
Contradictory Phylogenetic Signals in the Laurasiatheria Anomaly Zone

 , , , , , , ,
, , , , , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Retrotransposon Analyses
2.1.1. Genome Assemblies and 2-Way Alignments
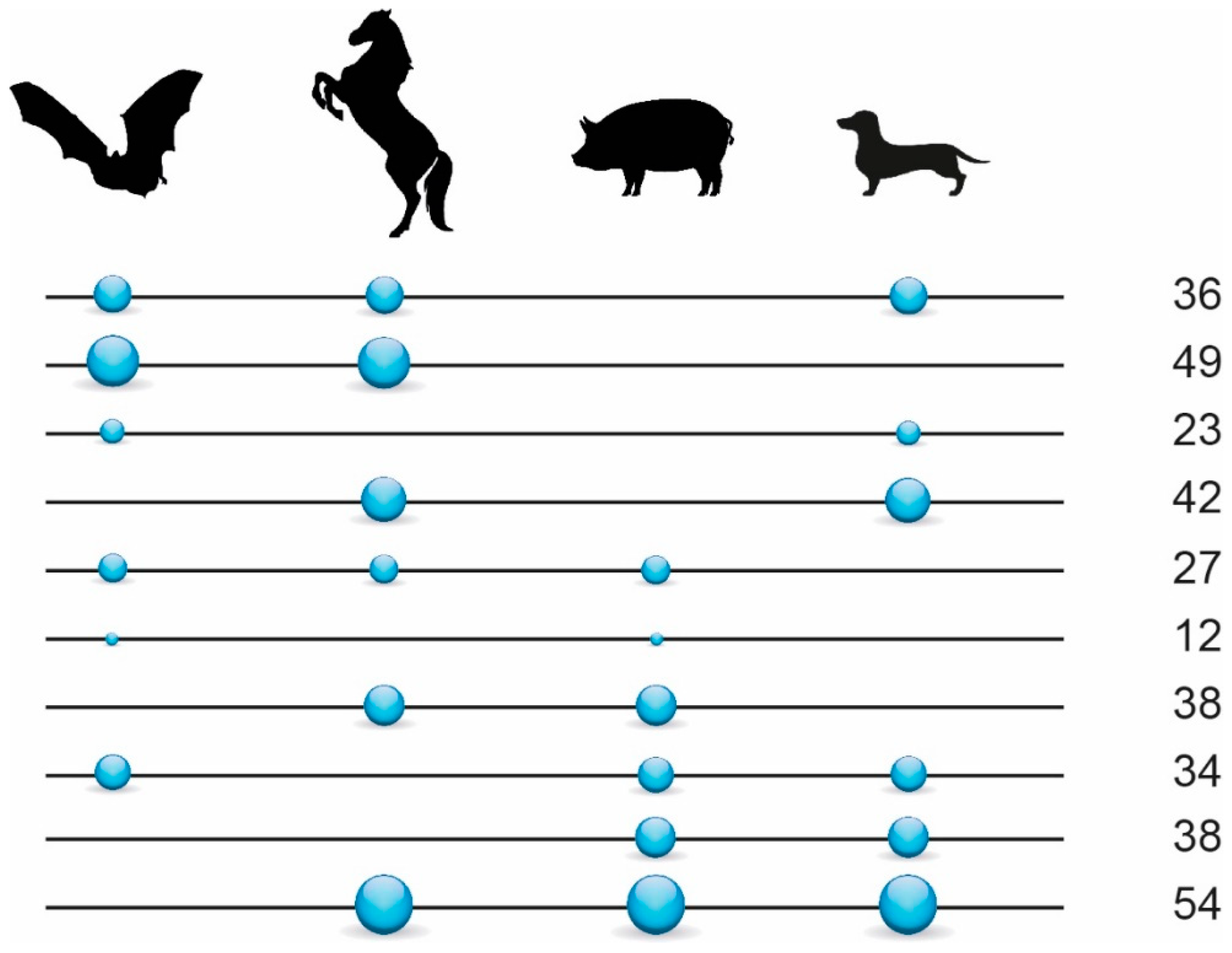
2.1.2. Screening for Phylogenetically Informative Retrotransposons
2.1.3. Phylogenetic Reconstructions
2.2. Coding Exon Data Analyses
2.2.1. Generating Mammalian Exon Alignments
2.2.2. Inference of Best-Fit Models of Sequence Evolution and Exon Tree
2.2.3. Species Tree Inference Using Concatenation and Coalescence Methods
3. Results
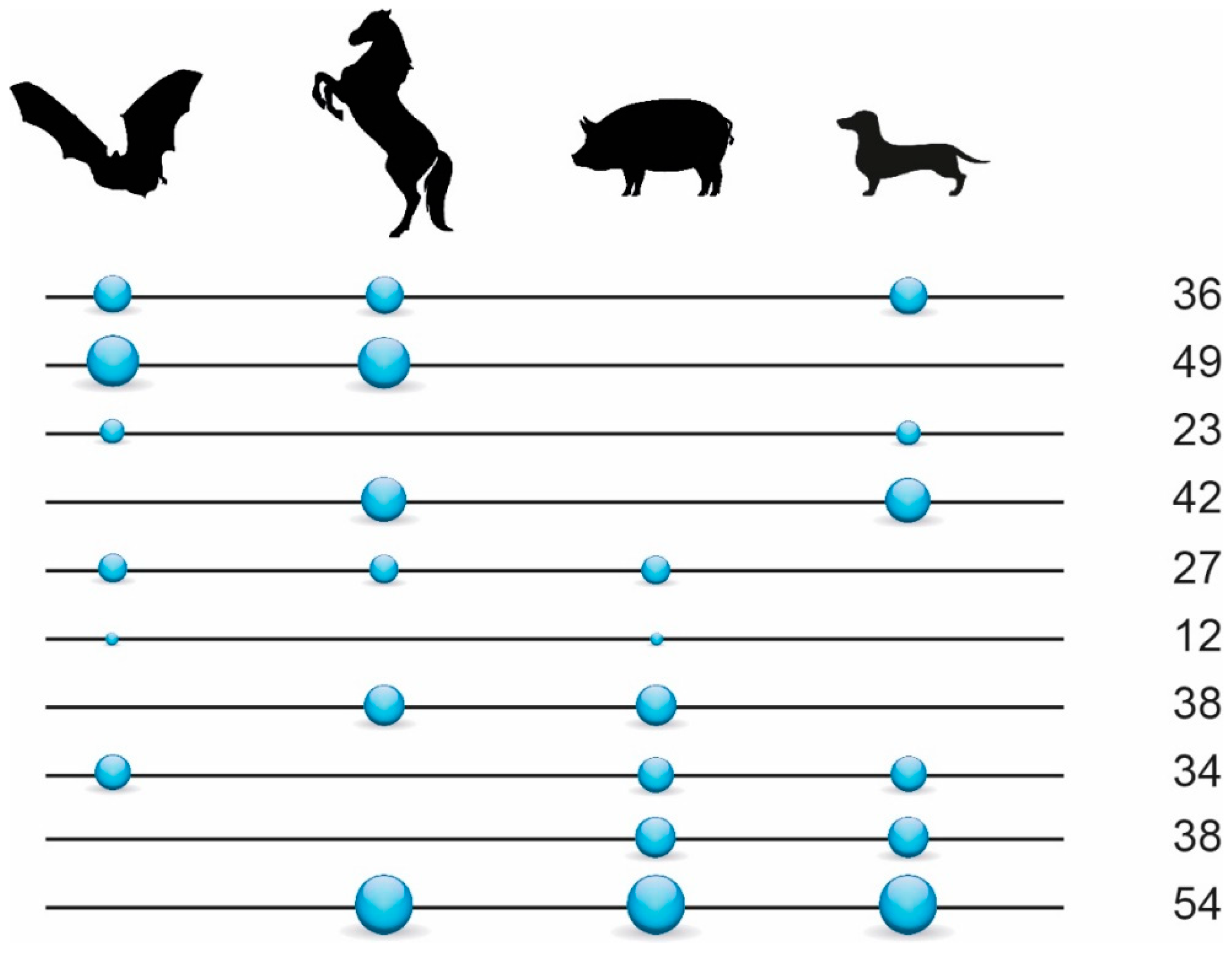
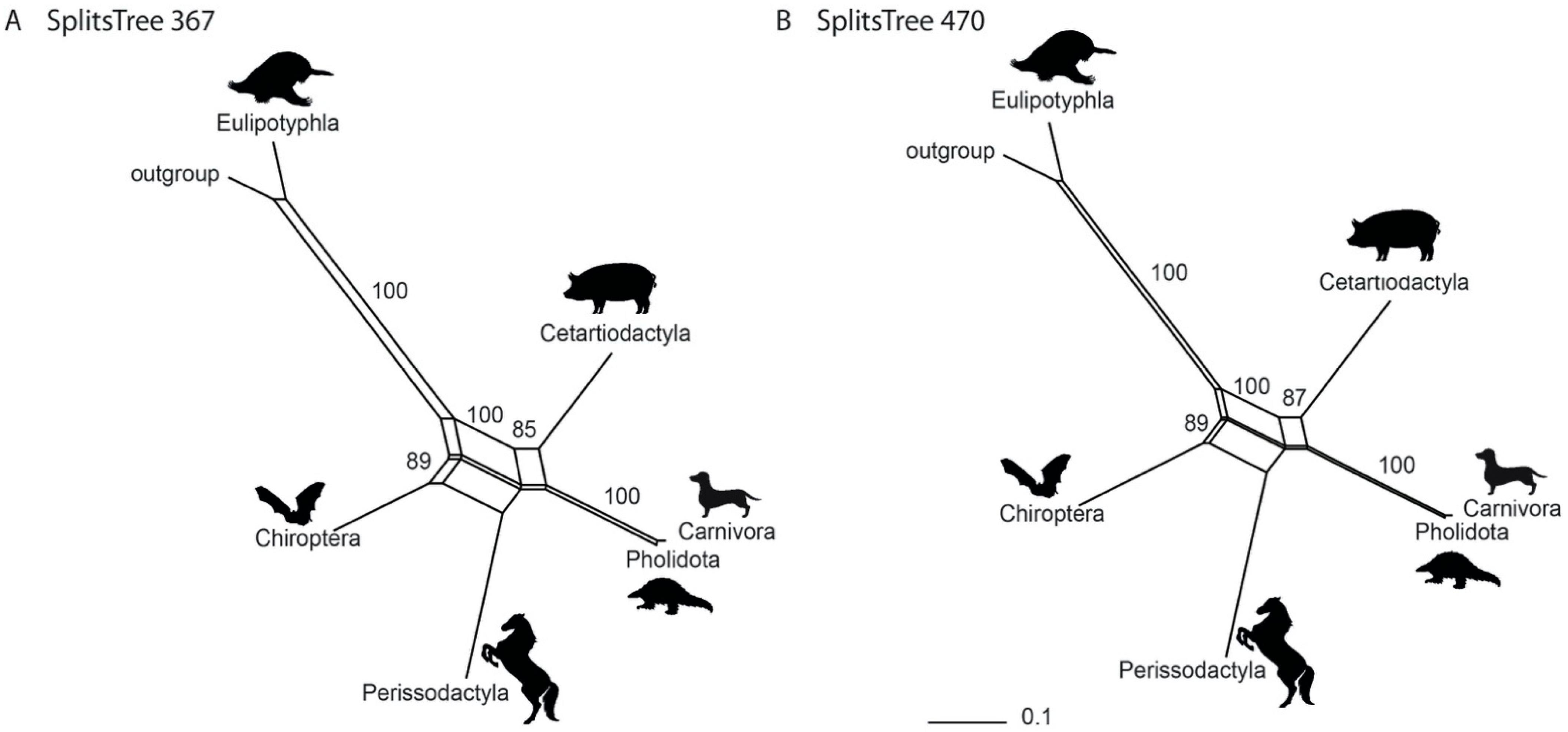
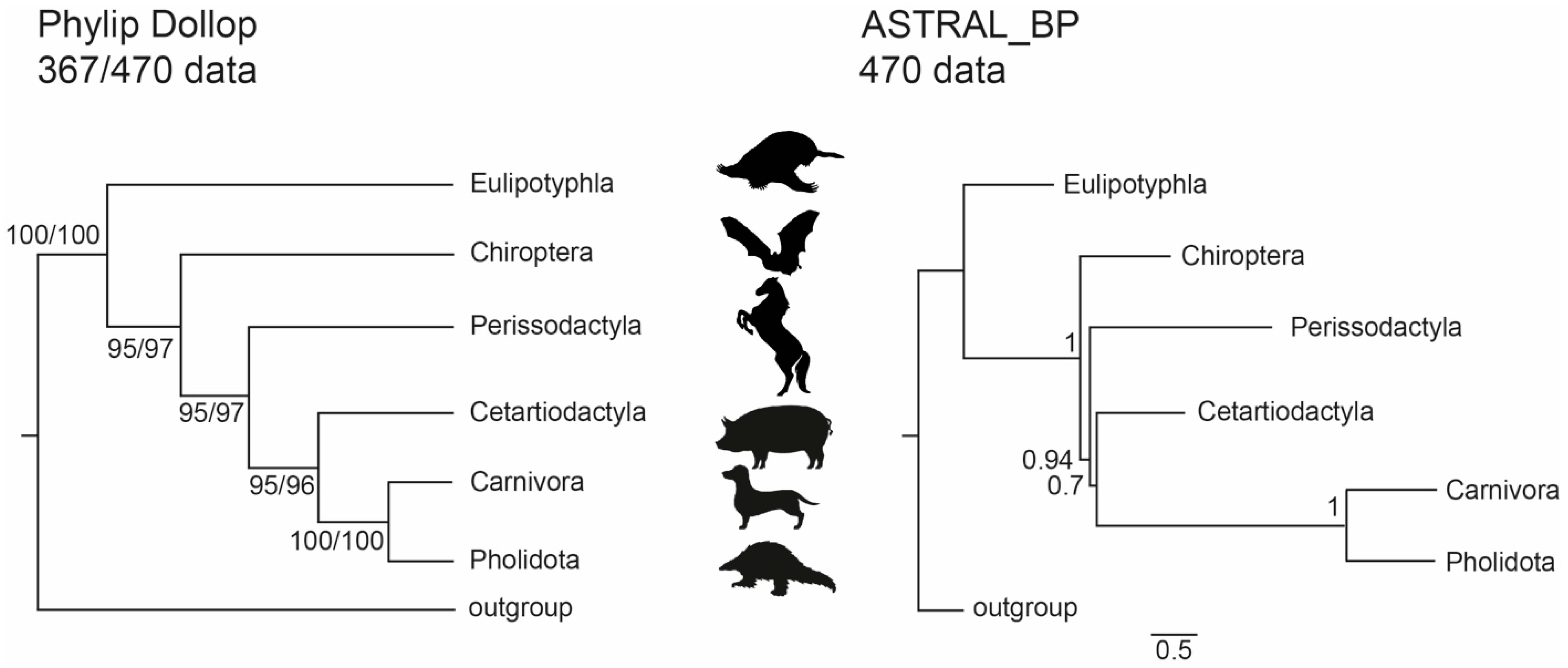
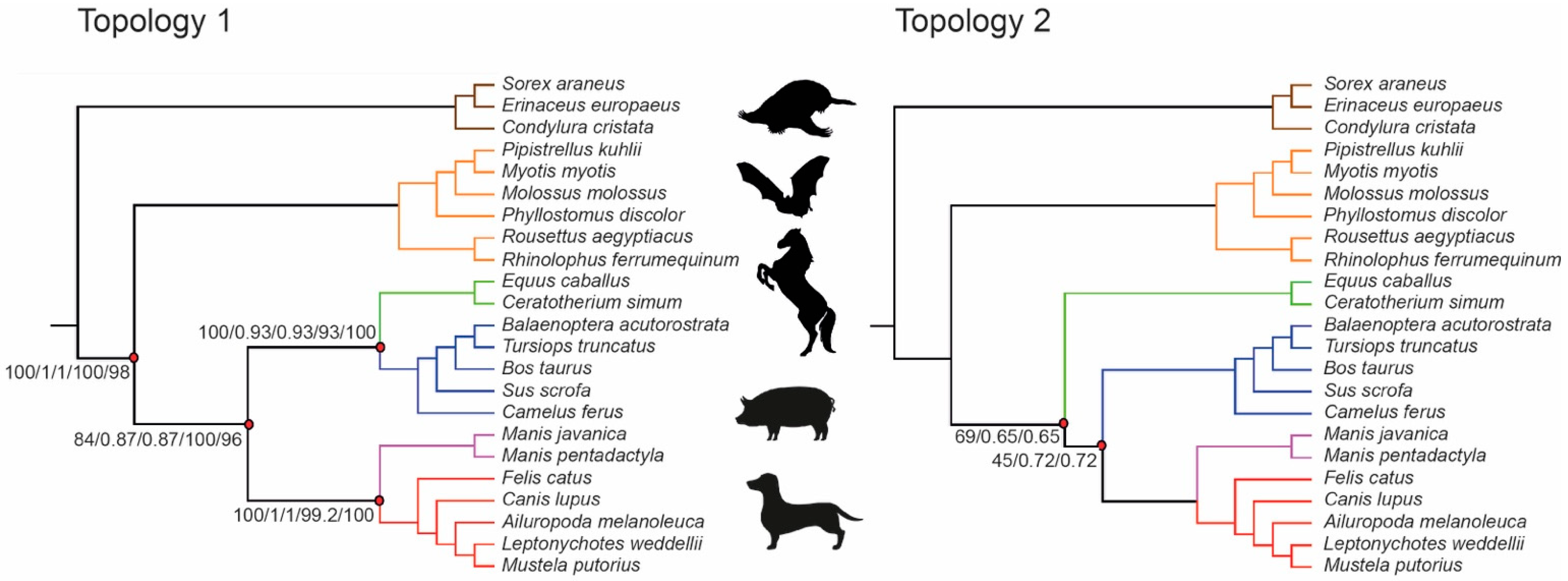
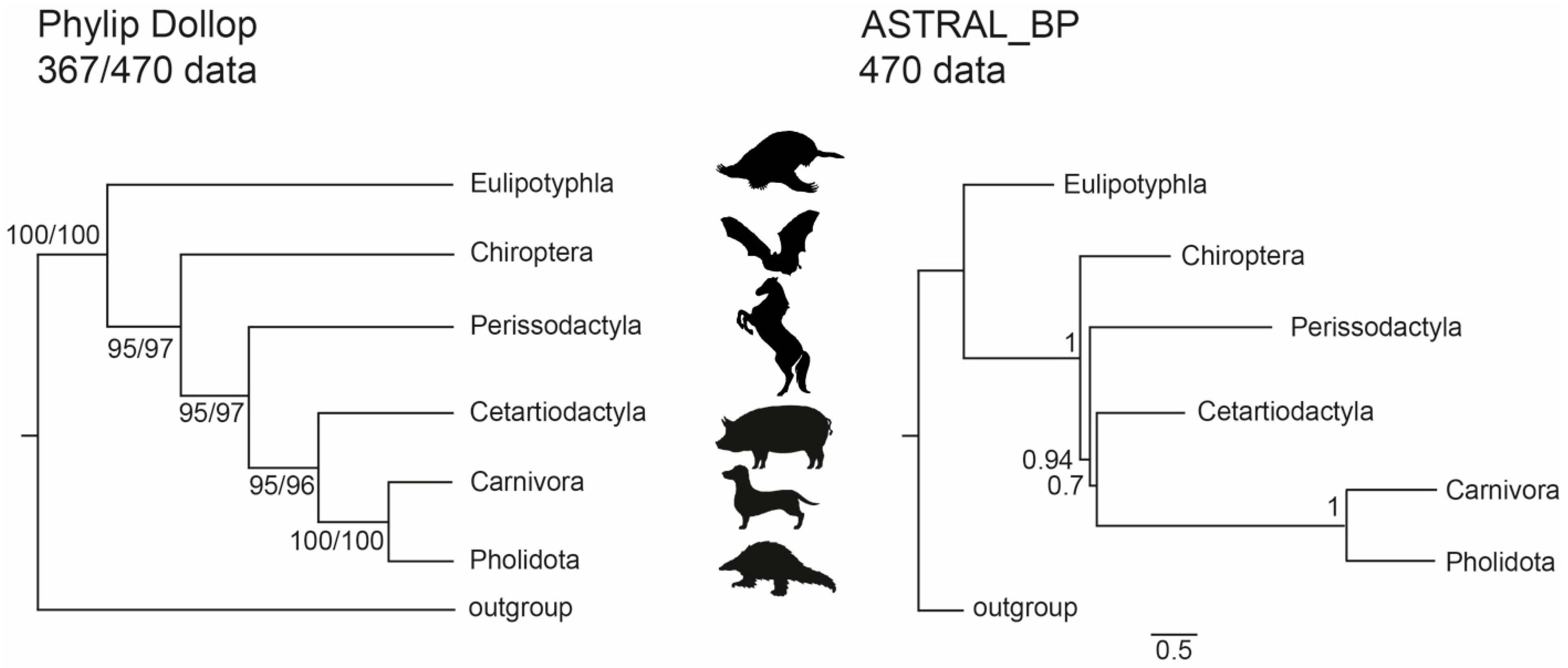
3.1. Phylogenetic Inference Using Retrotransposon Presence/Absence Data
3.2. Phylogenetic Inference Using Exon Data
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Degnan, J.H.; Rosenberg, N.A. Discordance of species trees with their most likely gene trees. PLoS Genet. 2006, 2, e68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuritzin, A.; Kischka, T.; Schmitz, J.; Churakov, G. Incomplete lineage sorting and hybridization statistics for large-scale retroposon insertion data. PLoS Comput. Biol. 2016, 12, e1004812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doronina, L.; Churakov, G.; Shi, J.; Brosius, J.; Baertsch, R.; Clawson, H.; Schmitz, J. Exploring massive incomplete lineage sorting in arctoids (Laurasiatheria, Carnivora). Mol. Biol. Evol. 2015, 32, 3194–3204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doronina, L.; Feigin, C.Y.; Schmitz, J. Reunion of Australasian possums by shared SINE insertions. Syst. Biol. 2022, syac025. [Google Scholar] [CrossRef]
- Feigin, C.Y.; Newton, A.H.; Doronina, L.; Schmitz, J.; Hipsley, C.A.; Mitchell, K.J.; Gower, G.; Llamas, B.; Soubrier, J.; Heider, T.N.; et al. Genome of the Tasmanian tiger provides insights into the evolution and demography of an extinct marsupial carnivore. Nat. Ecol. Evol. 2018, 2, 182–192. [Google Scholar] [CrossRef]
- Suh, A.; Paus, M.; Kiefmann, M.; Churakov, G.; Franke, F.A.; Brosius, J.; Kriegs, J.O.; Schmitz, J. Mesozoic retroposons reveal parrots as the closest living relatives of passerine birds. Nat. Commun. 2011, 2, 443. [Google Scholar] [CrossRef]
- Matzke, A.; Churakov, G.; Berkes, P.; Arms, E.M.; Kelsey, D.; Brosius, J.; Kriegs, J.O.; Schmitz, J. Retroposon insertion patterns of neoavian birds: Strong evidence for an extensive incomplete lineage sorting era. Mol. Biol. Evol. 2012, 29, 1497–1501. [Google Scholar] [CrossRef] [Green Version]
- Koblmüller, S.; Egger, B.; Sturmbauer, C.; Sefc, K.M. Rapid radiation, ancient incomplete lineage sorting and ancient hybridization in the endemic Lake Tanganyika cichlid tribe Tropheini. Mol. Phylogenet. Evol. 2010, 55, 318–334. [Google Scholar] [CrossRef]
- Alexander, A.M.; Su, Y.C.; Oliveros, C.H.; Olson, K.V.; Travers, S.L.; Brown, R.M. Genomic data reveals potential for hybridization, introgression, and incomplete lineage sorting to confound phylogenetic relationships in an adaptive radiation of narrow-mouth frogs. Evolution 2017, 71, 475–488. [Google Scholar] [CrossRef]
- Suh, A.; Smeds, L.; Ellegren, H. The dynamics of incomplete lineage sorting across the ancient adaptive radiation of neoavian birds. PLoS Biol. 2015, 13, e1002224. [Google Scholar] [CrossRef] [Green Version]
- Murphy, W.J.; Eizirik, E.; O’Brien, S.J.; Madsen, O.; Scally, M.; Douady, C.J.; Teeling, E.; Ryder, O.A.; Stanhope, M.J.; de Jong, W.W.; et al. Resolution of the early placental mammal radiation using Bayesian phylogenetics. Science 2001, 294, 2348–2351. [Google Scholar] [CrossRef] [PubMed]
- dos Reis, M.; Inoue, J.; Hasegawa, M.; Asher, R.J.; Donoghue, P.C.J.; Yang, Z. Phylogenomic datasets provide both precision and accuracy in estimating the timescale of placental mammal phylogeny. Proc. R. Soc. B 2012, 279, 3491–3500. [Google Scholar] [CrossRef] [PubMed]
- Emerling, C.A.; Huynh, H.T.; Nguyen, M.A.; Meredith, R.W.; Springer, M.S. Spectral shifts of mammalian ultraviolet-sensitive pigments (short wavelength-sensitive opsin 1) are associated with eye length and photic niche evolution. Proc. R. Soc. B 2015, 282, 20151817. [Google Scholar] [CrossRef] [Green Version]
- Foley, N.M.; Springer, M.S.; Teeling, E.C. Mammal madness: Is the mammal tree of life not yet resolved. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2016, 371, 20150140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nery, M.F.; Gonzalez, D.J.; Hoffmann, F.G.; Opazo, J.C. Resolution of the laurasiatherian phylogeny: Evidence from genomic data. Mol. Phylogenet. Evol. 2012, 64, 685–689. [Google Scholar] [CrossRef] [PubMed]
- Tarver, J.E.; Dos Reis, M.; Mirarab, S.; Moran, R.J.; Parker, S.; O’Reilly, J.E.; King, B.L.; O’Connell, M.J.; Asher, R.J.; Warnow, T.; et al. The interrelationships of placental mammals and the limits of phylogenetic inference. Genome Biol. Evol. 2016, 8, 330–344. [Google Scholar] [CrossRef]
- Chen, M.-Y.; Liang, D.; Zhang, P. Phylogenomic resolution of the phylogeny of laurasiatherian mammals: Exploring phylogenetic signals within coding and noncoding sequences. Genome Biol. Evol. 2017, 9, 1998–2012. [Google Scholar] [CrossRef] [Green Version]
- Lv, X.; Hu, J.; Hu, Y.; Li, Y.; Xu, D.; Ryder, O.A.; Irwin, D.M.; Yu, L. Diverse phylogenomic datasets uncover a concordant scenario of laurasiatherian interordinal relationships. Mol. Phylogenet. Evol. 2021, 157, 107065. [Google Scholar] [CrossRef]
- Chan, K.O.; Hutter, C.R.; Wood, P.L.; Grismer, L.L.; Brown, R.M. Larger, unfiltered datasets are more effective at resolving phylogenetic conflict: Introns, exons, and UCEs resolve ambiguities in Golden-backed frogs (Anura: Ranidae; genus Hylarana). Mol. Phylogenet. Evol. 2020, 151, 106899. [Google Scholar] [CrossRef]
- Springer, M.S.; Foley, N.M.; Brady, P.L.; Gatesy, J.; Murphy, W.J. Evolutionary models for the diversification of placental mammals across the KPg boundary. Front. Genet. 2019, 10, 1241. [Google Scholar] [CrossRef]
- Esselstyn, J.A.; Oliveros, C.H.; Swanson, M.T.; Faircloth, B.C. Investigating difficult nodes in the placental mammal tree with expanded taxon sampling and thousands of ultraconserved elements. Genome Biol. Evol. 2017, 9, 2308–2321. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Zhang, J.; Rheindt, F.E.; Lei, F.; Qu, Y.; Wang, Y.; Zhang, Y.; Sullivan, C.; Nie, W.; Wang, J.; et al. Genomic evidence reveals a radiation of placental mammals uninterrupted by the KPg boundary. Proc. Natl. Acad. Sci. USA 2017, 114, E7282–E7290. [Google Scholar] [CrossRef] [Green Version]
- Gatesy, J.; Springer, M.S. Phylogenomic red flags: Homology errors and zombie lineages in the evolutionary diversification of placental mammals. Proc. Natl. Acad. Sci. USA 2017, 114, E9431–E9432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, Y.; Wu, S.; Edwards, S.V.; Liu, L. The effect of alignment uncertainty, substitution models and priors in building and dating the mammal tree of life. BMC Evol. Biol. 2019, 19, 203. [Google Scholar] [CrossRef]
- Jebb, D.; Huang, Z.; Pippel, M.; Hughes, G.M.; Lavrichenko, K.; Devanna, P.; Winkler, S.; Jermiin, L.S.; Skirmuntt, E.C.; Katzourakis, A.; et al. Six reference-quality genomes reveal evolution of bat adaptations. Nature 2020, 583, 578–584. [Google Scholar] [CrossRef] [PubMed]
- Murphy, W.J.; Foley, N.M.; Bredemeyer, K.R.; Gatesy, J.; Springer, M.S. Phylogenomics and the genetic architecture of the placental mammal radiation. Annu. Rev. Anim. Biosci. 2021, 9, 29–53. [Google Scholar] [CrossRef] [PubMed]
- Springer, M.S.; Gatesy, J. The gene tree delusion. Mol. Phylogenet. Evol. 2016, 94, 1–33. [Google Scholar] [CrossRef] [PubMed]
- Scornavacca, C.; Galtier, N. Incomplete lineage sorting in mammalian phylogenomics. Syst. Biol. 2017, 66, 112–120. [Google Scholar] [CrossRef]
- Liu, L.; Yu, L.; Kubatko, L.; Pearl, D.K.; Edwards, S.V. Coalescent methods for estimating phylogenetic trees. Mol. Phyl. Evol. 2009, 53, 320–328. [Google Scholar] [CrossRef]
- Song, S.; Liu, L.; Edwards, S.V.; Wu, S. Resolving conflict in eutherian mammal phylogeny using phylogenomics and the multispecies coalescent model. Proc. Natl. Acad. Sci. USA 2012, 109, 14942–14947. [Google Scholar] [CrossRef] [Green Version]
- Springer, M.S.; Gatesy, J. Delimiting coalescence genes (c-genes in phylogenomic datasets). Genes 2018, 9, 123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shedlock, A.M.; Takahashi, K.; Okada, N. SINEs of speciation: Tracking lineages with retroposons. Trends Ecol. Evol. 2004, 19, 545–553. [Google Scholar] [CrossRef] [PubMed]
- Doronina, L.; Reising, O.; Clawson, H.; Ray, D.A.; Schmitz, J. True homoplasy of retrotransposon insertions in primates. Syst. Biol. 2019, 68, 482–493. [Google Scholar] [CrossRef] [PubMed]
- Springer, M.S.; Molloy, E.K.; Sloan, D.B.; Simmons, M.P.; Gatesy, J. ILS-aware analysis of low-homoplasy retroelement insertions: Inference of species trees and introgression using quartets. J. Hered. 2020, 111, 147–168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ray, D.A.; Xing, J.; Salem, A.H.; Batzer, M.A. SINEs of a nearly perfect character. Syst. Biol. 2006, 55, 928–935. [Google Scholar] [CrossRef] [PubMed]
- Doronina, L.; Churakov, G.; Kuritzin, A.; Shi, J.; Baertsch, R.; Clawson, H.; Schmitz, J. Speciation network in Laurasiatheria: Retrophylogenomic signals. Genome Res. 2017, 27, 997–1003. [Google Scholar] [CrossRef] [PubMed]
- Nishihara, H.; Hasegawa, M.; Okada, N. Pegasoferae, an unexpected mammalian clade revealed by tracking ancient retroposon insertions. Proc. Natl. Acad. Sci. USA 2006, 103, 9929–9934. [Google Scholar] [CrossRef] [Green Version]
- Churakov, G.; Kuritzin, A.; Chukharev, K.; Zhang, F.; Wünnemann, F.; Ulyantsev, V.; Schmitz, J. A 4-lineage statistical suite to evaluate the support of large-scale retrotransposon insertion data to reconstruct evolutionary trees. BioRxiv 2020. [Google Scholar] [CrossRef]
- Harris, R.S. Improved Pairwise Alignment of Genomic DNA. Ph.D. Thesis, Pennsylvania State University, State College, PA, USA, 2007. [Google Scholar]
- Kent, W.J.; Baertsch, R.; Hinrichs, A.; Miller, W.; Haussler, D. Evolution’s cauldron: Duplication, deletion, and rearrangement in the mouse and human genomes. Proc. Natl. Acad. Sci. USA 2003, 100, 11484–11489. [Google Scholar] [CrossRef] [Green Version]
- Osipova, E.; Hecker, N.; Hiller, M. RepeatFiller newly identifies megabases of aligning repetitive sequences and improves annotations of conserved non-exonic elements. Gigascience 2019, 8, giz132. [Google Scholar] [CrossRef] [Green Version]
- Suarez, H.G.; Langer, B.E.; Ladde, P.; Hiller, M. chainCleaner improves genome alignment specificity and sensitivity. Bioinformatics 2017, 33, 1596–1603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Churakov, G.; Zhang, F.; Grundmann, N.; Makalowski, W.; Noll, A.; Doronina, L.; Schmitz, J. The multicomparative 2-n-way genome suite. Genome Res. 2020, 30, 1508–1516. [Google Scholar] [CrossRef] [PubMed]
- Meredith, R.W.; Janečka, J.E.; Gatesy, J.; Ryder, O.A.; Fisher, C.A.; Teeling, E.C.; Goodbla, A.; Eizirik, E.; Simão, T.L.; Stadler, T.; et al. Impacts of the cretaceous terrestrial revolution and KPg extinction on mammal diversification. Science 2011, 334, 521–524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huson, D.H.; Bryant, D. Application of phylogenetic networks in evolutionary studies. Mol. Biol. Evol. 2006, 23, 254–267. [Google Scholar] [CrossRef] [PubMed]
- Felsenstein, J. PHYLIP—Phylogeny inference package (version 3.2). Cladistics 1989, 5, 164–166. [Google Scholar]
- Molloy, E.K.; Gatesy, J.; Springer, M.S. Theoretical and practical considerations when using retroelement insertions to estimate species trees in the anomaly zone. Syst. Biol. 2021, syab086. [Google Scholar] [CrossRef]
- Ronquist, F.; Teslenko, M.; van der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef] [Green Version]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; Von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [Green Version]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE2: New models and efficient methods for phylogenetic inference in the genomic era. Mol. Ecol. Evol. 2020, 36, 1530–1534. [Google Scholar]
- Kück, P.; Meusemann, K. FASconCAT: Convenient handling of data matrices. Mol. Phylogenet. Evol. 2010, 56, 1115–1118. [Google Scholar] [CrossRef]
- Minh, B.Q.; Nguyen, M.A.T.; von Haeseler, A. Ultrafast approximation for phylogenetic bootstrap. Mol. Biol. Evol. 2013, 30, 1188–1195. [Google Scholar] [CrossRef] [PubMed]
- Mirarab, S.; Reaz, R.; Bayzid, M.S.; Zimmermann, T.; Swensom, M.S.; Warnow, T.A. ASTRAL: Genome-scale coalescent-based species tree estimation. Bioinformatics 2014, 30, 541–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chifman, J.; Kubatko, L. Quartet inference from SNP data under the coalescent model. Bioinformatics 2014, 30, 3317–3324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paradis, E.; Claude, J.; Strimmer, K. APE: Analyses of phylogenetics and evolution in R language. Bioinformatics 2004, 2, 289–290. [Google Scholar] [CrossRef] [Green Version]
- Schliep, K.P. Phangorn: Phylogenetic analysis in R. Bioinformatics 2011, 27, 592–593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jermiin, L.S.; Poladian, L.; Charleston, M.A. Is the “big bang” in animal evolution real? Science 2005, 310, 1910–1911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsagkogeorga, G.; Parker, J.; Stupka, E.; Cotton, J.A.; Rossiter, S.J. Phylogenomic analyses elucidate the evolutionary relationships of bats. Curr. Biol. 2013, 23, 2262–2267. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
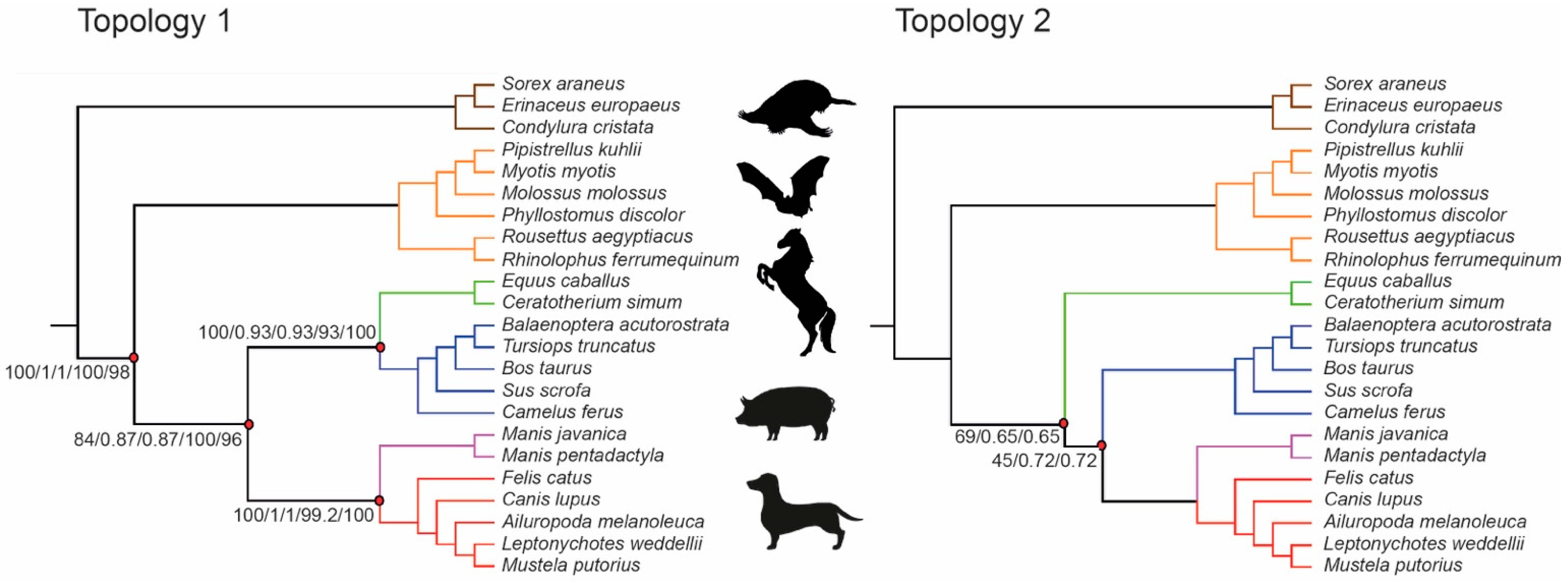
| Seq Type | Data Type | Exon Dataset | Method | Topology | Lowest Bootstrap/Local Posterior Probability Values |
|---|---|---|---|---|---|
| NT | Concatenated SM | 9266 | ML | 1 | 84 (Fer(Per,Cet)) |
| NT | Coalescence, b.n.c. | 9266 | ASTRAL | 1 | 0.87 (Fer(Per,Cet)) |
| NT | Concatenated SM | 9266 | SVDquartets | 1 | 93 (Per,Cet) |
| NT | Concatenated SM | 157 | ML | 2 | 45 (Fer,Cet) |
| NT | Coalescence, b.n.c. | 157 | ASTRAL | 2 | 0.65 (Per(Fer,Cet)) |
| NT | Concatenated SM | 157 | SVDquartets | 5 | 65.8 (Chi,Eul) |
| AA | Concatenated SM | 9266 | ML | 1 | 96 (Per,Cet) |
| AA | Coalescence, b.n.c. | 9266 | ASTRAL | 6 | 0.5 (Car,Pho) |
| AA | Concatenated SM | 157 | ML | 3 | 46 (Cet,Chi) |
| AA | Coalescence, b.n.c. | 157 | ASTRAL | 4 | 0.35 (Cet(Pho(Per,Car))) |
| NT | Coalescence, p.c. | 9266 | ASTRAL | 1 | 0.59 (Fer(Per,Cet)) |
| AA | Coalescence, p.c. | 9266 | ASTRAL | 6 | 0.59 (Per,Fer) |
| NT | Coalescence, p.c. | 157 | ASTRAL | 6 | 0.46 (Cet(Per,Fer)) |
| AA | Coalescence, p.c. | 157 | ASTRAL | 7 | 0.24 (Cet,Pho) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Doronina, L.; Hughes, G.M.; Moreno-Santillan, D.; Lawless, C.; Lonergan, T.; Ryan, L.; Jebb, D.; Kirilenko, B.M.; Korstian, J.M.; Dávalos, L.M.; et al. Contradictory Phylogenetic Signals in the Laurasiatheria Anomaly Zone. Genes 2022, 13, 766. https://doi.org/10.3390/genes13050766
Doronina L, Hughes GM, Moreno-Santillan D, Lawless C, Lonergan T, Ryan L, Jebb D, Kirilenko BM, Korstian JM, Dávalos LM, et al. Contradictory Phylogenetic Signals in the Laurasiatheria Anomaly Zone. Genes. 2022; 13(5):766. https://doi.org/10.3390/genes13050766
Chicago/Turabian StyleDoronina, Liliya, Graham M. Hughes, Diana Moreno-Santillan, Colleen Lawless, Tadhg Lonergan, Louise Ryan, David Jebb, Bogdan M. Kirilenko, Jennifer M. Korstian, Liliana M. Dávalos, and et al. 2022. "Contradictory Phylogenetic Signals in the Laurasiatheria Anomaly Zone" Genes 13, no. 5: 766. https://doi.org/10.3390/genes13050766
APA StyleDoronina, L., Hughes, G. M., Moreno-Santillan, D., Lawless, C., Lonergan, T., Ryan, L., Jebb, D., Kirilenko, B. M., Korstian, J. M., Dávalos, L. M., Vernes, S. C., Myers, E. W., Teeling, E. C., Hiller, M., Jermiin, L. S., Schmitz, J., Springer, M. S., & Ray, D. A. (2022). Contradictory Phylogenetic Signals in the Laurasiatheria Anomaly Zone. Genes, 13(5), 766. https://doi.org/10.3390/genes13050766