
Transcriptomic Profile Analysis of Populus talassica × Populus euphratica Response and Tolerance under Salt Stress Conditions



Abstract
:1. Introduction
2. Materials and Methods
2.1. Plants and Stress Treatment
2.2. RNA Extraction, Library Construction, and Sequencing
2.3. Data Quality Control and Reference Genome Alignment
2.4. Quantification of Gene Expression and Differential Expression Analysis
2.5. Novel Gene Prediction
2.6. Gene Ontology (GO) Enrichment Analysis and Kyoto Encyclopedia of Genes and Genomes (KEGG) Pathway Enrichment Analysis
2.7. Mutation Locus Analysis
3. Results
3.1. Sequencing Quality Analysis
3.2. Reference Genome Alignment Results Were Analyzed
3.3. Quantitative Analysis of Gene Expression
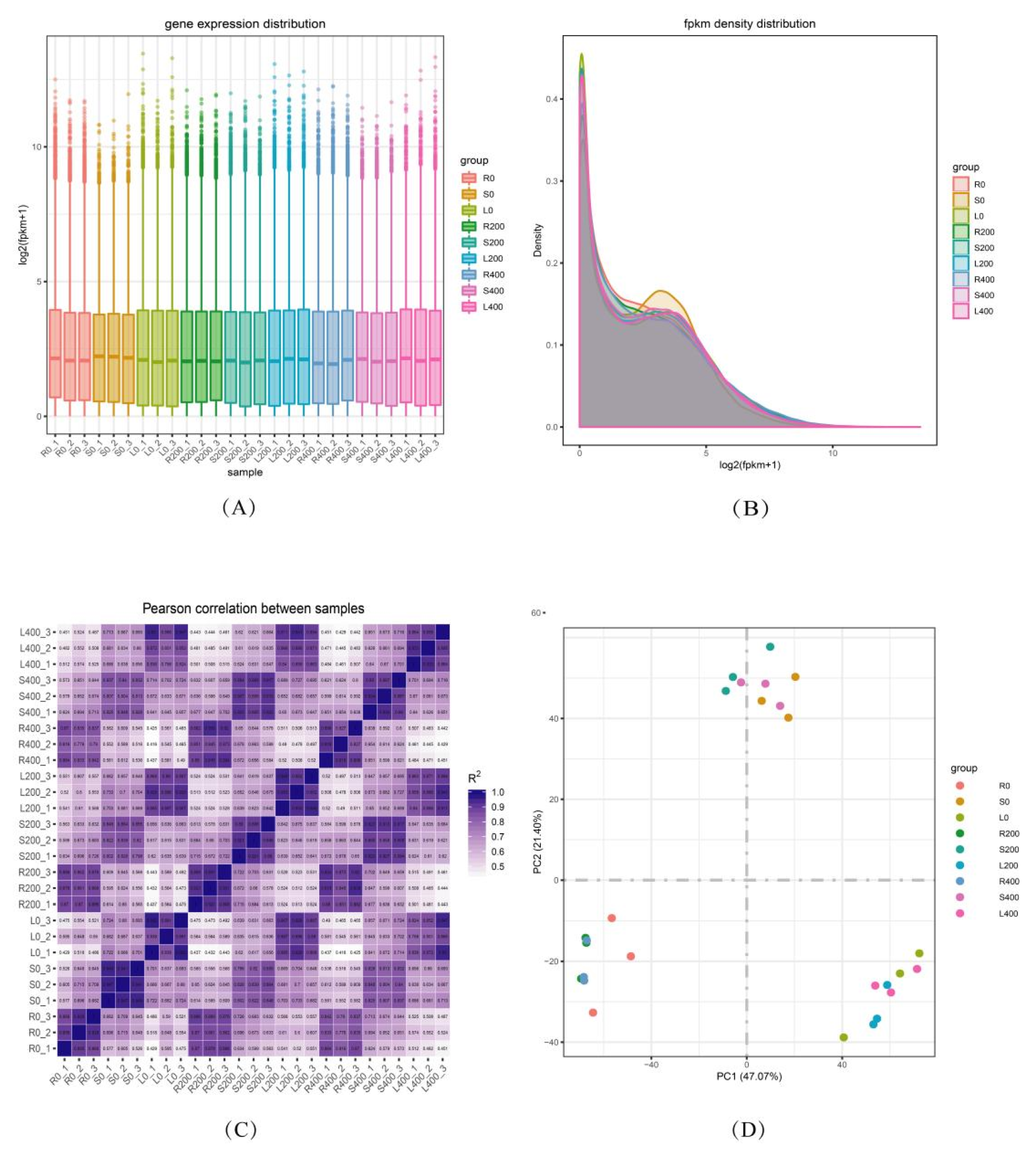
3.3.1. Gene Expression Distribution
3.3.2. FPKM Density Distribution
3.3.3. Correlation Analysis between Samples
3.4. Analysis of Differential Gene Expression Levels
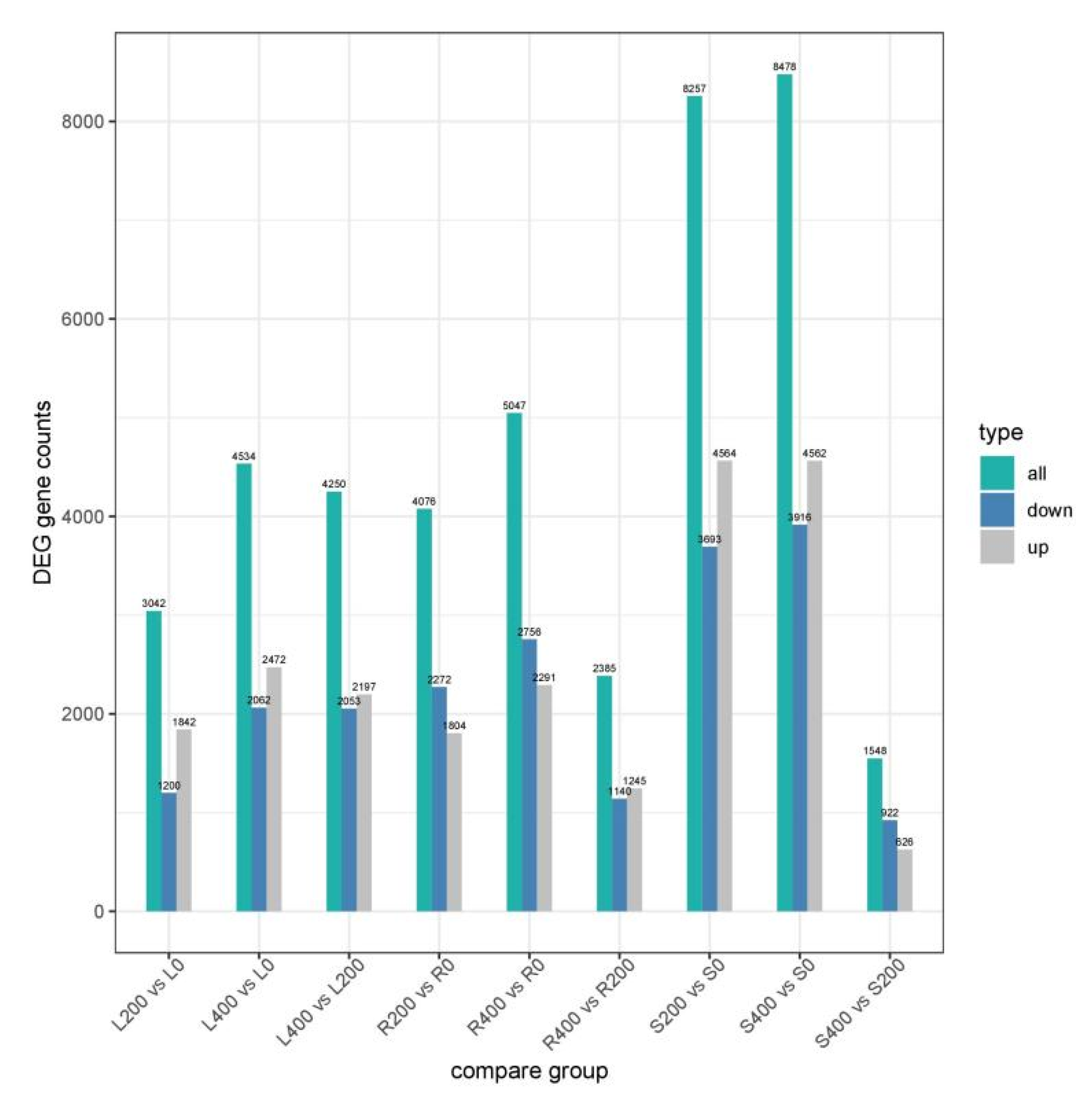
3.4.1. DEGs in Different Comparison Groups
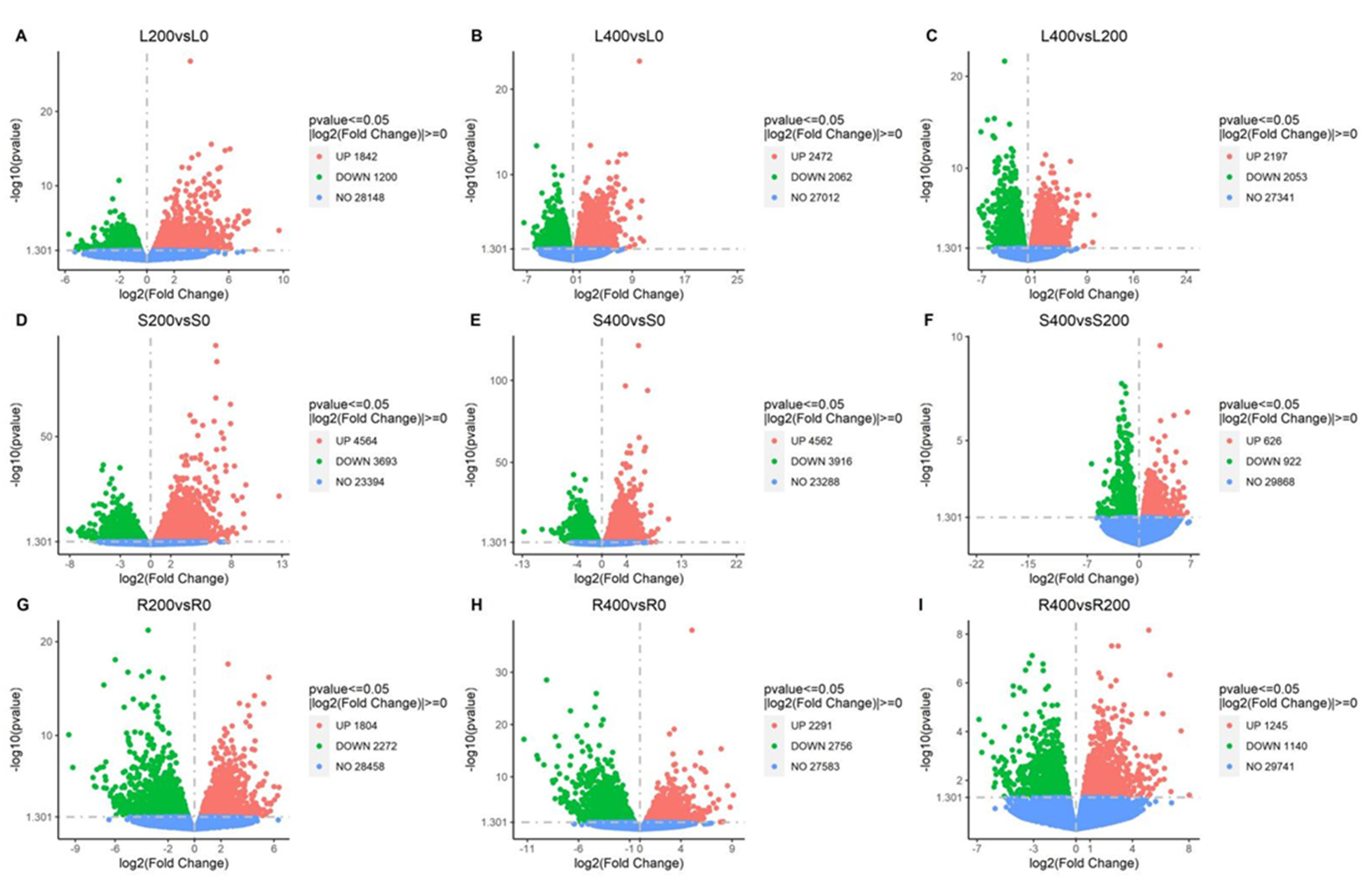
3.4.2. Differential Gene Volcano Map
Volcano Map of Differential Gene Expression Levels in Leaf Comparison Groups
Volcano Map of Differential Gene Expression Levels in Stem Comparison Groups
Volcanic Map of Differential Gene Expression Levels in Root Comparison Groups
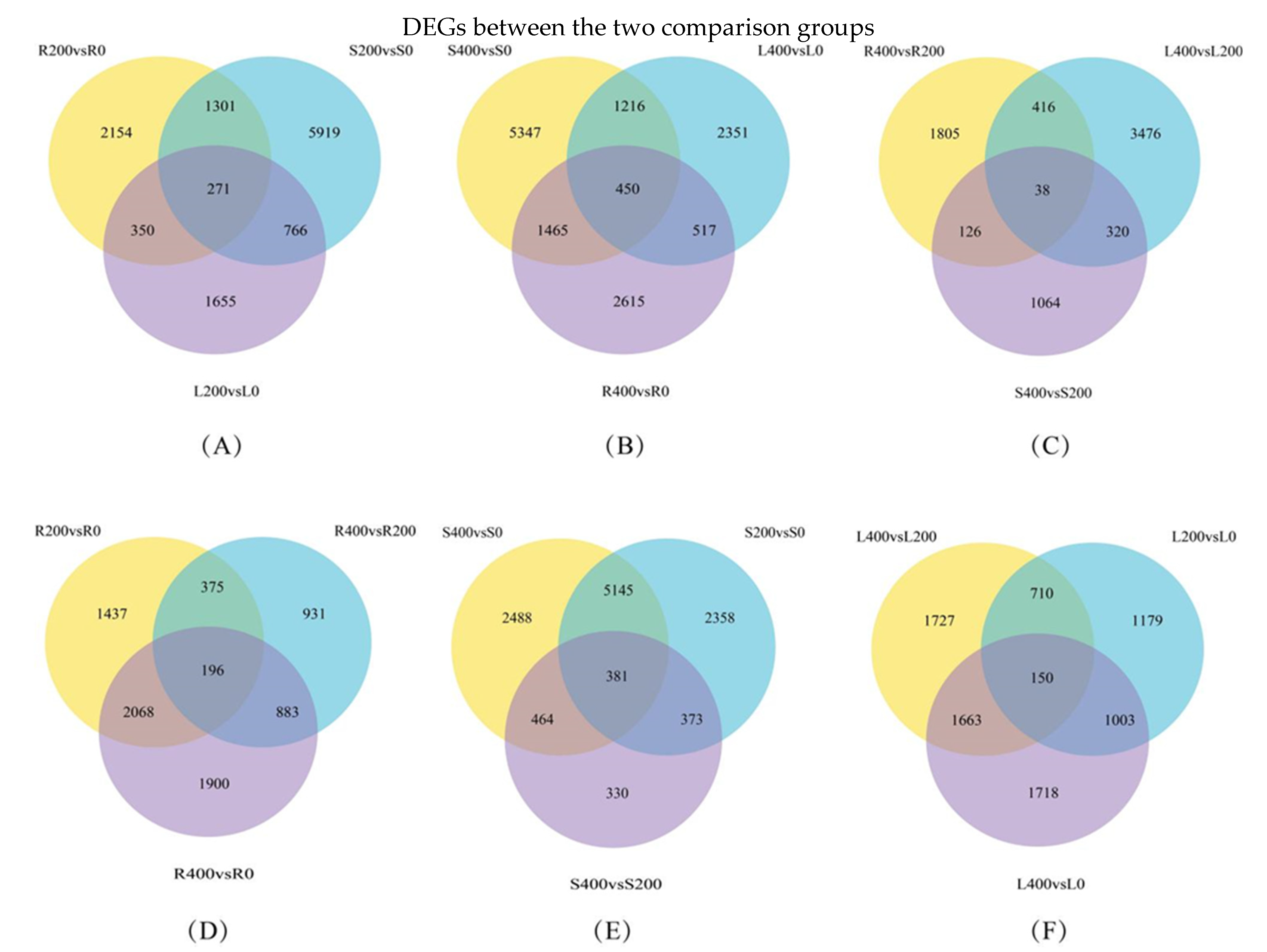
3.4.3. Differential Gene Venn Diagram
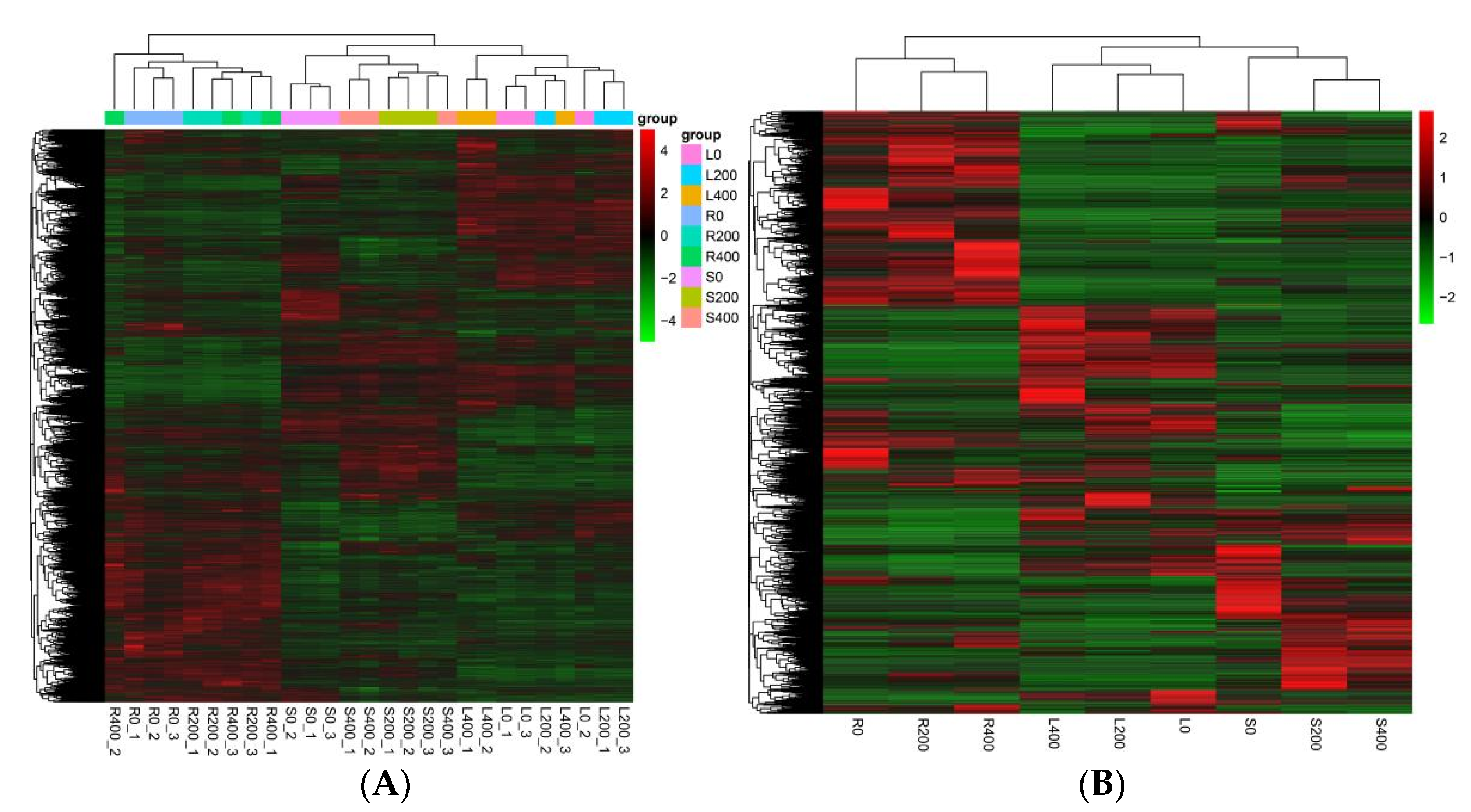
3.4.4. Cluster Analysis of DEGs
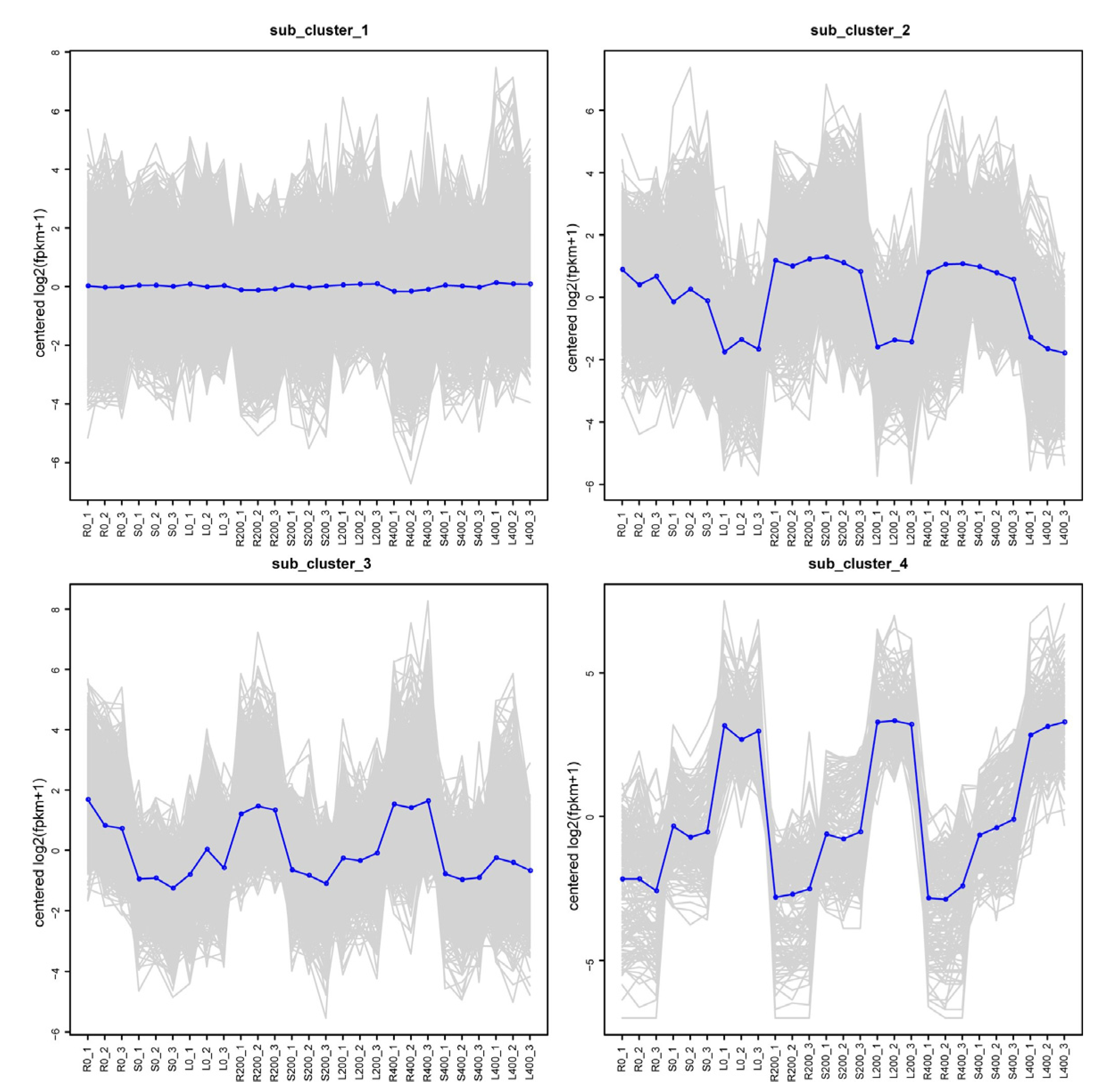
3.4.5. Trend Analysis
3.4.6. Novel Gene Prediction Results
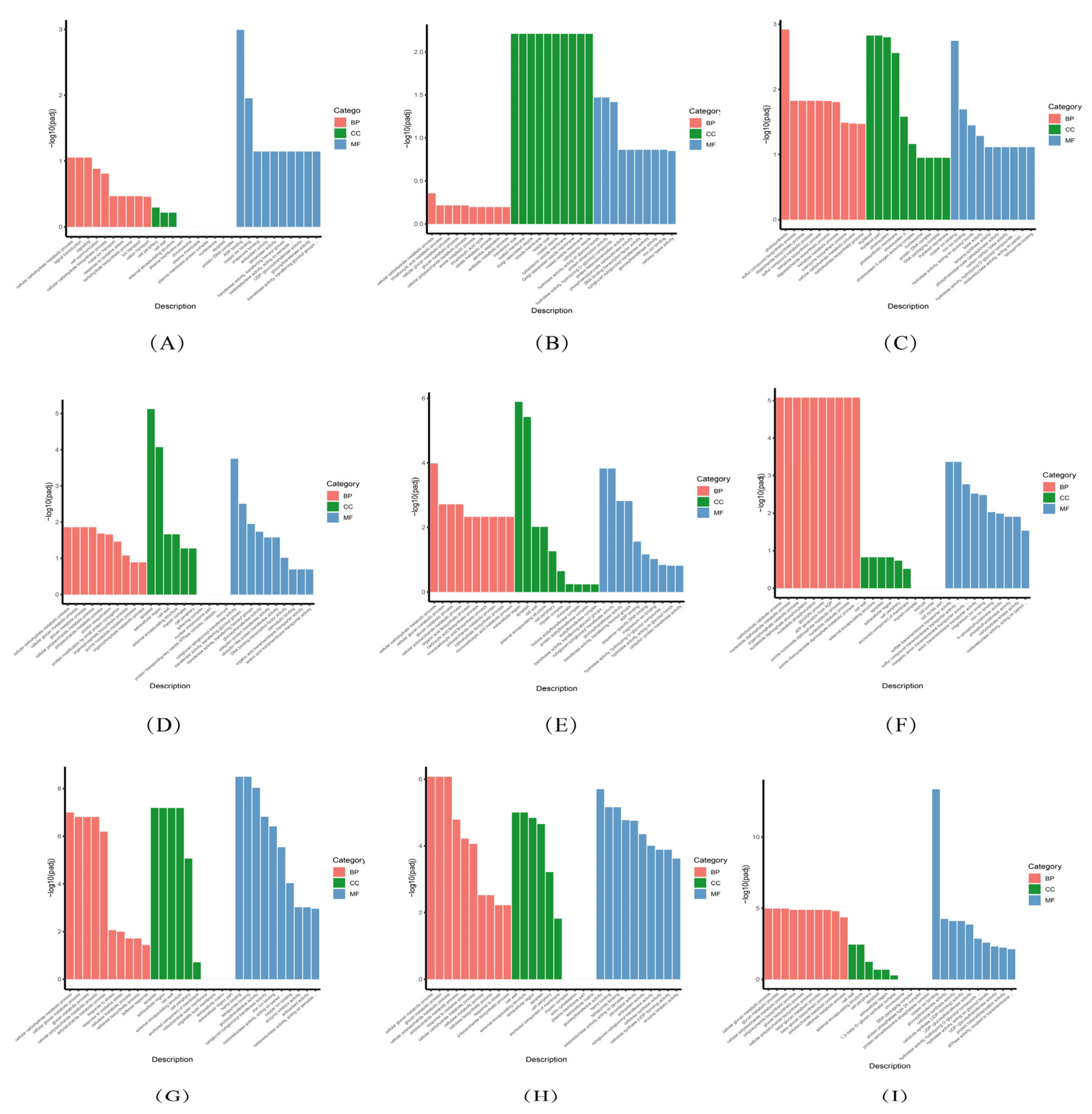
3.5. GO Enrichment Analysis of DEGs
3.5.1. GO Enrichment Analysis Diagram of DEGs in Each Leaf Comparison Group
3.5.2. GO Enrichment Analysis Diagram of DEGs in Each Stem Comparison Group
3.5.3. GO Enrichment Analysis Diagram of DEGs in Each Root Comparison Group
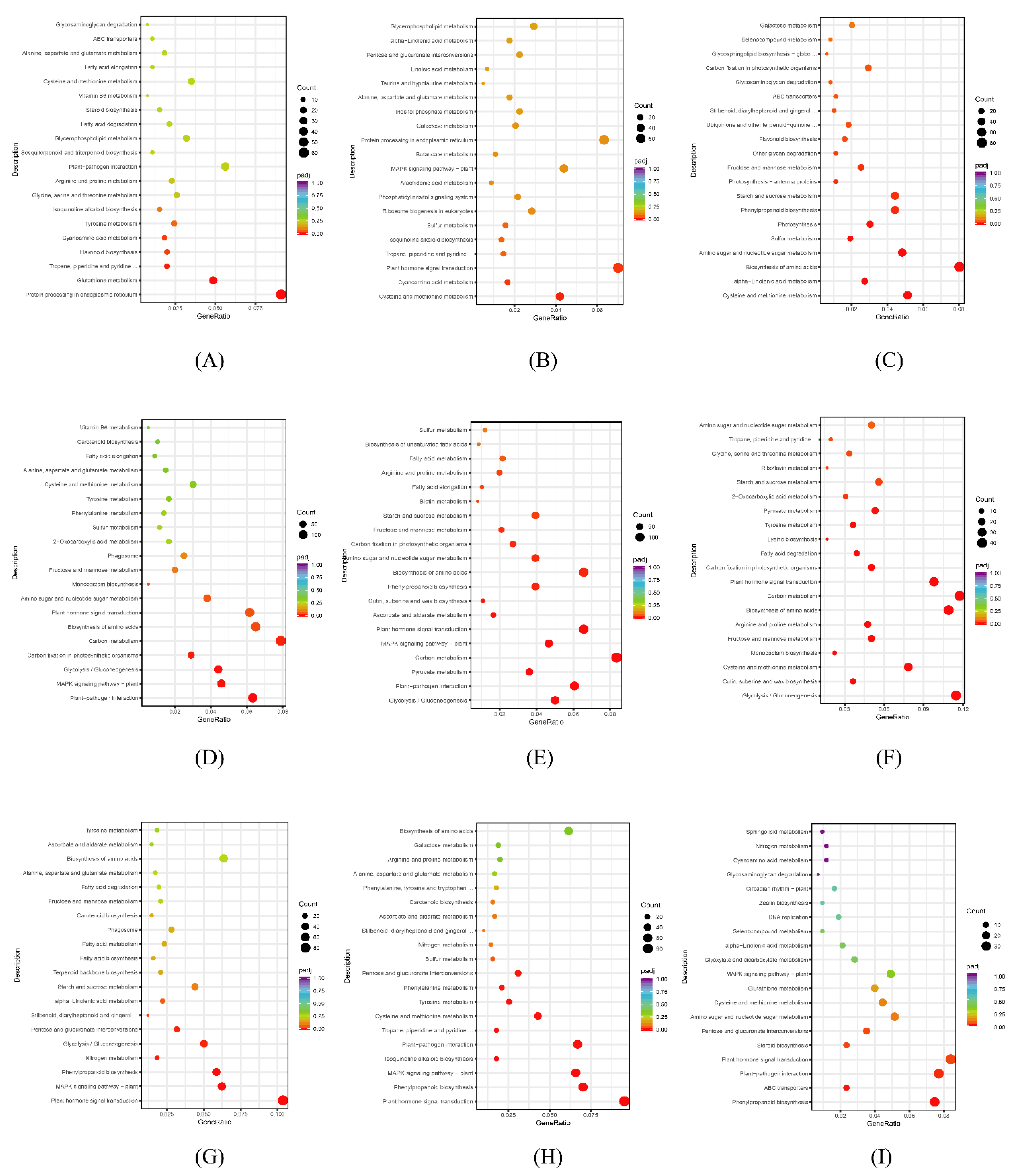
3.6. KEGG Pathway Enrichment Analysis of DEGs
3.6.1. KEGG Pathway Enrichment Analysis Diagram of DEGs in Each Leaf Comparison Group
3.6.2. KEGG Pathway Enrichment Analysis Diagram of DEGs in Each Comparison Group of Stem
3.6.3. KEGG Pathway Enrichment Analysis Diagram of DEGs in Each Root Comparison Group
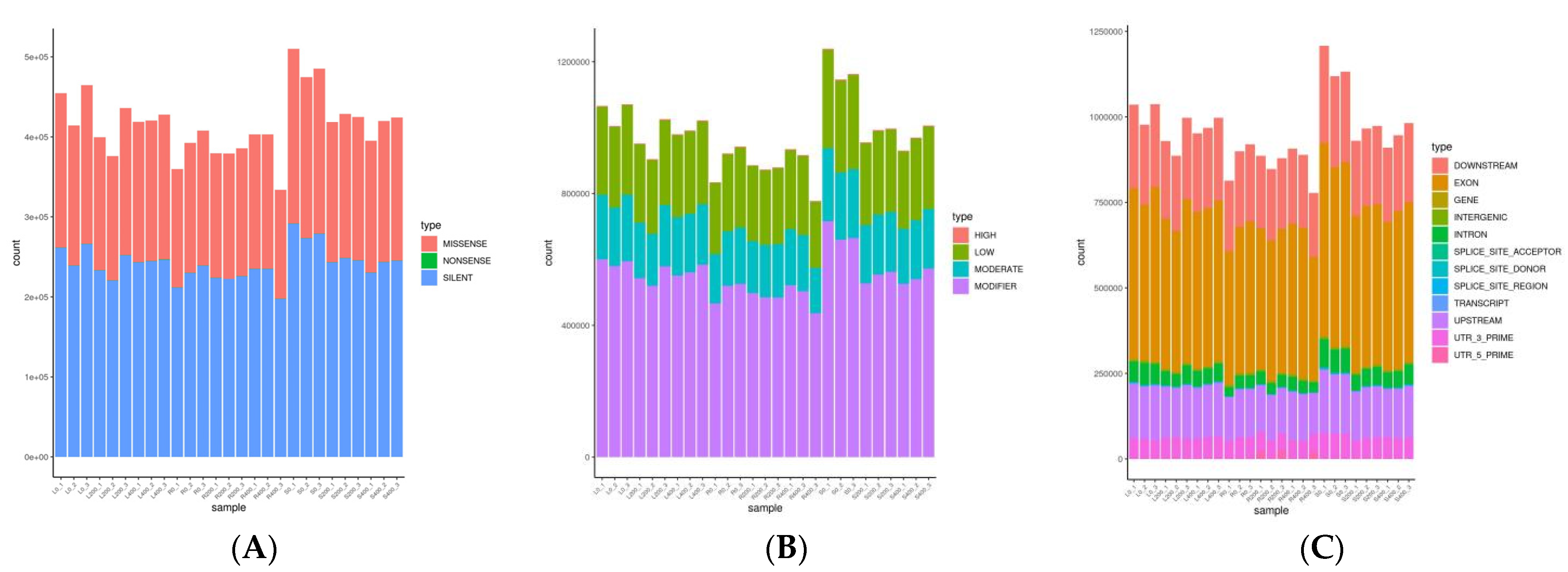
3.7. Mutation Loci Analysis
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| DEGs | Difffferentially expressed genes |
| BP | Biological process |
| CC | Cellular component |
| MF | Molecular function |
| RNA | Ribonucleic acid |
| FPKM | Fragments per kilobase of exon model per million mapped fragments |
| mRNA | Messenger RNA |
| SNP | Single-nucleotide polymorphism |
| PCR | Polymerase chain reaction |
| DNA | Deoxyribonucleic acid |
| PCA | Principal component analysis |
| dNTP | Deoxy-ribonucleoside triphosphate |
References
- Mukhopadhyay, R.; Sarkar, B.; Jat, H.S.; Sharma, P.C.; Bolan, N.S. Soil salinity under climate change: Challenges for sustainable agriculture and food security. J. Environ. Manag. 2020, 280, 111736. [Google Scholar] [CrossRef] [PubMed]
- Pitman, M.G.; Läuchli, A. Global Impact of Salinity and Agricultural Ecosystems. In Salinity: Environment—Plants—Molecules; Läuchli, A., Lüttge, U., Eds.; Springer: Dordrecht, The Netherlands, 2002. [Google Scholar]
- Kafi, M.; Asadi, H.; Ganjeali, A. Possible utilization of high-salinity waters and application of low amounts of water for production of the halophyte Kochia scoparia as alternative fodder in saline agroecosystems. Agric. Water Manag. 2010, 97, 139–147. [Google Scholar] [CrossRef]
- Alcázar, R.; Bueno, M.; Tiburcio, A.F. Polyamines: Small Amines with Large Effects on Plant Abiotic Stress Tolerance. Cells 2020, 9, 2373. [Google Scholar] [CrossRef] [PubMed]
- Hurkman, W.J. Effect of salt stress on plant gene expression: A review. Plant Soil 1992, 146, 145–151. [Google Scholar] [CrossRef]
- Munns, R.; Gilliham, M. Salinity tolerance of crops—What is the cost? New Phytol. 2015, 208, 668–673. [Google Scholar] [CrossRef] [Green Version]
- Hasegawa, P.M.; Bressan, R.A.; Zhu, J.K.; Bohnert, H.J. Plant Cellular and Molecular Responses to High Salinity. Annu. Rev. Plant Physiol. Plant Mol. Biol. 2000, 51, 463–499. [Google Scholar] [CrossRef] [Green Version]
- Seki, M.; Narusaka, M.; Abe, H.; Kasuga, M.; Yamaguchi-Shinozaki, K.; Carninci, P.; Hayashizaki, Y.; Shinozaki, K. Monitoring the Expression Pattern of 1300 Arabidopsis Genes under Drought and Cold Stresses by Using a Full-Length cDNA Microarray. Plant Cell 2001, 13, 61–72. [Google Scholar] [CrossRef] [Green Version]
- Bartels, D.; Sunkar, R. Drought and Salt Tolerance in Plants. Crit. Rev. Plant Sci. 2005, 24, 23–58. [Google Scholar] [CrossRef]
- Yamaguchi-Shinozaki, K.; Shinozaki, K. Transcriptional regulatory networks in cellular responses and tolerance to dehydration and cold stresses. Annu. Rev. Plant Biol. 2006, 57, 781–803. [Google Scholar] [CrossRef] [Green Version]
- Munns, R.; Tester, M. Mechanisms of Salinity Tolerance. Annu. Rev. Plant Biol. 2008, 59, 651–681. [Google Scholar] [CrossRef] [Green Version]
- Stitt, M.; Sulpice, R.; Keurentjes, J. Metabolic networks: How to identify key components in the regulation of metabolism and growth. Plant Physiol. 2010, 152, 428–444. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J.K. Plant salt tolerance. Trends Plant Sci. 2001, 6, 66–71. [Google Scholar] [CrossRef]
- Ashraf, M.F.M.R.; Foolad, M.R. Roles of glycine betaine and proline in improving plant abiotic stress resistance. Environ. Exp. Bot. 2007, 59, 206–216. [Google Scholar] [CrossRef]
- Manchanda, G.; Garg, N. Salinity and its effects on the functional biology of legumes. Acta Physiol. Plant. 2008, 30, 595–618. [Google Scholar] [CrossRef]
- Shrivastava, P.; Kumar, R. Soil salinity: A serious environmental issue and plant growth promoting bacteria as one of the tools for its alleviation. Saudi J. Biol. Sci. 2015, 22, 123–131. [Google Scholar] [CrossRef] [Green Version]
- Yuan, F.; Guo, J.; Shabala, S.; Wang, B. Reproductive Physiology of Halophytes: Current Standing. Front. Plant Sci. 2019, 9, 1954. [Google Scholar] [CrossRef]
- Li, M.; Yuyama, N.; Luo, L.; Hirata, M.; Cai, H. In silico mapping of 1758 new SSR markers developed from public genomic sequences for sorghum. Mol. Breed. 2009, 24, 41–47. [Google Scholar] [CrossRef]
- Sudhakar, R.P.; Srinivas, R.D.; Sivasakthi, K.; Bhatnagar-Mathur, P.; Vadez, V.; Sharma, K.K. Evaluation of Sorghum [Sorghum bicolor (L.)] Reference Genes in Various Tissues and under Abiotic Stress Conditions for Quantitative Real-Time PCR Data Normalization. Front. Plant Sci. 2016, 7, 529. [Google Scholar] [CrossRef]
- Flowers, T.J.; Colmer, T.D. Plant salt tolerance: Adaptations in halophytes. Ann. Bot. 2015, 115, 327–331. [Google Scholar] [CrossRef] [Green Version]
- Gu, J.; Xia, Z.; Luo, Y.; Jiang, X.; Qian, B.; Xie, H.; Zhu, J.-K.; Xiong, L.; Zhu, J.; Wang, Z.Y. Spliceosomal protein U1A is involved in alternative splicing and salt stress tolerance in Arabidopsis thaliana. Nucleic Acids Res. 2018, 46, 1777–1792. [Google Scholar] [CrossRef] [Green Version]
- Yuan, J.; Li, J.; Yang, Y.; Tan, C.; Zhu, Y.; Hu, L.; Qi, Y.; Lu, Z.J. Stress-responsive regulation of long non-coding RNA polyadenylation in Oryza sativa. Plant J. 2017, 93, 814–827. [Google Scholar] [CrossRef] [Green Version]
- Cui, F.; Sui, N.; Duan, G.; Liu, Y.; Han, Y.; Liu, S.; Wan, S.; Li, G. Identification of Metabolites and Transcripts Involved in Salt Stress and Recovery in Peanut. Front. Plant Sci. 2018, 9, 217. [Google Scholar] [CrossRef]
- Skorupa, M.; Gołębiewski, M.; Kurnik, K.; Niedojadło, J.; Kęsy, J.; Klamkowski, K.; Wójcik, K.; Treder, W.; Tretyn, A.; Tyburski, J. Salt stress vs. salt shock—The case of sugar beet and its halophytic ancestor. BMC Plant Biol. 2019, 19, 57. [Google Scholar] [CrossRef] [Green Version]
- Zhang, F.; Zhu, G.; Du, L.; Shang, X.; Cheng, C.; Yang, B.; Hu, Y.; Cai, C.; Guo, W. Retraction Note: Genetic regulation of salt stress tolerance revealed by RNA-Seq in cotton diploid wild species, Gossypium davidsonii. Sci. Rep. 2019, 9, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Xie, R.; Pan, X.; Zhang, J.; Ma, Y.; He, S.; Zheng, Y.; Ma, Y. Effect of salt-stress on gene expression in citrus roots revealed by RNA-seq. Funct. Integr. Genom. 2017, 18, 155–173. [Google Scholar] [CrossRef]
- Xiong, Y.; Yan, H.; Liang, H.; Zhang, Y.; Guo, B.; Niu, M.; Jian, S.; Ren, H.; Zhang, X.; Li, Y.; et al. RNA-Seq analysis of Clerodendrum inerme (L.) roots in response to salt stress. BMC Genom. 2019, 20, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Panahi, B.; Frahadian, M.; Dums, J.T.; Hejazi, M.A. Integration of cross species RNA-seq meta-analysis and machine-learning models identifes the most important salt stress-responsive pathways in Microalga Dunaliella. Front. Genet. 2019, 10, 752. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Li, D.; Chen, A.; Tang, H.; Li, J.; Huang, S. RNA-seq for comparative transcript profiling of kenaf under salinity stress. J. Plant Res. 2017, 130, 365–372. [Google Scholar] [CrossRef] [Green Version]
- Bahieldin, A.; Atef, A.; Sabir, J.S.; Gadalla, N.O.; Edris, S.; Alzohairy, A.M.; Radhwan, N.A.; Baeshen, M.N.; Ramadan, A.M.; Eissa, H.F.; et al. RNA-Seq analysis of the wild barley (H. spontaneum) leaf transcriptome under salt stress. Comptes Rendus Biol. 2015, 338, 285–297. [Google Scholar] [CrossRef]
- Bowman, J.L.; Alvarez, J.; Weigel, D.; Meyerowitz, E.M.; Smyth, D.R. Control of flower development in Arabidopsis thaliana by APETALA1 and interacting genes. Development 1993, 119, 721–743. [Google Scholar] [CrossRef]
- Zhang, Z.; Dunwell, J.M.; Zhang, Y.M. An integrated omics analysis reveals molecular mechanisms that are associated with differences in seed oil content between Glycine max and Brassica napus. BMC Plant Biol. 2018, 18, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Schuster, S.C. Next-generation sequencing transforms today’s biology. Nat. Methods 2008, 5, 16. [Google Scholar] [CrossRef] [PubMed]
- Haseneyer, G.; Schmutzer, T.; Seidel, M.; Zhou, R.; Mascher, M.; Schön, C.C.; Taudien, S.; Scholz, U.; Stein, N.; Bauer, E. From RNA-seq to large-scale genotyping—Genomics resources for rye (Secale cereale L.). BMC Plant Biol. 2011, 11, 131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, C.; Zhang, Z.; Yan, G.; Wang, F.; Zhao, L.; Liu, N.; Abudurezike, A.; Li, Y.; Wang, W.; Shi, S. Salt-responsive transcriptome analysis of triticale reveals candidate genes involved in the key metabolic pathway in response to salt stress. Sci. Rep. 2020, 10, 20669. [Google Scholar] [CrossRef]
- Parkhomchuk, D.; Borodina, T.; Amstislavskiy, V.; Banaru, M.; Hallen, L.; Krobitsch, S.; Lehrach, H.; Soldatov, A. Transcriptome analysis by strand-specific sequencing of complementary DNA. Nucleic Acids Res. 2009, 37, e123. [Google Scholar] [CrossRef] [Green Version]
- Qiu, Q.; Ma, T.; Hu, Q.; Liu, B.; Wu, Y.; Zhou, H.; Wang, Q.; Wang, J.; Liu, J. Genome-scale transcriptome analysis of the desert poplar, Populus euphratica. Tree Physiol. 2011, 31, 452. [Google Scholar] [CrossRef] [Green Version]
- Xu, P.; Liu, Z.; Fan, X.; Gao, J.; Zhang, X.; Zhang, X.; Shen, X. De novo transcriptome sequencing and comparative analysis of differentially expressed genes in Gossypium aridum under salt stress. Gene 2013, 525, 26–34. [Google Scholar] [CrossRef]
- Rong, L.; Li, Q.; Li, S.; Tang, L.; Wen, J. De novo transcriptome sequencing of Acer palmatum and comprehensive analysis of differentially expressed genes under salt stress in two contrasting genotypes. Mol. Genet. Genom. 2016, 291, 575–586. [Google Scholar] [CrossRef]
- Ji, F.; Tang, L.; Yang, Z.; Li, Y.; Wang, W.; Xu, Y.; Li, S.; Li, X. Transcriptome sequencing and comparative analysis of differentially expressed genes in the roots of Musa Paradisiaca under salt stress. Plant Biotechnol. Rep. 2021, 15, 389–401. [Google Scholar] [CrossRef]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef]
- Yang, L.; Smyth, G.K.; Wei, S. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar]
- Garber, M.; Grabherr, M.G.; Guttman, M.; Trapnell, C. Computational methods for transcriptome annotation and quantification using RNA-seq. Nat. Methods 2011, 8, 469–477. [Google Scholar] [CrossRef] [Green Version]
- Bray, N.L.; Pimentel, H.; Melsted, P.; Pachter, L. Near-optimal RNA-Seq quantification. arXiv 2015, arXiv:1505.02710. [Google Scholar]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139. [Google Scholar]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Method Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biol. 2010, 11, R14. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Q.; Wang, X.J. GOEAST: A web-based software toolkit for Gene Ontology enrichment analysis. Nucleic Acids Res. 2008, 36 (Suppl. 2), W358–W363. [Google Scholar] [CrossRef] [Green Version]
- Zhen, S.; Xin, Z. EasyGO: Gene Ontology-based annotation and functional enrichment analysis tool for agronomical species. BMC Genom. 2007, 8, 1–4. [Google Scholar]
- Ong, E.; He, Y. GOfox: Semantics-based simplified hierarchical classification and interactive visualization to support GO enrichment analysis. In Proceedings of the 2015 International Conference on Biomedical Ontologies (ICBO-2015), Lisbon, Portugal, 27–30 July 2015; pp. 121–122. [Google Scholar]
- Hung, K.S.; Hsiao, C.C.; Pai, T.W.; Hu, C.H.; Tzou, W.S.; Wang, W.D.; Chen, Y.R. Functional enrichment analysis based on long noncoding RNA associations. BMC Syst. Biol. 2018, 12, 45. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Chen, L.; Kong, X.; Huang, T.; Cai, Y.D. Analysis of Tumor Suppressor Genes Based on Gene Ontology and the KEGG Pathway. PLoS ONE 2014, 9, e107202. [Google Scholar]
- Sun, C.G.; Cao, X.J.; Zhou, C.; Liu, L.J.; Feng, F.B.; Liu, R.J.; Zhuang, J.; Li, Y.J. Construction of gene/protein interaction networks for primary myelofibrosis and KEGG pathway-enrichment analysis of molecular compounds. Genet. Mol. Res. GMR 2015, 14, 16126–16132. [Google Scholar] [CrossRef]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; Van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [Green Version]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; DePristo, M.A. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [Green Version]
- Gangaraj, K.P.; Rajesh, M.K. Dataset of dual RNA-sequencing of Phytophthora palmivora infecting coconut (Cocos nucifera L.). Data Brief 2020, 30, 105455. [Google Scholar] [CrossRef]
- Han, Z.J.; Sun, Y.; Zhang, M.; Zhai, J.T. Transcriptomic profile analysis of the halophyte Suaeda rigida response and tolerance under NaCl stress. Sci. Rep. 2020, 10, 15148. [Google Scholar] [CrossRef]
- Aoki-Kinoshita, K.F.; Kanehisa, M. Gene Annotation and Pathway Mapping in KEGG. In Comparative Genomics. Methods in Molecular Biology; Bergman, N.H., Ed.; Humana Press: New York, NY, USA, 2007; Volume 396. [Google Scholar]
- Yao, L.; Wang, J.; Li, B.; Meng, Y.; Ma, X.; Si, E.; Ren, P.; Yang, K.; Shang, K.; Wang, H. Transcriptome sequencing and comparative analysis of differentially-expressed isoforms in the roots of Halogeton glomeratus under salt stress. Gene 2018, 646, 159–168. [Google Scholar] [CrossRef]
- Zou, J.; Jin, H. Transcriptome sequencing and gene expression profiling of Populus wutunensis, a natural hybrid, during salinity stress. Trees 2020, 34, 1427–1438. [Google Scholar] [CrossRef]
- Zhou, Y.; Yang, P.; Cui, F.; Zhang, F.; Luo, X.; Xie, J. Transcriptome Analysis of Salt Stress Responsiveness in the Seedlings of Dongxiang Wild Rice (Oryza rufipogon Griff.). PLoS ONE 2016, 11, e0146242. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Lin, J.; Yang, Q.S.; Li, X.G.; Chang, Y.H. Comprehensive analysis of differentially expressed genes under salt stress in pear (Pyrus betulaefolia) using RNA-Seq. Plant Growth Regul. 2017, 82, 409–420. [Google Scholar] [CrossRef]
- Yong, H.Y.; Zou, Z.; Kok, E.P.; Kwan, B.H.; Chow, K.; Nasu, S.; Nanzyo, M.; Kitashiba, H.; Nishio, T. Comparative Transcriptome Analysis of Leaves and Roots in Response to Sudden Increase in Salinity in Brassica napus by RNA-seq. J. Biomed. Biotechnol. 2014, 2014, 467395. [Google Scholar]
- Sun, L.; Huang, L.; Hong, Y.; Zhang, H.; Song, F.; Li, D. Comprehensive Analysis Suggests Overlapping Expression of Rice ONAC Transcription Factors in Abiotic and Biotic Stress Responses. Int. J. Mol. Sci. 2015, 16, 4306–4326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eulgem, T.; Rushton, P.J.; Robatzek, S.; Somssich, I.E. The WRKY superfamily of plant transcription factors. Trends Plant Sci. 2000, 5, 199–206. [Google Scholar] [CrossRef]
- Maleck, K.; Levine, A.; Eulgem, T.; Morgan, A.; Schmid, J.; Lawton, K.A.; Dangl, J.L.; Dietrich, R.A. The transcriptome of Arabidopsis thaliana during systemic acquired resistance. Nat. Genet. 2000, 26, 403–410. [Google Scholar] [CrossRef]
- Dong, J.; Chen, C.; Chen, Z. Expression profiles of the Arabidopsis WRKY gene superfamily during plant defense response. Plant Mol. Biol. 2003, 51, 21–37. [Google Scholar] [CrossRef]
- Ülker, B.; Somssich, I.E. WRKY transcription factors: From DNA binding towards biological function. Curr. Opin. Plant Biol. 2004, 7, 491–498. [Google Scholar] [CrossRef] [Green Version]
- Hu, R.; Qi, G.; Kong, Y.; Kong, D.; Gao, Q.; Zhou, G. Comprehensive Analysis of NAC Domain Transcription Factor Gene Family in Populus trichocarpa. BMC Plant Biol. 2010, 10, 145. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Z.; Zhang, X.; Zhao, K.; Zhou, B.; Jiang, T. Ectopic expression of a poplar gene NAC13 confers enhanced tolerance to salinity stress in transgenic Nicotiana tabacum. J. Plant Res. 2020, 133, 727–737. [Google Scholar] [CrossRef]
- Movahedi, A.; Zhang, J.; Gao, P.; Yang, Y.; Wang, L.; Yin, T.; Kadkhodaei, S.; Ebrahimi, M.; Zhuge, Q. Expression of the chickpeaCarNAC3gene enhances salinity and drought tolerance in transgenic poplars. Plant Cell Tissue Organ. Cult. 2015, 120, 141–154. [Google Scholar] [CrossRef]
- Nguyen, K.H.; Mostofa, M.G.; Li, W.; Van Ha, C.; Watanabe, Y.; Le, D.T.; Thao, N.P.; Tran, L.S.P. The soybean transcription factor GmNAC085 enhances drought tolerance in Arabidopsis. Environ. Exp. Bot. 2018, 151, 12–20. [Google Scholar] [CrossRef]
- Hao, Y.J.; Wei, W.; Song, Q.X.; Chen, H.W.; Zhang, Y.Q.; Wang, F.; Zou, H.F.; Lei, G.; Tian, A.G.; Chen, S.Y. Soybean NAC transcription factors promote abiotic stress tolerance and lateral root formation in transgenic plants. Plant J. Cell Mol. Biol. 2011, 68, 302–313. [Google Scholar] [CrossRef]
- Wang, X.E.; Basnayake, B.V.S.; Zhang, H.; Li, G.; Li, W.; Virk, N.; Mengiste, T.; Song, F. The Arabidopsis ATAF1, a NAC transcription factor, is a negative regulator of defense responses against necrotrophic fungal and bacterial pathogens. Mol. Plant-Microbe Interact. 2009, 22, 1227–1238. [Google Scholar] [CrossRef] [Green Version]
- Welner, D.H.; Lindemose, S.; Grossmann, J.G.; Møllegaard, N.E.; Olsen, A.N.; Helgstrand, C.; Skriver, K.; Lo Leggio, L. DNA binding by the plant-specific NAC transcription factors in crystal and solution: A firm link to WRKY and GCM transcription factors. Biochem. J. 2012, 444, 395. [Google Scholar] [CrossRef]
- Hsieh, T.H.; Li, C.W.; Su, R.C.; Cheng, C.P.; Tsai, Y.C.; Chan, M.T. A tomato bZIP transcription factor, SlAREB, is involved in water deficit and salt stress response. Planta 2010, 231, 1459–1473. [Google Scholar] [CrossRef]
- Nakano, T.; Suzuki, K.; Fujimura, T.; Shinshi, H. Genome-Wide Analysis of the ERF Gene Family in Arabidopsis and Rice. Plant Physiol. 2006, 140, 411. [Google Scholar] [CrossRef] [Green Version]
- Dietz, K.J.; Vogel, M.O.; Viehhauser, A. AP2/EREBP transcription factors are part of gene regulatory networks and integrate metabolic, hormonal and environmental signals in stress acclimation and retrograde signalling. Protoplasma 2010, 245, 3–14. [Google Scholar] [CrossRef]
- Zhuang, J.; Cai, B.; Peng, R.H.; Zhu, B.; Jin, X.F.; Xue, Y.; Gao, F.; Fu, X.Y.; Tian, Y.S.; Yao, Q.H. Genome-wide analysis of the AP2/ERF gene family in Populus trichocarpa. Biochem. Biophys. Res. Commun. 2008, 371, 468–474. [Google Scholar] [CrossRef]
- Zhuang, J.; Chen, J.M.; Yao, Q.H.; Xiong, F.; Sun, C.C.; Zhou, X.R.; Zhang, J.; Xiong, A.S. Discovery and expression profile analysis of AP2/ERF family genes from Triticum aestivum. Mol. Biol. Rep. 2011, 38, 745–753. [Google Scholar] [CrossRef]
- Ishiguro, S.; Nakamura, K. Characterization of a cDNA encoding a novel DNA-binding protein, SPF1, that recognizes SP8 sequences in the 5′ upstream regions of genes coding for sporamin and β-amylase from sweet potato. Mol. Gen. Genet. MGG 1994, 244, 563–571. [Google Scholar] [CrossRef]
- Rushton, P.J.; Macdonald, H.; Huttly, A.K.; Lazarus, C.M.; Hooley, R. Members of a new family of DNA-binding proteins bind to a conserved cis-element in the promoters of α-Amy2 genes. Plant Mol. Biol. 1995, 29, 691–702. [Google Scholar] [CrossRef]
- de Pater, S.; Greco, V.; Pham, K.; Memelink, J.; Kijne, J. Characterization of a zinc-dependent transcriptional activator from Arabidopsis. Nucleic Acids Res. 1996, 24, 4624–4631. [Google Scholar] [CrossRef] [Green Version]
- Rushton, D.L.; Tripathi, P.; Rabara, R.C.; Lin, J.; Ringler, P.; Boken, A.K.; Langum, T.J.; Smidt, L.; Boomsma, D.D.; Rushton, P.J. WRKY transcription factors: Key components in abscisic acid signalling. Plant Biotechnol. J. 2011, 10, 2–11. [Google Scholar] [CrossRef]
- Li, H.L.; Zhang, L.B.; Guo, D.; Li, C.Z.; Peng, S.Q. Identification and expression profiles of the WRKY transcription factor family in Ricinus communis. Gene 2012, 503, 248–253. [Google Scholar] [CrossRef]
- Jiang, Y.; Duan, Y.; Yin, J.; Ye, S.; Zhu, J.; Zhang, F.; Lu, W.; Fan, D.; Luo, K. Genome-wide identification and characterization of the Populus WRKY transcription factor family and analysis of their expression in response to biotic and abiotic stresses. J. Exp. Bot. 2014, 22, 6629–6644. [Google Scholar] [CrossRef]
- Karim, A.; Jiang, Y.; Guo, L.; Ling, Z.; Ye, S.; Duan, Y.; Li, C.; Luo, K. Isolation and characterization of a subgroup IIa WRKY transcription factor PtrWRKY40 from Populus trichocarpa. Tree Physiol. 2015, 35, 1129–1139. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Chen, N.; Chen, F.; Cai, B.; Dal Santo, S.; Tornielli, G.B.; Pezzotti, M.; Cheng, Z.M.M. Genome-wide analysis and expression profile of the bZIP transcription factor gene family in grapevine (Vitis vinifera). BMC Genom. 2014, 15, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Chen, W.; Zhou, J.; He, H.; Chen, L.; Chen, H.; Deng, X.W. Basic leucine zipper transcription factor OsbZIP16 positively regulates drought resistance in rice. Plant Sci. Int. J. Exp. Plant Biol. 2012, 193–194, 8–17. [Google Scholar] [CrossRef]
- Yoshida, T.; Fujita, Y.; Sayama, H.; Kidokoro, S.; Maruyama, K.; Mizoi, J.; Shinozaki, K.; Yamaguchi-Shinozaki, K. AREB1, AREB2, and ABF3 are master transcription factors that cooperatively regulate ABRE-dependent ABA signaling involved in drought stress tolerance and require ABA for full activation. Plant J. 2010, 61, 672–685. [Google Scholar] [CrossRef]
- Fan, W. In Vivo Interaction between NPR1 and Transcription Factor TGA2 Leads to Salicylic Acid-Mediated Gene Activation in Arabidopsis. Plant Cell Online 2002, 14, 1377–1389. [Google Scholar] [CrossRef] [Green Version]
- Despres, C. The Arabidopsis NPR1 Disease Resistance Protein Is a Novel Cofactor That Confers Redox Regulation of DNA Binding Activity to the Basic Domain/Leucine Zipper Transcription Factor TGA1. Plant Cell Online 2003, 15, 2181–2191. [Google Scholar] [CrossRef] [Green Version]
- Uno, Y.; Furihata, T.; Abe, H.; Yoshida, R.; Shinozaki, K.; Yamaguchi-Shinozaki, K. Arabidopsis basic leucine zipper transcription factors involved in an abscisic acid-dependent signal transduction pathway under drought and high-salinity conditions. Proc. Natl. Acad. Sci. USA 2000, 97, 11632–11637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jakoby, M.; Weisshaar, B.; Dröge-Laser, W.; Vicente-Carbajosa, J.; Tiedemann, J.; Kroj, T.; Parcy, F. bZIP transcription factors in Arabidopsis. Trends Plant Sci. 2002, 7, 106–111. [Google Scholar] [CrossRef]
- Dang, Z.H.; Zheng, L.L.; Wang, J.; Gao, Z.; Wu, S.B.; Qi, Z.; Wang, Y.C. Transcriptomic profiling of the salt-stress response in the wild recretohalophyte Reaumuria trigyna. BMC Genom. 2013, 14, 1–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, H.; Dong, D.; Yang, Q.; Zhu, D. Salt-Responsive Transcriptome Profiling of Suaeda glauca via RNA Sequencing. PLoS ONE 2016, 11, e0150504. [Google Scholar] [CrossRef]
- Zhang, H.; Zhou, C. Signal transduction in leaf senescence. Plant Mol. Biol. 2013, 82, 539–545. [Google Scholar] [CrossRef]
- Strohm, A.K.; Baldwin, K.L.; Masson, P.H. Molecular mechanisms of root gravity sensing and signal transduction. Wiley Interdiscip. Rev. Dev. Biol. 2012, 1, 276–285. [Google Scholar] [CrossRef]
- Seyfferth, C.; Tsuda, K. Salicylic acid signal transduction: The initiation of biosynthesis, perception and transcriptional reprogramming. Front. Plant Sci. 2014, 5, 697. [Google Scholar] [CrossRef] [Green Version]
- Huang, G.T.; Ma, S.L.; Bai, L.P.; Zhang, L.; Ma, H.; Jia, P.; Liu, J.; Zhong, M.; Guo, Z.F. Signal transduction during cold, salt, and drought stresses in plants. Mol. Biol. Rep. 2012, 39, 969–987. [Google Scholar] [CrossRef]
- Abreu, I.A.; Farinha, A.P.; Negrão, S.; Gonçalves, N.; Fonseca, C.; Rodrigues, M.; Batista, R.; Saibo, B.J.M.; Oliveira, M.M. Coping with abiotic stress: Proteome changes for crop improvement. J. Proteom. 2013, 93, 145–168. [Google Scholar] [CrossRef]
- Geng, Y.; Wu, R.; Wee, C.W.; Xie, F.; Wei, X.; Chan, P.M.Y.; Tham, C.; Duan, L.; Dinneny, J.R. A Spatio-Temporal Understanding of Growth Regulation during the Salt Stress Response in Arabidopsis. Plant Cell 2013, 25, 2132–2154. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Library | Raw_Reads | Clean_Reads | Clean_Bases | Error_Rate | Q20 | Q30 | GC_Pct |
|---|---|---|---|---|---|---|---|---|
| L0_1 | FRAS210141894-1r | 43,229,878 | 41,916,070 | 6.29 G | 0.03 | 97.72 | 93.28 | 43.80 |
| L0_2 | FRAS210141895-1r | 43,503,400 | 41,412,972 | 6.21 G | 0.03 | 97.72 | 93.30 | 42.92 |
| L0_3 | FRAS210141896-1r | 41,700,782 | 40,131,530 | 6.02 G | 0.03 | 97.64 | 93.04 | 43.93 |
| L200_1 | FRAS210141903-1r | 42,337,890 | 41,376,760 | 6.21 G | 0.03 | 97.75 | 93.37 | 44.00 |
| L200_2 | FRAS210141904-1r | 41,637,272 | 40,851,878 | 6.13 G | 0.03 | 97.72 | 93.30 | 43.90 |
| L200_3 | FRAS210141905-1r | 41,808,814 | 40,466,298 | 6.07 G | 0.03 | 97.77 | 93.38 | 43.56 |
| L400_1 | FRAS210141912-1r | 43,477,378 | 41,505,660 | 6.23 G | 0.03 | 97.72 | 93.27 | 43.25 |
| L400_2 | FRAS210141913-1r | 40,334,862 | 39,717,696 | 5.96 G | 0.03 | 97.58 | 92.95 | 43.92 |
| L400_3 | FRAS210141914-1r | 43,098,376 | 42,022,164 | 6.30 G | 0.03 | 97.71 | 93.32 | 43.79 |
| R0_1 | FRAS210141888-1r | 41,219,840 | 38,942,918 | 5.84 G | 0.03 | 97.31 | 92.50 | 44.26 |
| R0_2 | FRAS210141889-1r | 40,474,488 | 39,722,168 | 5.96 G | 0.03 | 97.66 | 93.17 | 43.25 |
| R0_3 | FRAS210141890-1r | 41,990,470 | 41,326,130 | 6.20 G | 0.03 | 97.70 | 93.25 | 43.58 |
| R200_1 | FRAS210141897-1r | 41,346,852 | 40,288,012 | 6.04 G | 0.03 | 97.60 | 93.07 | 43.87 |
| R200_2 | FRAS210141898-1r | 41,922,834 | 40,528,650 | 6.08 G | 0.03 | 97.59 | 93.08 | 44.29 |
| R200_3 | FRAS210141899-1r | 42,625,060 | 41,322,596 | 6.20 G | 0.03 | 97.54 | 93.02 | 44.55 |
| R400_1 | FRAS210141906-2r | 47,618,458 | 46,825,304 | 7.02 G | 0.03 | 97.39 | 92.57 | 44.77 |
| R400_2 | FRAS210141907-2r | 44,838,230 | 43,672,332 | 6.55 G | 0.03 | 97.40 | 92.61 | 44.64 |
| R400_3 | FRAS210141908-1r | 40,153,594 | 38,188,658 | 5.73 G | 0.03 | 97.58 | 93.05 | 44.60 |
| S0_1 | FRAS210141891-1r | 42,998,830 | 41,953,190 | 6.29 G | 0.03 | 97.68 | 93.21 | 43.01 |
| S0_2 | FRAS210141892-1r | 41,626,198 | 40,928,406 | 6.14 G | 0.03 | 97.67 | 93.14 | 42.92 |
| S0_3 | FRAS210141893-1r | 42,162,498 | 40,897,322 | 6.13 G | 0.03 | 97.75 | 93.32 | 43.04 |
| S200_1 | FRAS210141900-1r | 41,365,530 | 39,960,594 | 5.99 G | 0.03 | 97.58 | 93.11 | 43.65 |
| S200_2 | FRAS210141901-1r | 41,693,852 | 40,826,972 | 6.12 G | 0.03 | 97.50 | 92.84 | 43.60 |
| S200_3 | FRAS210141902-1r | 43,498,006 | 42,704,462 | 6.41 G | 0.03 | 97.74 | 93.33 | 43.13 |
| S400_1 | FRAS210141909-1r | 43,083,076 | 41,548,528 | 6.23 G | 0.03 | 97.65 | 93.17 | 43.24 |
| S400_2 | FRAS210141910-1r | 42,854,862 | 41,521,356 | 6.23 G | 0.03 | 97.70 | 93.27 | 43.29 |
| S400_3 | FRAS210141911-1r | 40,153,198 | 38,880,092 | 5.83 G | 0.03 | 97.44 | 92.77 | 42.81 |
| Sample | Total_Reads | Total_Map | Unique_Map | Multi_Map | Read1_Map | Read2_Map | Positive_Map | Negative_Map | Splice_Map | Unsplice_Map | Proper_Map |
|---|---|---|---|---|---|---|---|---|---|---|---|
| L0_1 | 41,916,070 | 34,099,861 | 31,929,576 | 2,170,285 | 16,031,977 | 15,897,599 | 15,964,215 | 15,965,361 | 14,181,366 | 17,748,210 | 28,988,606 |
| (81.35%) | (76.18%) | (5.18%) | (38.25%) | (37.93%) | (38.09%) | (38.09%) | (33.83%) | (42.34%) | (69.16%) | ||
| L0_2 | 41,412,972 | 32,664,958 | 30,703,326 | 1,961,632 | 15,406,956 | 15,296,370 | 15,331,819 | 15,371,507 | 11,989,541 | 18,713,785 | 27,601,396 |
| (78.88%) | (74.14%) | (4.74%) | (37.20%) | (36.94%) | (37.02%) | (37.12%) | (28.95%) | (45.19%) | (66.65%) | ||
| L0_3 | 40,131,530 | 32,731,445 | 30,693,348 | 2,038,097 | 15,420,588 | 15,272,760 | 15,336,379 | 15,356,969 | 13,627,713 | 17,065,635 | 27,583,972 |
| (81.56%) | (76.48%) | (5.08%) | (38.43%) | (38.06%) | (38.22%) | (38.27%) | (33.96%) | (42.52%) | (68.73%) | ||
| L200_1 | 41,376,760 | 32,898,394 | 30,611,563 | 2,286,831 | 15,356,087 | 15,255,476 | 15,298,246 | 15,313,317 | 12,888,498 | 17,723,065 | 27,751,170 |
| (79.51%) | (73.98%) | (5.53%) | (37.11%) | (36.87%) | (36.97%) | (37.01%) | (31.15%) | (42.83%) | (67.07%) | ||
| L200_2 | 40,851,878 | 32,073,201 | 29,883,288 | 2,189,913 | 14,986,813 | 14,896,475 | 14,936,879 | 14,946,409 | 12,214,282 | 17,669,006 | 27,036,076 |
| (78.51%) | (73.15%) | (5.36%) | (36.69%) | (36.46%) | (36.56%) | (36.59%) | (29.90%) | (43.25%) | (66.18%) | ||
| L200_3 | 40,466,298 | 32,754,785 | 30,638,096 | 2,116,689 | 15,357,668 | 15,280,428 | 15,310,291 | 15,327,805 | 12,668,641 | 17,969,455 | 27,793,958 |
| (80.94%) | (75.71%) | (5.23%) | (37.95%) | (37.76%) | (37.83%) | (37.88%) | (31.31%) | (44.41%) | (68.68%) | ||
| L400_1 | 41,505,660 | 33,238,571 | 31,112,328 | 2,126,243 | 15,607,894 | 15,504,434 | 15,538,117 | 15,574,211 | 12,673,945 | 18,438,383 | 28,325,300 |
| (80.08%) | (74.96%) | (5.12%) | (37.60%) | (37.35%) | (37.44%) | (37.52%) | (30.54%) | (44.42%) | (68.24%) | ||
| L400_2 | 39,717,696 | 31,919,467 | 29,885,359 | 2,034,108 | 15,027,743 | 14,857,616 | 14,941,370 | 14,943,989 | 12,497,015 | 17,388,344 | 26,970,556 |
| (80.37%) | (75.24%) | (5.12%) | (37.84%) | (37.41%) | (37.62%) | (37.63%) | (31.46%) | (43.78%) | (67.91%) | ||
| L400_3 | 42,022,164 | 33,553,507 | 31,383,445 | 2,170,062 | 15,742,857 | 15,640,588 | 15,688,348 | 15,695,097 | 13,645,448 | 17,737,997 | 28,539,948 |
| (79.85%) | (74.68%) | (5.16%) | (37.46%) | (37.22%) | (37.33%) | (37.35%) | (32.47%) | (42.21%) | (67.92%) | ||
| R0_1 | 38,942,918 | 29,175,374 | 27,383,231 | 1,792,143 | 13,782,210 | 13,601,021 | 13,664,279 | 13,718,952 | 9,546,470 | 17,836,761 | 24,479,086 |
| (74.92%) | (70.32%) | (4.60%) | (35.39%) | (34.93%) | (35.09%) | (35.23%) | (24.51%) | (45.80%) | (62.86%) | ||
| R0_2 | 39,722,168 | 30,801,737 | 28,995,952 | 1,805,785 | 14,556,757 | 14,439,195 | 14,473,119 | 14,522,833 | 10,181,574 | 18,814,378 | 26,065,798 |
| (77.54%) | (73.00%) | (4.55%) | (36.65%) | (36.35%) | (36.44%) | (36.56%) | (25.63%) | (47.36%) | (65.62%) | ||
| R0_3 | 41,326,130 | 32,315,221 | 30,390,025 | 1,925,196 | 15,250,293 | 15,139,732 | 15,172,739 | 15,217,286 | 11,044,773 | 19,345,252 | 27,505,356 |
| (78.20%) | (73.54%) | (4.66%) | (36.90%) | (36.63%) | (36.71%) | (36.82%) | (26.73%) | (46.81%) | (66.56%) | ||
| R200_1 | 40,288,012 | 31,451,369 | 29,490,159 | 1,961,210 | 14,795,186 | 14,694,973 | 14,720,654 | 14,769,505 | 11,114,536 | 18,375,623 | 26,546,638 |
| (78.07%) | (73.20%) | (4.87%) | (36.72%) | (36.47%) | (36.54%) | (36.66%) | (27.59%) | (45.61%) | (65.89%) | ||
| R200_2 | 40,528,650 | 31,057,737 | 29,136,961 | 1,920,776 | 14,612,559 | 14,524,402 | 14,547,707 | 14,589,254 | 10,636,348 | 18,500,613 | 26,202,788 |
| (76.63%) | (71.89%) | (4.74%) | (36.05%) | (35.84%) | (35.89%) | (36.00%) | (26.24%) | (45.65%) | (64.65%) | ||
| R200_3 | 41,322,596 | 30,250,140 | 28,378,130 | 1,872,010 | 14,241,200 | 14,136,930 | 14,165,430 | 14,212,700 | 10,488,271 | 17,889,859 | 25,446,492 |
| (73.20%) | (68.67%) | (4.53%) | (34.46%) | (34.21%) | (34.28%) | (34.39%) | (25.38%) | (43.29%) | (61.58%) | ||
| R400_1 | 46,825,304 | 34,091,693 | 31,999,767 | 2,091,926 | 16,001,871 | 15,997,896 | 15,983,812 | 16,015,955 | 11,468,760 | 20,531,007 | 28,565,350 |
| (72.81%) | (68.34%) | (4.47%) | (34.17%) | (34.17%) | (34.13%) | (34.20%) | (24.49%) | (43.85%) | (61.00%) | ||
| R400_2 | 43,672,332 | 34,574,074 | 32,289,809 | 2,284,265 | 16,166,727 | 16,123,082 | 16,116,947 | 16,172,862 | 11,606,074 | 20,683,735 | 29,067,328 |
| (79.17%) | (73.94%) | (5.23%) | (37.02%) | (36.92%) | (36.90%) | (37.03%) | (26.58%) | (47.36%) | (66.56%) | ||
| R400_3 | 38,188,658 | 25,409,066 | 23,861,327 | 1,547,739 | 11,973,526 | 11,887,801 | 11,908,929 | 11,952,398 | 8,480,579 | 15,380,748 | 21,370,942 |
| (66.54%) | (62.48%) | (4.05%) | (31.35%) | (31.13%) | (31.18%) | (31.30%) | (22.21%) | (40.28%) | (55.96%) | ||
| S0_1 | 41,953,190 | 32,267,320 | 30,360,095 | 1,907,225 | 15,242,528 | 15,117,567 | 15,157,535 | 15,202,560 | 11,350,223 | 19,009,872 | 27,252,422 |
| (76.91%) | (72.37%) | (4.55%) | (36.33%) | (36.03%) | (36.13%) | (36.24%) | (27.05%) | (45.31%) | (64.96%) | ||
| S0_2 | 40,928,406 | 31,190,534 | 29,382,790 | 1,807,744 | 14,736,558 | 14,646,232 | 14,660,622 | 14,722,168 | 10,667,910 | 18,714,880 | 26,398,080 |
| (76.21%) | (71.79%) | (4.42%) | (36.01%) | (35.79%) | (35.82%) | (35.97%) | (26.06%) | (45.73%) | (64.50%) | ||
| S0_3 | 40,897,322 | 31,534,019 | 29,548,587 | 1,985,432 | 14,809,974 | 14,738,613 | 14,739,097 | 14,809,490 | 11,417,118 | 18,131,469 | 26,865,924 |
| (77.11%) | (72.25%) | (4.85%) | (36.21%) | (36.04%) | (36.04%) | (36.21%) | (27.92%) | (44.33%) | (65.69%) | ||
| S200_1 | 39,960,594 | 31,613,489 | 29,619,742 | 1,993,747 | 14,855,726 | 14,764,016 | 14,775,203 | 14,844,539 | 11,488,947 | 18,130,795 | 26,757,178 |
| (79.11%) | (74.12%) | (4.99%) | (37.18%) | (36.95%) | (36.97%) | (37.15%) | (28.75%) | (45.37%) | (66.96%) | ||
| S200_2 | 40,826,972 | 32,184,287 | 30,214,916 | 1,969,371 | 15,174,062 | 15,040,854 | 15,076,017 | 15,138,899 | 11,759,858 | 18,455,058 | 27,384,426 |
| (78.83%) | (74.01%) | (4.82%) | (37.17%) | (36.84%) | (36.93%) | (37.08%) | (28.80%) | (45.20%) | (67.07%) | ||
| S200_3 | 42,704,462 | 32,736,990 | 30,757,053 | 1,979,937 | 15,435,697 | 15,321,356 | 15,343,424 | 15,413,629 | 12,076,016 | 18,681,037 | 27,631,548 |
| (76.66%) | (72.02%) | (4.64%) | (36.15%) | (35.88%) | (35.93%) | (36.09%) | (28.28%) | (43.74%) | (64.70%) | ||
| S400_1 | 41,548,528 | 31,715,603 | 29,766,573 | 1,949,030 | 14,916,876 | 14,849,697 | 14,844,254 | 14,922,319 | 11,238,002 | 18,528,571 | 26,809,282 |
| (76.33%) | (71.64%) | (4.69%) | (35.90%) | (35.74%) | (35.73%) | (35.92%) | (27.05%) | (44.60%) | (64.53%) | ||
| S400_2 | 41,521,356 | 32,538,126 | 30,505,240 | 2,032,886 | 15,313,023 | 15,192,217 | 15,209,470 | 15,295,770 | 12,225,058 | 18,280,182 | 27,518,922 |
| (78.36%) | (73.47%) | (4.90%) | (36.88%) | (36.59%) | (36.63%) | (36.84%) | (29.44%) | (44.03%) | (66.28%) | ||
| S400_3 | 38,880,092 | 30,215,448 | 28,441,954 | 1,773,494 | 14,304,860 | 14,137,094 | 14,181,255 | 14,260,699 | 10,674,997 | 17,766,957 | 25,446,248 |
| (77.71%) | (73.15%) | (4.56%) | (36.79%) | (36.36%) | (36.47%) | (36.68%) | (27.46%) | (45.70%) | (65.45%) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Y.; Han, Z.J.; Su, M.X.; Zhang, M. Transcriptomic Profile Analysis of Populus talassica × Populus euphratica Response and Tolerance under Salt Stress Conditions. Genes 2022, 13, 1032. https://doi.org/10.3390/genes13061032
Liu Y, Han ZJ, Su MX, Zhang M. Transcriptomic Profile Analysis of Populus talassica × Populus euphratica Response and Tolerance under Salt Stress Conditions. Genes. 2022; 13(6):1032. https://doi.org/10.3390/genes13061032
Chicago/Turabian StyleLiu, Ying, Zhan Jiang Han, Meng Xu Su, and Min Zhang. 2022. "Transcriptomic Profile Analysis of Populus talassica × Populus euphratica Response and Tolerance under Salt Stress Conditions" Genes 13, no. 6: 1032. https://doi.org/10.3390/genes13061032
APA StyleLiu, Y., Han, Z. J., Su, M. X., & Zhang, M. (2022). Transcriptomic Profile Analysis of Populus talassica × Populus euphratica Response and Tolerance under Salt Stress Conditions. Genes, 13(6), 1032. https://doi.org/10.3390/genes13061032