morine, Y.; Ishibashi, H.; Usui, N.; Shimada, M. Risk of carcinogenesis in the biliary epithelium of children with congenital biliary dilatation through epigenetic and genetic regulation. Surg. Today 2022, 52, 215–223. [Google Scholar] [CrossRef] [PubMed]
- Zhan, J.H.; Hu, X.L.; Dai, C.J.; Niu, J.; Gu, J.Q. Expressions of p53 and inducible nitric oxide synthase in congenital choledochal cysts. Hepatobiliary Pancreat. Dis. Int. 2004, 3, 120–123. [Google Scholar] [PubMed]
- Eraslan, G.; Avsec, Z.; Gagneur, J.; Theis, F.J. Deep learning: New computational modelling techniques for genomics. Nat. Rev. Genet. 2019, 20, 389–403. [Google Scholar] [CrossRef] [PubMed]
- Dias, R.; Torkamani, A. Artificial intelligence in clinical and genomic diagnostics. Genome Med. 2019, 11, 70. [Google Scholar] [CrossRef]
- Schmauch, B.; Romagnoni, A.; Pronier, E.; Saillard, C.; Maille, P.; Calderaro, J.; Kamoun, A.; Sefta, M.; Toldo, S.; Zaslavskiy, M.; et al. A deep learning model to predict RNA-Seq expression of tumours from whole slide images. Nat. Commun. 2020, 11, 3877. [Google Scholar] [CrossRef]


{kind=link}
| Chromosomal Anomaly | Localization | Phenotype Description | Type | Candidate Gene | References |
|---|---|---|---|---|---|
| Duplication | 17q12 | Diabetes, renal disorders, structural brain anomalies, etc. | Ia | HNF1B | [21] |
| Deletion | Microdeletion 17q12 | Diabetes, renal cysts, neurological and psychological anomalies, biliary or hepatic clinical phenotype | I, IV | HNF1B | [30] |
| Deletion | 2p15p16.1 | facial features, developmental delay Congenital microcephalyand intractable myoclonic epilepsy | II | EHBP1 | [34] |
| Deletion | 5q | Familial adenomatous polyposis | V | - | [14] |
| Translocation | t(3;8)(p23;q13) | Abdominal pain, jaundice | V | APC | [40] |
| Mutation | Disease | References |
|---|---|---|
| PKHD1 | CD + ARPKD | [16,20,79,84] |
| PKHD1 | CS + type II Abernethy malformation | [87] |
| PKD1, PKD2 | CD + ARPKD | [17,19,89,90,91,92] |
| WDR19/NPHP13 | CD + NPHP-RC | [95] |
| GLIS2/NPHP7 | CD + NPHP-RC | [95] |
| WDR19 | CD + NPHP 13 | [18] |
| NPHP3 | Ciliopathy malformation complex | [96] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ye, Y.; Lui, V.C.H.; Tam, P.K.H. Pathogenesis of Choledochal Cyst: Insights from Genomics and Transcriptomics. Genes 2022, 13, 1030. https://doi.org/10.3390/genes13061030
Ye Y, Lui VCH, Tam PKH. Pathogenesis of Choledochal Cyst: Insights from Genomics and Transcriptomics. Genes. 2022; 13(6):1030. https://doi.org/10.3390/genes13061030
Chicago/Turabian StyleYe, Yongqin, Vincent Chi Hang Lui, and Paul Kwong Hang Tam. 2022. "Pathogenesis of Choledochal Cyst: Insights from Genomics and Transcriptomics" Genes 13, no. 6: 1030. https://doi.org/10.3390/genes13061030
APA StyleYe, Y., Lui, V. C. H., & Tam, P. K. H. (2022). Pathogenesis of Choledochal Cyst: Insights from Genomics and Transcriptomics. Genes, 13(6), 1030. https://doi.org/10.3390/genes13061030