
Clinical Targeted Panel Sequencing Analysis in Clinical Evaluation of Children with Autism Spectrum Disorder in China

Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Participants and Case Review
2.2. Clinical Targeted Panel Sequencing, Data Processing, and Variant Classification
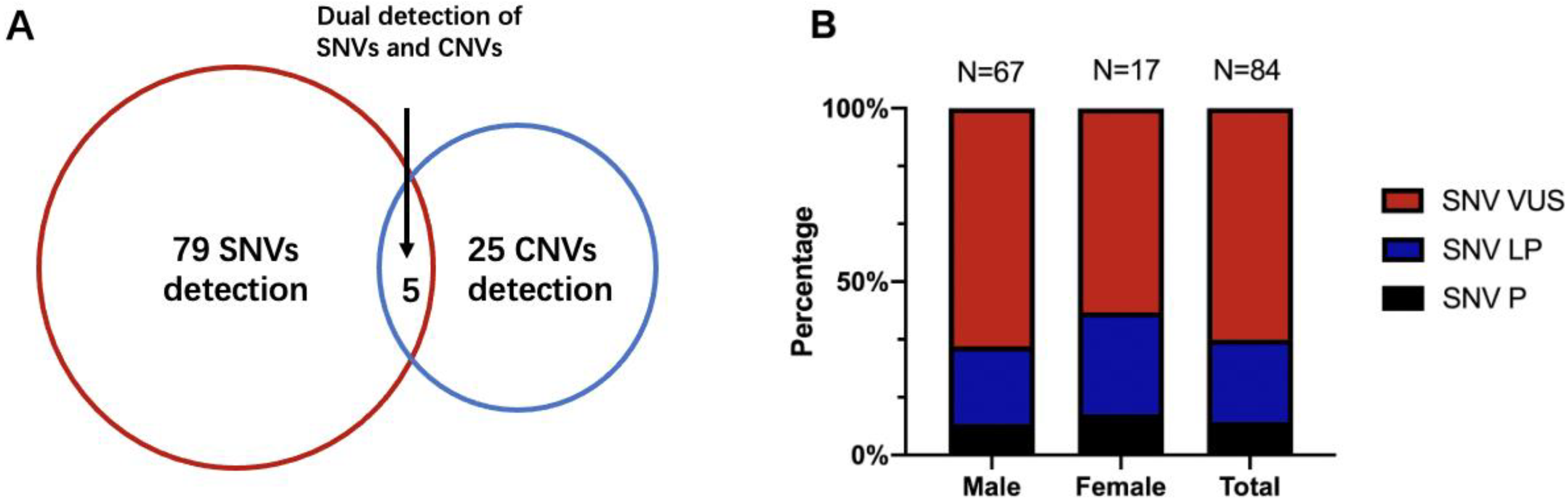
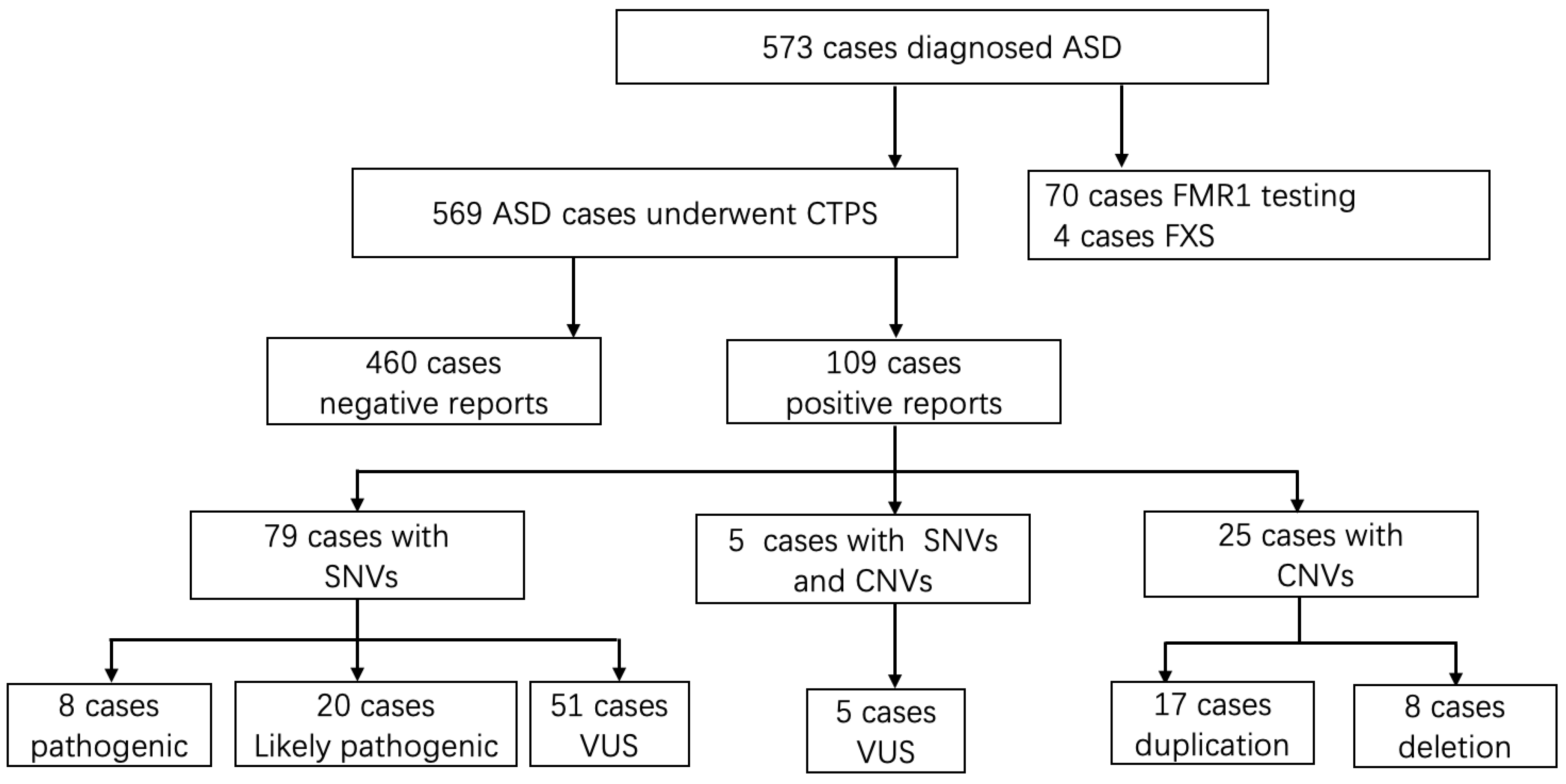
3. Results
3.1. Demographics and Clinical Files of Patients
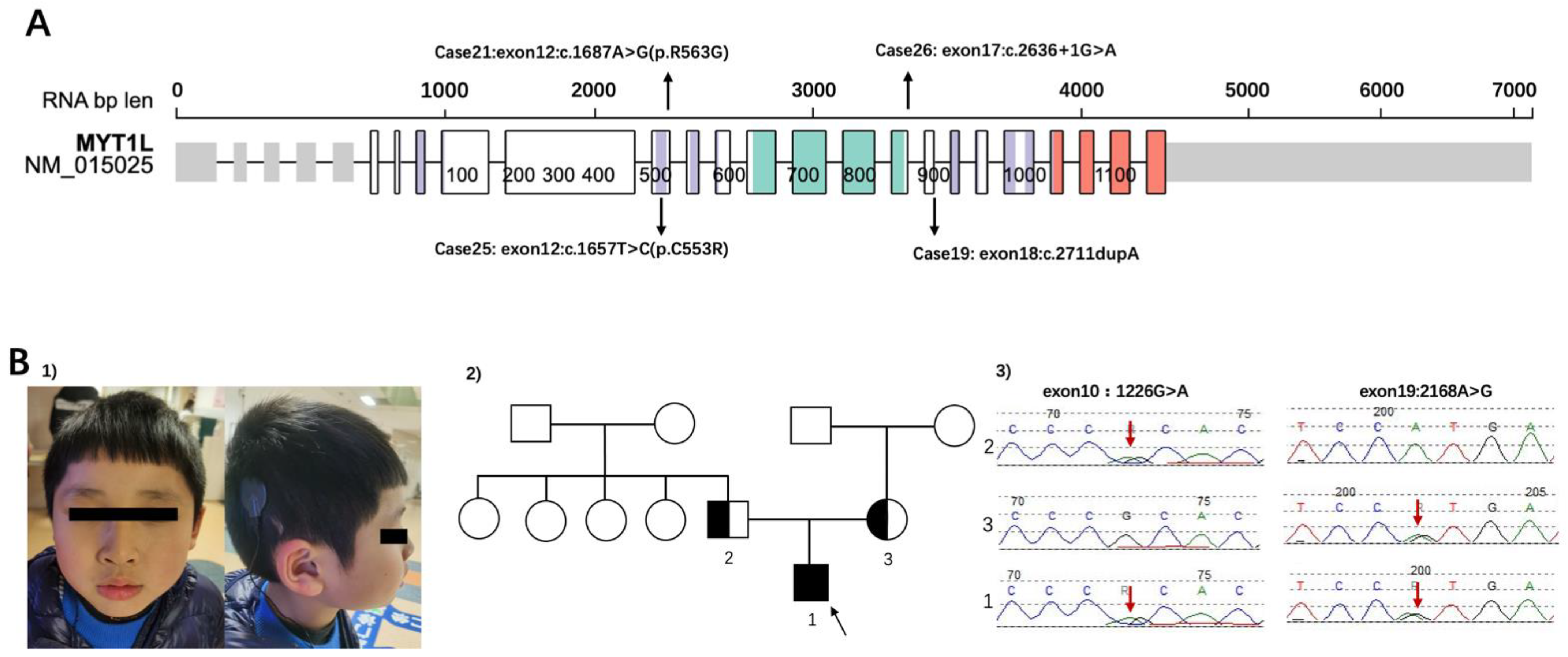
3.2. Representative Cases from CTPS
3.2.1. Summarized Cases: Analysis of Clinical Characteristics Classify Genetic Problems
3.2.2. Renewed Case: Genetic Diagnosis Should Also Focus on Clinical Manifestations
4. Discussion
4.1. Clinical Benefits and Limits of CTPS
4.2. Implications for Representative Cases from CTPS
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lord, C.; Elsabbagh, M.; Baird, G.; Veenstra-Vanderweele, J. Autism Spectrum Disorder. Lancet 2018, 392, 508–520. [Google Scholar] [CrossRef]
- Brugha, T.S.; Spiers, N.; Bankart, J.; Cooper, S.A.; McManus, S.; Scott, F.J.; Smith, J.; Tyrer, F. Epidemiology of Autism in Adults across Age Groups and Ability Levels. Br. J. Psychiatry 2016, 209, 498–503. [Google Scholar] [CrossRef] [Green Version]
- Lord, C.; Brugha, T.S.; Charman, T.; Cusack, J.; Dumas, G.; Frazier, T.; Jones, E.J.H.; Jones, R.M.; Pickles, A.; State, M.W.; et al. Autism Spectrum Disorder. Nat. Rev. Dis. Primers 2020, 6, 5. [Google Scholar] [CrossRef] [PubMed]
- Tick, B.; Bolton, P.; Happe, F.; Rutter, M.; Rijsdijk, F. Heritability of Autism Spectrum Disorders: A Meta-Analysis of Twin Studies. J. Child Psychol. Psychiatry 2016, 57, 585–595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woodbury-Smith, M.; Scherer, S.W. Progress in the Genetics of Autism Spectrum Disorder. Dev. Med. Child Neurol. 2018, 60, 445–451. [Google Scholar] [CrossRef] [PubMed]
- Sandin, S.; Lichtenstein, P.; Kuja-Halkola, R.; Larsson, H.; Hultman, C.M.; Reichenberg, A. The Familial Risk of Autism. JAMA 2014, 311, 1770–1777. [Google Scholar] [CrossRef]
- He, X.; Sanders, S.J.; Liu, L.; de Rubeis, S.; Lim, E.T.; Sutcliffe, J.S.; Schellenberg, G.D.; Gibbs, R.A.; Daly, M.J.; Buxbaum, J.D.; et al. Integrated Model of De Novo and Inherited Genetic Variants Yields Greater Power to Identify Risk Genes. PLoS Genet. 2013, 9, e1003671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hyman, S.L.; Levy, S.E.; Myers, S.M.; Section On Developmental Council On Children With Disabilities, and Pediatrics Behavioral. Identification, Evaluation, and Management of Children with Autism Spectrum Disorder. Pediatrics 2020, 145. [Google Scholar] [CrossRef] [Green Version]
- Volkmar, F.; Siegel, M.; Woodbury-Smith, M.; King, B.; McCracken, J.; State, M. Child American Academy of, and Issues Adolescent Psychiatry Committee on Quality. Practice Parameter for the Assessment and Treatment of Children and Adolescents with Autism Spectrum Disorder. J. Am. Acad. Child Adolesc. Psychiatry 2014, 53, 237–257. [Google Scholar] [CrossRef] [Green Version]
- Schaefer, G.B.; Mendelsohn, N.J. Professional Practice and Guidelines Committee. Clinical Genetics Evaluation in Identifying the Etiology of Autism Spectrum Disorders: 2013 Guideline Revisions. Genet. Med. 2013, 15, 399–407. [Google Scholar] [CrossRef] [Green Version]
- O’Roak, B.J.; Vives, L.; Fu, W.; Egertson, J.D.; Stanaway, I.B.; Phelps, I.G.; Carvill, G.; Kumar, A.; Lee, C.; Ankenman, K.; et al. Multiplex Targeted Sequencing Identifies Recurrently Mutated Genes in Autism Spectrum Disorders. Science 2012, 338, 1619–1622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srivastava, S.; Love-Nichols, J.A.; Dies, K.A.; Ledbetter, D.H.; Martin, C.L.; Chung, W.K.; Firth, H.V.; Frazier, T.; Hansen, R.L.; Prock, L.; et al. Correction: Meta-Analysis and Multidisciplinary Consensus Statement: Exome Sequencing Is a First-Tier Clinical Diagnostic Test for Individuals with Neurodevelopmental Disorders. Genet. Med. 2020, 22, 1731–1732. [Google Scholar] [CrossRef] [PubMed]
- Lowther, C.; Valkanas, E.; Giordano, J.L.; Wang, H.Z.; Currall, B.B.; O’Keefe, K.; Collins, R.L.; Zhao, X.; Austin-Tse, C.A.; Evangelista, E.; et al. Systematic Evaluation of Genome Sequencing as a First-Tier Diagnostic Test for Prenatal and Pediatric Disorders. bioRxiv 2020. [Google Scholar] [CrossRef]
- Coury, D. Medical Treatment of Autism Spectrum Disorders. Curr. Opin. Neurol. 2010, 23, 131–136. [Google Scholar] [CrossRef] [PubMed]
- APAd American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, 5th ed.; American Psychiatric Association: Washington, DC, USA, 2013. [Google Scholar]
- Dong, X.; Liu, B.; Yang, L.; Wang, H.; Wu, B.; Liu, R.; Chen, H.; Chen, X.; Yu, S.; Chen, B.; et al. Clinical Exome Sequencing as the First-Tier Test for Diagnosing Developmental Disorders Covering Both Cnv and Snv: A Chinese Cohort. J. Med. Genet. 2020, 57, 558–566. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Kong, Y.; Dong, X.; Hu, L.; Lin, Y.; Chen, X.; Ni, Q.; Lu, Y.; Wu, B.; Wang, H.; et al. Clinical and Genetic Spectrum of a Large Cohort of Children with Epilepsy in China. Genet. Med. 2019, 21, 564–571. [Google Scholar] [CrossRef]
- McLaren, W.; Pritchard, B.; Rios, D.; Chen, Y.; Flicek, P.; Cunningham, F. Deriving the Consequences of Genomic Variants with the Ensembl Api and Snp Effect Predictor. Bioinformatics 2010, 26, 2069–2070. [Google Scholar] [CrossRef]
- Wang, K.; Li, M.; Hakonarson, H. Annovar: Functional Annotation of Genetic Variants from High-Throughput Sequencing Data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef]
- Qian, Q.I.N.; Bo, L.I.U.; Lin, Y.A.N.G.; Bing-bing, W.U.; Hui-jun, W.A.N.G.; Xin-Ran, D.O.N.G.; Yu-lan, L.U.; Wen-Hao, Z.H.O.U. Application of Copy Number Variation Screening Analysis Process Based on High Throughput Sequencing Technology. Chin. J. Evid. Based Pediatrics 2018, 13, 275–279. [Google Scholar]
- Lord, C.M.; Rutter, P.D.; Labore, S.; Risi, K.; Gotham, B.S. Autism Diagnostic Observation Schedule, (Ados-2), 2nd ed.; Western Pschological Services: Los Angeles, CA, USA, 2012. [Google Scholar]
- David, W. Wechsler Preschool and Primary Scale of Intelligence, 4th ed.; Pearson: London, UK, 2012. [Google Scholar]
- Kim, Y.; An, J.Y. Spatio-Temporal Roles of Asd-Associated Variants in Human Brain Development. Genes 2020, 11, 535. [Google Scholar] [CrossRef]
- Sanders, S.J.; He, X.; Willsey, A.J.; Ercan-Sencicek, A.G.; Samocha, K.E.; Cicek, A.E.; Murtha, M.T.; Bal, V.H.; Bishop, S.L.; Dong, S.; et al. Insights into Autism Spectrum Disorder Genomic Architecture and Biology from 71 Risk Loci. Neuron 2015, 87, 1215–1233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- An, J.Y.; Sanders, S.J. Appreciating the Population-Wide Impact of Copy Number Variants on Cognition. Biol. Psychiatry 2017, 82, 78–80. [Google Scholar] [CrossRef] [PubMed]
- Satterstrom, F.K.; Kosmicki, J.A.; Wang, J.; Breen, M.S.; De Rubeis, S.; An, J.Y.; Peng, M.; Collins, R.; Grove, J.; Klei, L.; et al. Large-Scale Exome Sequencing Study Implicates Both Developmental and Functional Changes in the Neurobiology of Autism. Cell 2020, 180, 568–584.e23. [Google Scholar] [CrossRef]
- Choi, L.; An, J.Y. Genetic Architecture of Autism Spectrum Disorder: Lessons from Large-Scale Genomic Studies. Neurosci. Biobehav. Rev. 2021, 128, 244–257. [Google Scholar] [CrossRef]
- Werling, D.M.; Brand, H.; An, J.Y.; Stone, M.R.; Zhu, L.; Glessner, J.T.; Collins, R.L.; Dong, S.; Layer, R.M.; Markenscoff-Papadimitriou, E.; et al. An Analytical Framework for Whole-Genome Sequence Association Studies and Its Implications for Autism Spectrum Disorder. Nat. Genet. 2018, 50, 727–736. [Google Scholar] [CrossRef]
- Tammimies, K.; Marshall, C.R.; Walker, S.; Kaur, G.; Thiruvahindrapuram, B.; Lionel, A.C.; Yuen, R.; Uddin, M.; Roberts, W.; Weksberg, R.; et al. Molecular Diagnostic Yield of Chromosomal Microarray Analysis and Whole-Exome Sequencing in Children with Autism Spectrum Disorder. JAMA 2015, 314, 895–903. [Google Scholar] [CrossRef]
- Ho, K.S.; Wassman, E.R.; Baxter, A.L.; Hensel, C.H.; Martin, M.M.; Prasad, A.; Twede, H.; Vanzo, R.J.; Butler, M.G. Chromosomal Microarray Analysis of Consecutive Individuals with Autism Spectrum Disorders Using an Ultra-High Resolution Chromosomal Microarray Optimized for Neurodevelopmental Disorders. Int. J. Mol. Sci. 2016, 17, 2070. [Google Scholar] [CrossRef] [Green Version]
- Rossi, M.; El-Khechen, D.; Black, M.H.; Hagman, K.D.F.; Tang, S.; Powis, Z. Outcomes of Diagnostic Exome Sequencing in Patients with Diagnosed or Suspected Autism Spectrum Disorders. Pediatr. Neurol. 2017, 70, 34–43.e2. [Google Scholar] [CrossRef] [Green Version]
- Stefanski, A.; Calle-López, Y.; Leu, C.; Pérez-Palma, E.; Pestana-Knight, E.; Lal, D. Clinical Sequencing Yield in Epilepsy, Autism Spectrum Disorder, and Intellectual Disability: A Systematic Review and Meta-Analysis. Epilepsia 2021, 62, 143–151. [Google Scholar] [CrossRef]
- Aspromonte, M.C.; Bellini, M.; Gasparini, A.; Carraro, M.; Bettella, E.; Polli, R.; Cesca, F.; Bigoni, S.; Boni, S.; Carlet, O.; et al. Characterization of Intellectual Disability and Autism Comorbidity through Gene Panel Sequencing. Hum. Mutat. 2020, 41, 1183. [Google Scholar] [CrossRef]
- Kim, J.G.; Armstrong, R.C.; Agoston, D.V.; Robinsky, A.; Wiese, C.; Nagle, J.; Hudson, L.D. Myelin Transcription Factor 1 (Myt1) of the Oligodendrocyte Lineage, Along with a Closely Related Cchc Zinc Finger, Is Expressed in Developing Neurons in the Mammalian Central Nervous System. J. Neurosci. Res. 1997, 50, 272–290. [Google Scholar] [CrossRef] [Green Version]
- Werling, D.M.; Pochareddy, S.; Choi, J.; An, J.-Y.; Sheppard, B.; Peng, M.; Li, Z.; Dastmalchi, C.; Santpere, G.; Sousa, A.M.; et al. Whole-Genome and Rna Sequencing Reveal Variation and Transcriptomic Coordination in the Developing Human Prefrontal Cortex. Cell Rep. 2020, 31, 107489. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Shao, Q.; Li, Z.; Gonzalez, G.A.; Lu, F.; Wang, D.; Pu, Y.; Huang, A.; Zhao, C.; He, C.; et al. Myt1l Promotes Differentiation of Oligodendrocyte Precursor Cells and Is Necessary for Remyelination after Lysolecithin-Induced Demyelination. Neurosci. Bull. 2018, 34, 247–260. [Google Scholar] [CrossRef] [PubMed]
- Coursimault, J.; Guerrot, A.M.; Morrow, M.M.; Schramm, C.; Zamora, F.M.; Shanmugham, A.; Liu, S.; Zou, F.; Bilan, F.; Le Guyader, G.; et al. Myt1l-Associated Neurodevelopmental Disorder: Description of 40 New Cases and Literature Review of Clinical and Molecular Aspects. Hum. Genet. 2021, 141, 65–80. [Google Scholar] [CrossRef] [PubMed]
- De Rocker, N.; Vergult, S.; Koolen, D.; Jacobs, E.; Hoischen, A.; Zeesman, S.; Bang, B.; Béna, F.; Bockaert, N.; Bongers, E.M.; et al. Refinement of the Critical 2p25.3 Deletion Region: The Role of Myt1l in Intellectual Disability and Obesity. Genet. Med. 2015, 17, 460–466. [Google Scholar] [CrossRef] [Green Version]
- Windheuser, I.C.; Becker, J.; Cremer, K.; Hundertmark, H.; Yates, L.M.; Mangold, E.; Peters, S.; Degenhardt, F.; Ludwig, K.U.; Zink, A.M.; et al. Nine Newly Identified Individuals Refine the Phenotype Associated with Myt1l Mutations. Am. J. Med. Genet. A 2020, 182, 1021–1031. [Google Scholar] [CrossRef] [Green Version]
- Blanchet, P.; Bebin, M.; Bruet, S.; Cooper, G.M.; Thompson, M.L.; Duban-Bedu, B.; Gerard, B.; Piton, A.; Suckno, S.; Deshpande, C.; et al. Myt1l Mutations Cause Intellectual Disability and Variable Obesity by Dysregulating Gene Expression and Development of the Neuroendocrine Hypothalamus. PLoS Genet. 2017, 13, e1006957. [Google Scholar] [CrossRef] [Green Version]
- Carvalho, L.M.; D’Angelo, C.S.; Mustacchi, Z.; da Silva, I.T.; Krepischi, A.C.V.; Koiffmann, C.P.; Rosenberg, C. A Novel Myt1l Mutation in a Boy with Syndromic Obesity: Case Report and Literature Review. Obes. Res. Clin. Pract. 2021, 15, 124–132. [Google Scholar] [CrossRef]
- Loid, P.; Makitie, R.; Costantini, A.; Viljakainen, H.; Pekkinen, M.; Makitie, O. A Novel Myt1l Mutation in a Patient with Severe Early-Onset Obesity and Intellectual Disability. Am. J. Med. Genet. A 2018, 176, 1972–1975. [Google Scholar] [CrossRef]
- Al Tuwaijri, A.; Alfadhel, M. Myt1l Mutation in a Patient Causes Intellectual Disability and Early Onset of Obesity: A Case Report and Review of the Literature. J. Pediatr. Endocrinol. Metab. 2019, 32, 409–413. [Google Scholar] [CrossRef]
- Everett, L.A.; Glaser, B.; Beck, J.C.; Idol, J.R.; Buchs, A.; Heyman, M.A.; Adawi, F.; Hazani, E.; Nassir, E.; Baxevanis, A.D.; et al. Pendred Syndrome Is Caused by Mutations in a Putative Sulphate Transporter Gene (Pds). Nat. Genet. 1997, 17, 411–422. [Google Scholar] [CrossRef] [PubMed]
- Fagerberg, L.; Hallström, B.M.; Oksvold, P.; Kampf, C.; Djureinovic, D.; Odeberg, J.; Habuka, M.; Tahmasebpoor, S.; Danielsson, A.; Edlund, K.; et al. Analysis of the Human Tissue-Specific Expression by Genome-Wide Integration of Transcriptomics and Antibody-Based Proteomics. Mol. Cell. Proteom. 2014, 13, 397–406. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
{kind=link}
| Case | Sex | Classification | Gene | Position | Mutation | Zygosity | Inherited/ de Novo | Inheritance Pattern | Population Frequency |
|---|---|---|---|---|---|---|---|---|---|
| Pathogenic | |||||||||
| 1 | M | P | SHANK3 | chr22:51159988 | NM_033517:exon21:c.3727C>T(p.Q1243X) | Het | AD | De novo | NA |
| 2 | M | P | TRIP12 | chr2:230744756 | NM_004238:exon2:c.40C>T(p.R14X) | Het | AD | De novo | NA |
| 3 | M | P | NF1 | chr17:29552152 | NM_000267:exon17:c.1885G>A(p.G629R) | Het | AD | De novo | 3.98 × 10−6 |
| 4 | M | P | RAB39B/ NDST1/ NDST1 | chrX:154493531/ chr5:149907553/ chr5:149929268 | NM_171998:exon1:c.43G>C(p.G15R)/ NM_001543:exon3:c.701C>T(p.T234I)/ NM_001543:exon13:c.2345G>T(p.R782L) | Hemi/ Het/ Het | X-linked/ AR/ AR | NA | NA/ NA/ 4.24 × 10-5 |
| 5 | M | P | TCF20 | chr22:42608728 | NM_005650:exon1:c.2582_2583del | Het | AD | De novo | NA |
| 6 | M | P | SLC26A4/ SLC26A4 | chr7:107330645/ chr7:107350577 | NM_000441:exon10:c.1226G>A(p.R409H)/NM_0004411:exon19:c.2168A>G(p.H723R) | Het/ Het | AR/ AR | Paternal/ Maternal | 9.57 × 10−5/ 1.13 × 10−4 |
| 7 | F | P | SHANK3 | chr22:51158733 | NM_033517:exon21:c.2475dupC | Het | AD | De novo | NA |
| 8 | F | P | MECP2 | chrX:153296399 | NM_004992:exon4:c.880C>T(p.R294X) | Het | XLD/XLR | NA | NA/ |
| Likely pathogenic | |||||||||
| 9 | M | LP | BRAF | chr7:140453146 | NM_004333:exon15:c.1789C>G(p.L597V) | Het | AD | NA | NA |
| 10 | M | LP | DIP2B | chr12:51128855 | NM_173602:exon34:c.4044-1G>A | Het | AD | NA | NA |
| 11 | M | LP | KDM6B | chr17:7752387_7752400 | NM_001080424:exon11:c.2781_2794del14;p.(Val928Hisfs*2) | Het | AD | NA | NA |
| 12 | M | LP | SCN2A | chr2:166201068 | NM_021007:exon16:c.2566C>T(p.R856X) | Het | AD | NA | NA |
| 13 | M | LP | STXBP1 | chr9:130432185 | NM_003165:exon11:c.913dupC | Het | AD/AR | Maternal | NA |
| 14 | M | LP | CHD8 | chr14:21871817 | NM_001170629:exon16:c.3312delT | Het | AD | De novo | NA |
| 15 | M | LP | DEAF1 | chr11:687941 | NM_021008:exon4:c.634G>A(p.G212S) | Het | AD/AR | Maternal | NA |
| 16 | M | LP | AP1S2 | chrX:15870644 | NM_003916:exon2:c.4C>T(p.Q2X) | Hemi | XLR | Maternal | NA |
| 17 | M | LP | EHMT1/ FGFR3 | chr9:140678599/ chr4:1807288 | NM_001145527:exon16:c.2424G>A(p.W808X)/ NM_000142:exon12:c.1537G>A(p.D513N) | Het/ Het | AD/ AD/AR | Maternal/ Maternal | NA/ 4.61 × 10−5 |
| 18 | M | LP | ABCD1 | chrX:152991143 | NM_000033:exon1:c.422C>T(p.A141V) | Hemi | XLR | NA | NA |
| 19 | M | LP | MYT1L | chr2:1890310 | NM_015025:exon18:c.2711dupA | Het | AD | De novo | NA |
| 20 | M | LP | DYRK1A | chr21:38865398 | NM_001396:exon7:c.1031_1037del | Het | AD | NA | NA |
| 21 | M | LP | MYT1L | chr2:1915808 | NM_015025:exon12:c.1687A>G(p.R563G) | Het | AD | De novo | NA |
| 22 | M | LP | BCL11A | chr2:60689252 | NM_022893:exon4:c.793_794delCT | Het | AD | De novo | NA |
| 23 | M | LP | ARID2 | chr12:46244889 | NM_152641:exon15:c.2983C>T(p.Q995X) | Het | AD | De novo | NA |
| 24* | F | LP | MECP2 | chrX:153296399 | NM_004992:exon4:c.880C>T(p.R294X) | Het | XLD/XLR | De novo | 1.64 × 10−5 |
| 25 | F | LP | MYT1L | chr2:1915838 | NM_015025:exon12:c.1657T>C(p.C553R) | Het | AD | De novo | NA |
| 26 | F | LP | MYT1L | chr2:1891259 | NM_015025:exon17:c.2636+1G>A | Het | AD | De novo | NA |
| 27 | F | LP | SHANK3 | chr22:51159684 | NM_033517:exon21:c.3424_3425delCT | Het | AD | NA | NA |
| 28 | F | LP | SLC2A1 | chr1:43395422 | NM_006516:exon6:c.709G>A(p.V237M) | Het | AD/AR | NA | 7.95 × 10-6 |
| Variant of unknown significance (VUS) | |||||||||
| 29 | M | VUS | TRIO | chr5:14508475 | NM_007118:exon57:c.9238C>T(p.R3080X) | Het | AD | Paternal | NA |
| 30 | M | VUS | TRIO | chr5:14474205 | NM_007118:exon40:c.6082G>A(p.D2028N) | Het | AD | NA | 3.98 × 10−6 |
| 31 | M | VUS | DCLRE1C/DCLRE1C/KANK1/ KIF1A | chr10:14976777/ chr10:14981850/ chr9:712617/ chr2:241702608 | NM_001033855:exon7:c.465-3C>T/ NM_001033855:exon4:c.265A>G(p.T89A)/ NM_015158:exon3:c.1852del/ NM_004321:exon19:c.1897G>A(p.D633N) | Het/ Het/ Het/ Het | AR/ AR/ AD/ AD/AR | NA | NA/ 2.85 × 10−5/ NA/ NA |
| 32 | M | VUS | SCN8A | chr12:52080904 | NM_014191:exon5:c.515A>T(p.E172V) | Het | AD | Maternal | NA |
| 33 | M | VUS | KIF7 | chr15:90172648 | NM_198525:exon17:c.3472_3474delAAG | Hom | AR | NA | 8.13 × 10−6 |
| 34 | M | VUS | SMARCA4 | chr19:11105513 | NM_001128849:exon9:c.1429A>G(p.N477D) | Het | AD | De novo | NA |
| 35 | M | VUS | MED13L | chr12:116418709 | NM_015335:exon23:c.5210A>G(p.K1737R) | Het | AD | NA | NA |
| 36 | M | VUS | SKI/ WDR81/ WDR81 | chr1:2161179/ chr17:1633670/ chr17:1636940 | NM_003036:exon1:c.969+5G>C/ NM_001163809:exon2:c.3668-4G>A/ NM_001163809:exon7:c.4609G>A(p.G1537S) | Het/ Het/ Het | AD/ AR/ AR | De novo/ Maternal/ Paternal | NA/ 4.34 × 10−5/ 4.14 × 10−5 |
| 37 | M | VUS | MAOA | chrX:43552577 | NM_000240:exon3:c.208G>A(p.V70M) | Hemi | XLR | Maternal | NA |
| 38 | M | VUS | NCAPD3/NCAPD3 | chr11:134022951/ chr11:134051019 | NM_015261:exon35:c.4389-4C>G/ NM_015261:exon20:c.2512A>G(p.I838V) | Het/ Het | AR/ AR | Paternal/ Maternal | NA/ 1.59 × 10−5 |
| 39 | M | VUS | IQSEC2 | chrX:53285098 | NM_001111125:exon3:c.883C>T(p.R295W) | Hemi | XLD | NA | NA |
| 40 | M | VUS | DYRK1A | chr21:38884272 | NM_001396:exon11:c.1730T>A(p.V577D) | Het | AD | NA | NA |
| 41 | M | VUS | PIGV/ PIGV | chr1:27121024/ chr1:27121133 | NM_017837:exon3:c.499G>A(p.G167S)/ NM_017837:exon3:c.608G>T(p.R203L) | Het/ Het | AR/ AR | NA | 3.54 × 10−5/ NA |
| 42 | M | VUS | FOXP2 | chr7:114174736 | NM_014491:exon3:c.233G>C(p.S78T) | Het | AD | NA | NA |
| 43 | M | VUS | HCFC1 | chrX:153219744 | NM_005334:exon17:c.4106T>C(p.M1369T) | Hemi | XLR | Maternal | NA |
| 44 | M | VUS | DYRK1A | chr21:38850489 | NM_001396:exon3:c.214C>G(p.P72A) | Het | AD | NA | NA |
| 45 | M | VUS | PTEN | chr10:89717672 | NM_000314:exon7:c.697C>T(p.R233X) | Het | AD/AR | NA | NA |
| 46 | M | VUS | SLITRK1 | chr13:84455351 | NM_052910:exon1:c.292G>A(p.V98I) | Het | AD | NA | NA |
| 47 | M | VUS | PLA2G6/ PLA2G6 | chr22:38516880/ chr22:38528924 | NM_003560:exon12:c.1628G>A(p.R543H)/ NM_003560:exon7:c.991G>T(p.D331Y) | Het/ Het | AR/ AR | NA | 2.83 × 10−5/ 3.57 × 10−5 |
| 48 | M | VUS | PHIP | chr6:79727249 | NM_017934:exon11:c.1046T>A(p.F349Y) | Het | AD | Paternal | NA |
| 49 | M | VUS | CTNNB1/ HCFC1/ SOS1 | chr3:41266085/ chrX:153217162/ chr2:39216457 | NM_001904:exon3:c.82C>G(p.Q28E)/ NM_005334:exon21:c.5261-4C>T/ NM_005633:exon21:c.3347-2A>G | Het/ Hemi/ Het | AD/ XLR/ AD | NA | NA/ 1.85 × 10−5/ NA |
| 50 | M | VUS | GRIA3 | chrX:122318409 | NM_000828:exon1:c.22G>A(p.G8R) | Hemi | XLR | NA | NA |
| 51 | M | VUS | COL4A3BP | chr5:74712810 | NM_001130105:exon8:c.1112G>A(p.G371E) | Het | AD | NA | NA |
| 52 | M | VUS | DPP6 | chr7:154585802 | NM_001936:exon11:c.964A>G(P.T322A) | Het | AD | NA | NA |
| 53 | M | VUS | PHIP/ ACVR1 | chr6:79672916/ chr2:158626989 | NM_017934:exon30:c.3433A>G(p.R1145G)/ NM_001105:exon7:c.681G>A(p.W227X) | Het/ Het | AD/ AD | NA | NA/ NA |
| 54 | M | VUS | SETBP1 | chr18:42531731 | NM_015559:exon4:c.2426A>G(p.Q809R) | Het | AD | Paternal | NA |
| 55 | M | VUS | FOXP1 | chr3:71015071 | NM_032682:exon20:c.1859G>A(p.S620N) | Het | AD | NA | NA |
| 56 | M | VUS | PHIP | chr6:79665392 | NM_017934:exon33:c.3790A>G(p.T1264A) | Het | AD | Paternal | NA |
| 57 | M | VUS | DDX3X/ DLG3 | chrX:41196685/ chrX:69719742 | NM_001193416:exon2:c.70T>G(p.S24A)/ NM_021120:exon16:c.1988G>A(p.R663Q) | Hemi/ Hemi | XL/ XL | Maternal/ Maternal | NA/ NA |
| 58 | M | VUS | GNAI3/ USP27X | chr1:110121866/ chrX:49645815 | NM_006496:exon4:c.344A>G(p.E115G)/ NM_001145073:exon1:c.905T>C(p.L302S) | Het/ Hemi | AD/ XL | NA | NA/ NA |
| 59 | M | VUS | TMLHE | chrX:154743783 | NM_018196:exon4:c.502C>T(p.Q168X) | Hemi | XLR | NA | NA |
| 60 | M | VUS | BRWD3 | chrX:80064545 | NM_153252:exon3:c.91-4T>C | Hemi | XLR | NA | NA |
| 61 | M | VUS | USP9X | chrX:41029747 | NM_001039590:exon20:c.2902A>C(p.I968L) | Hemi | dominant/XLR | Maternal | NA |
| 62 | M | VUS | FOXP1 | chr3:71037180 | NM_032682:exon14:c.1111G>A(p.V371M) | Het | AD | NA | NA |
| 63 | M | VUS | DIP2B | chr12:51074491 | NM_173602:exon9:c.1151C>T(p.T384I) | Het | AD | NA | 2.12 × 10−5 |
| 64 | M | VUS | L1CAM | chrX:153133875 | NM_000425:exon13:c.1585G>A(p.E529K) | Hemi | XLR | NA | NA |
| 65 | M | VUS | SHANK3 | chr22:51169394 | NM_033517:exon22:c.4850C>T(p.P1617L) | Het | AD | NA | NA |
| 66 | M | VUS | DIP2B | chr12:51068356 | NM_173602:exon6:c.740T>C(p.I247T) | Het | AD | NA | 3.18 × 10−5 |
| 67 | M | VUS | GRIA3 | chrX:122551611 | NM_000828:exon11:c.1859G>C(p.G620A) | Hemi | XLR | NA | NA |
| 68 | M | VUS | AFF2/ TRIO | chrX:147743835/ chr5:14387875 | NM_002025:exon3:c.587T>C(p.F196S)/ NM_007118:exon23:c.3800G>A(p.S1267N) | Hemi/ Het | XLR/AD | NA | NA/ 3.98 × 10−6 |
| 69 | M | VUS | FOXP2 | chr7:114303569 | NM_014491:exon15:c.1834T>A(p.L612M) | Het | AD | NA | NA |
| 70 | M | VUS | ARID1B/ CHD7 | chr6:157521844/ chr8:61654295 | NM_020732:exon18:c.4116C>A(p.Y1372X)/ NM_017780:exon2:c.304C>T(p.H102Y) | Het/ Het | AD/ AD | De novo/ Paternal | NA/ NA |
| 71 * | M | VUS | CTNNB1/ KLHL15 | chr3:41279547/ chrX:24006703 | NM_001904:exon14:c.2117C>A(p.P706H)/ NM_030624:exon4:c.1150G>A(p.V384I) | Het/ Hemi | AD/ XLR | NA | NA/ 5.48 × 10−6 |
| 72 * | M | VUS | RPS6KA3 | chrX:20284690 | NM_004586:exon1:c.61A>G(p.S21G) | Hemi | XLD | NA | NA |
| 73 * | M | VUS | KIF1A/ ZC4H2 | chr2:241700653/ chrX:64137775 | NM_004321:exon22:c.2231A>G(p.K744R)/ NM_018684:exon5:c.563C>T(p.A188V) | Het/ Hemi | AD/AR/ XLR | De novo/ Maternal | NA/ NA |
| 74 * | M | VUS | FBN2/ SPTAN1 | chr5:127625581/ chr9:131389713 | NM_001999:exon51:c.6503delC/ NM_001130483:exon50:c.6625G>A(p.D2209N) | Het/ Het | AD/ AD | NA | NA/ 6.34 × 10−6 |
| 75 | F | VUS | SHANK3 | chr22:51142293 | NM_033517:exon13:c.1618C>T(p.R540W) | Het | AD | NA | NA |
| 76 | F | VUS | GPR98/ GPR98 | chr5:89954046/ chr5:90074281 | NM_032119:exon21:c.4703G>A(p.S1568N)/ NM_032119:exon63:c.12704A>G(p.Y4235C) | Het/ Het | AD/AR/Digenic/ AD/AR/Digenic | NA | 5.46 × 10−5/ 1.61 × 10−4 |
| 77 | F | VUS | PPP2R5D | chr6:42974971 | NM_006245:exon5:c.560C>T(p.S187L) | Het | AD | NA | NA |
| 78 | F | VUS | MECP2 | chrX:153296071 | NM_004992:exon4:c.1158_1201del | Het | XLD/XLR | NA | NA |
| 79 | F | VUS | SETBP1 | chr18:42529856 | NM_015559:exon4:c.551G>T(p.R184M) | Het | AD | NA | NA |
| 80 | F | VUS | SETD5 | chr3:9512347 | NM_001080517:exon19:c.2929T>A(p.F977I) | Het | AD | Paternal | NA |
| 81 | F | VUS | HUWE1/ LRP2/ LRP2 | chrX:53574690/ chr2:170030607/ chr2:170081950 | NM_031407:exon68:c.10580T>C(p.V3527A)/ NM_004525:exon56:c.10836G>T(p.Q3612H)/ NM_004525:exon33:c.5406_5407del | Het/ Het/ Het | XL/ AR/ AR | NA | NA/ NA NA |
| 82 | F | VUS | ERCC2/ ERCC2/ ASXL3 | chr19:45868096-45868099/ chr19:45856520/ chr18:31322948 | NM_000400:exon7:c.591_594del/ NM_000400:exon18:c.1738G>A(p.A508T)/ NM_030632:exon12:c.3136G>A(p.G1046R) | Het/ Het/ Het | AR/ AR/ AD | Paternal/Maternal/Maternal | 1.20 × 10−5/ 1.59 × 10−5/ 1.61 × 10−5 |
| 83 | F | VUS | FOXP1 | chr3:71247424 | NM_032682:exon6:c.109T>C(p.S37P) | Het | AD | NA | NA |
| 84 * | F | VUS | DCX | chrX:110574270 | NM_178153:exon5:c.809-1G>C | Het | XL | NA | NA |
| Case | Sex | Band | Chr | Start(hg19) | Stop(hg19) | Size(kb) | Deletion/Duplication |
|---|---|---|---|---|---|---|---|
| 1 * | M | 15q13.2–15q13.3 | chr15 | 30,653,442 | 32,464,722 | 1,811,280 | deletion |
| 2 * | M | 20p12.1–20p13 | chr20 | 740,723 | 13,799,067 | 13,058,344 | duplication |
| 3 * | M | 16p11.2 | chr16 | 29,802,039 | 30,200,397 | 398,358 | deletion |
| 4 * | M | Xq28 | chrX | 153,576,898 | 153,780,404 | 203,506 | duplication |
| 5 * | F | 15q11.2–15q13.1 | chr15 | 23,043,276 | 28,327,041 | 5,283,765 | duplication |
| 6 | M | 7p13–7p14.1 | chr7 | 41,724,711 | 44,748,665 | 3,023,954 | duplication |
| 7 | M | 15q13.3 | chr15 | 32,064,983 | 32,443,563 | 378,580 | duplication |
| 8 | M | 15q11.2–15q13.1 | chr15 | 23,043,276 | 28,327,041 | 5,283,765 | duplication |
| 9 | M | 2q24.3–2q25.1 | chr2 | 9,628,275 | 16,087,129 | 6,458,854 | duplication |
| 10 | M | 15q11.2–15q13.1 | chr15 | 23,043,276 | 28,327,041 | 5,283,765 | duplication |
| 11 | M | 3q29 | chr3 | 196,195,653 | 197,024,106 | 828,453 | deletion |
| 12 | M | 1p21.2–1p21.3 | chr1 | 97,543,298 | 100,715,390 | 3,172,092 | duplication |
| 13 | M | 15q13.2–15q13.3 | chr15 | 30,659,620 | 32,464,722 | 1,805,102 | duplication |
| 14 | M | 7q36.1–7q36.3 | chr7 | 150,642,048 | 157,210,133 | 6,568,085 | deletion |
| 15 | M | Xp21.1 | chrX | 32,235,032 | 32,235,180 | 148 | deletion |
| 16 | M | 19p13.2–q13.3 | chr19 | 43,370,615 | 43,530,621 | 160,006 | deletion |
| 17 | M | 10q22.3–10q23.2 | chr10 | 81,697,495 | 88,854,623 | 7,157,128 | duplication |
| 18 | M | 4q35.1–q 35.2 | chr4 | 186,421,813 | 190,873,442 | 4,451,629 | deletion |
| 19 | M | 6q16.1–6q16.3 | chr6 | 97,337,188 | 105,307,794 | 7,970,606 | deletion |
| 20 | M | 15q11.2–15q13.1 | chr15 | 23,043,276 | 28,327,041 | 5,283,765 | duplication |
| 21 | M | 3q29 | chr3 | 195,776,154 | 197,300,194 | 1,524,040 | deletion |
| 22 | M | 1p34.3 | chr1 | 36,974,539 | 38,129,928 | 1,155,389 | duplication |
| 23 | F | 22q13.31–22q13.33 | chr22 | 45,680,862 | 51,171,726 | 5,490,864 | deletion |
| 24 | F | 14q21.1 | chr14 | 39,559,493 | 39,665,452 | 105,959 | duplication |
| 25 | F | 2q37.3 | chr2 | 240,016,194 | 242,708,226 | 2,692,032 | deletion |
| 26 | F | 17p11.2 | chr17 | 16,664,738 | 20,370,848 | 3,706,110 | duplication |
| 27 | F | 10q22.3–q23.2 | chr10 | 81,697,495 | 88,854,623 | 7,157,128 | duplication |
| 28 | F | 22q11.21 | chr22 | 18,900,293 | 21,245,506 | 2,345,213 | duplication |
| 29 | F | 2q37.12q37.3 | chr2 | 234,408,524 | 242,844,702 | 8,436,178 | deletion |
| 30 | F | p21.1 | chrX | 32,305,645 | 32,632,570 | 326,925 | duplication |
| Autism Spectrum Disorder | Language Delay | Motor Delay | Developmental Disorder/ Intellectual Disability | Stereotypic Hand Movements | Abnormal Sensory Processing | Hypotonia | Overweight/ Obesity | |
|---|---|---|---|---|---|---|---|---|
| Our study | 100% (4/4) | 100% (4/4) | 100% (4/4) | 100% (4/4) | 75% (3/4) | 100% (4/4) | 75% (3/4) | 75% (3/4) |
| Coursimault et al.‘s [ [37] | 43% (17/40) | 95% (38/40) | 78% (31/40) | 70% (21/30) | - | - | 47% (18/38) | 58% (23/40) |
| De Rocker et al.’s [38] | 32% (7/22) | 100% (22/22) | - | 100% (22/22) | 14% (3/22) | - | - | 74% (14/19) |
| Windheuser et al.’s [39] | 22% (2/9) | - | 87% (7/8) | 100% (8/8) | - | Mentioned in 1 patient | 78% (7/9) | 33% (3/9) |
| Blanchet et al.’s [40] | 44% (4/9) | 100% (9/9) | 100% (8/8) | - | - | - | Mentioned in 2 patients | 66.7% (6/9) |
| Carvalho et al.’s [41] | 0 | 100% (1/1) | - | 100% (1/1) | - | - | - | 100% (1/1) |
| Loid et al.’s [42] | 0 | 100% (1/1) | 100% (1/1) | 100% (1/1) | - | - | - | 100% (1/1) |
| Al Tuwaijri et al.’s [43] | 100% (1/1) | 100% (1/1) | 100% (1/1) | 100% (1/1) | - | - | 100% (1/1) | 100% (1/1) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hu, C.; He, L.; Li, H.; Ding, Y.; Zhang, K.; Li, D.; Zhu, G.; Wu, B.; Xu, X.; Xu, Q. Clinical Targeted Panel Sequencing Analysis in Clinical Evaluation of Children with Autism Spectrum Disorder in China. Genes 2022, 13, 1010. https://doi.org/10.3390/genes13061010
Hu C, He L, Li H, Ding Y, Zhang K, Li D, Zhu G, Wu B, Xu X, Xu Q. Clinical Targeted Panel Sequencing Analysis in Clinical Evaluation of Children with Autism Spectrum Disorder in China. Genes. 2022; 13(6):1010. https://doi.org/10.3390/genes13061010
Chicago/Turabian StyleHu, Chunchun, Linlin He, Huiping Li, Yanhua Ding, Kaifeng Zhang, Dongyun Li, Guoqing Zhu, Bingbing Wu, Xiu Xu, and Qiong Xu. 2022. "Clinical Targeted Panel Sequencing Analysis in Clinical Evaluation of Children with Autism Spectrum Disorder in China" Genes 13, no. 6: 1010. https://doi.org/10.3390/genes13061010
APA StyleHu, C., He, L., Li, H., Ding, Y., Zhang, K., Li, D., Zhu, G., Wu, B., Xu, X., & Xu, Q. (2022). Clinical Targeted Panel Sequencing Analysis in Clinical Evaluation of Children with Autism Spectrum Disorder in China. Genes, 13(6), 1010. https://doi.org/10.3390/genes13061010