
Rif1-Dependent Control of Replication Timing


Abstract
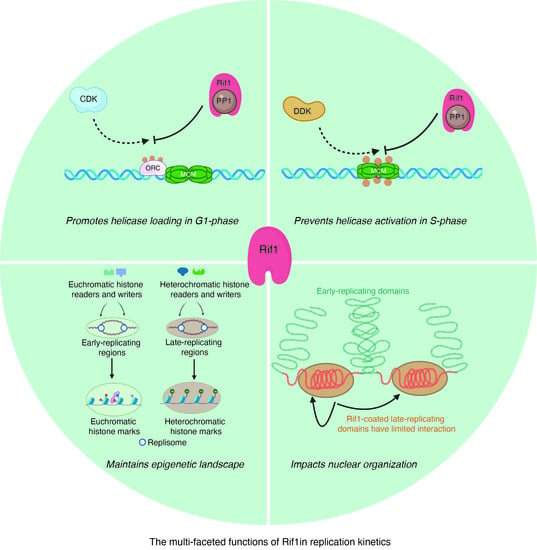
{kind=link}
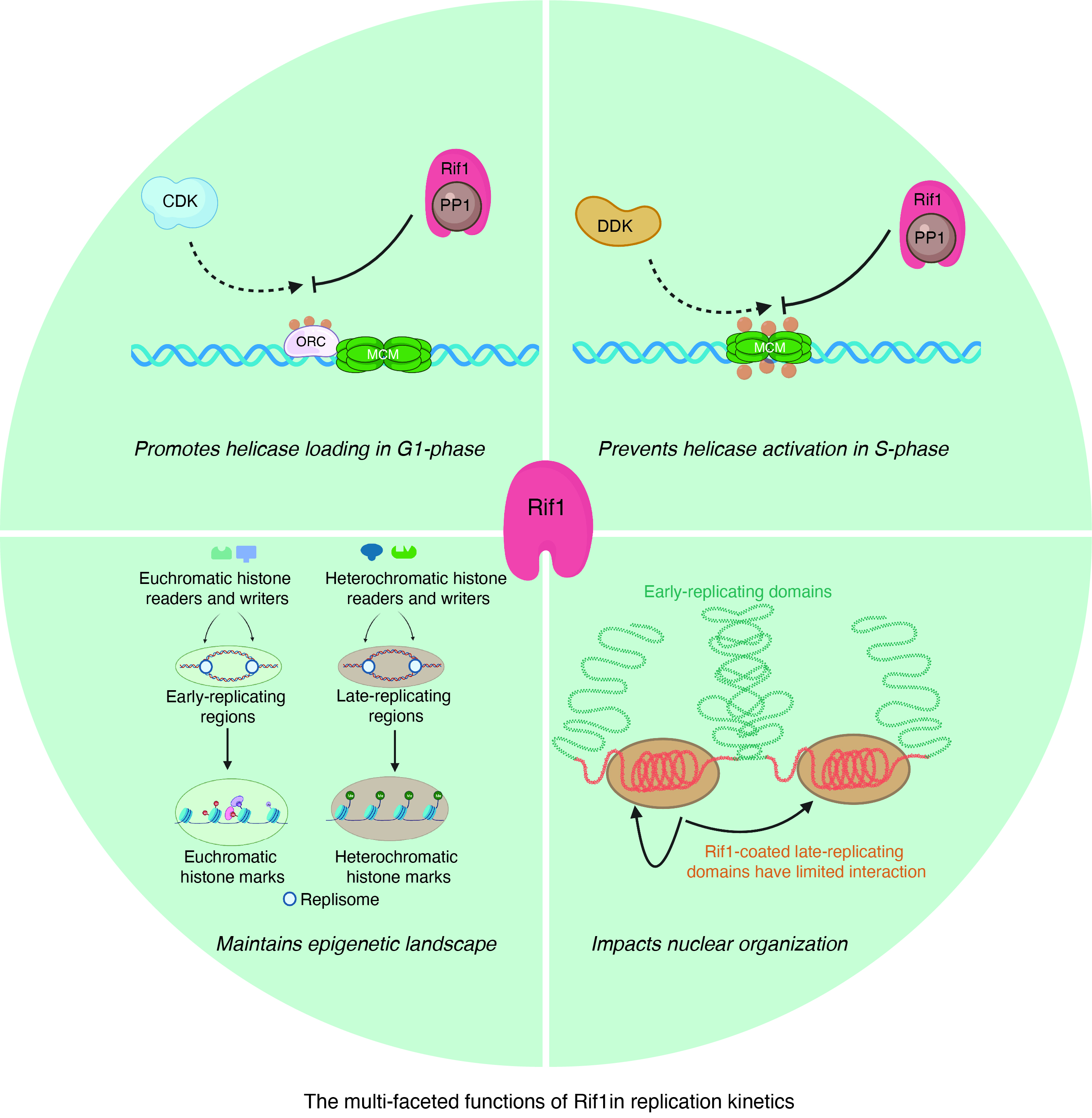
{kind=link}
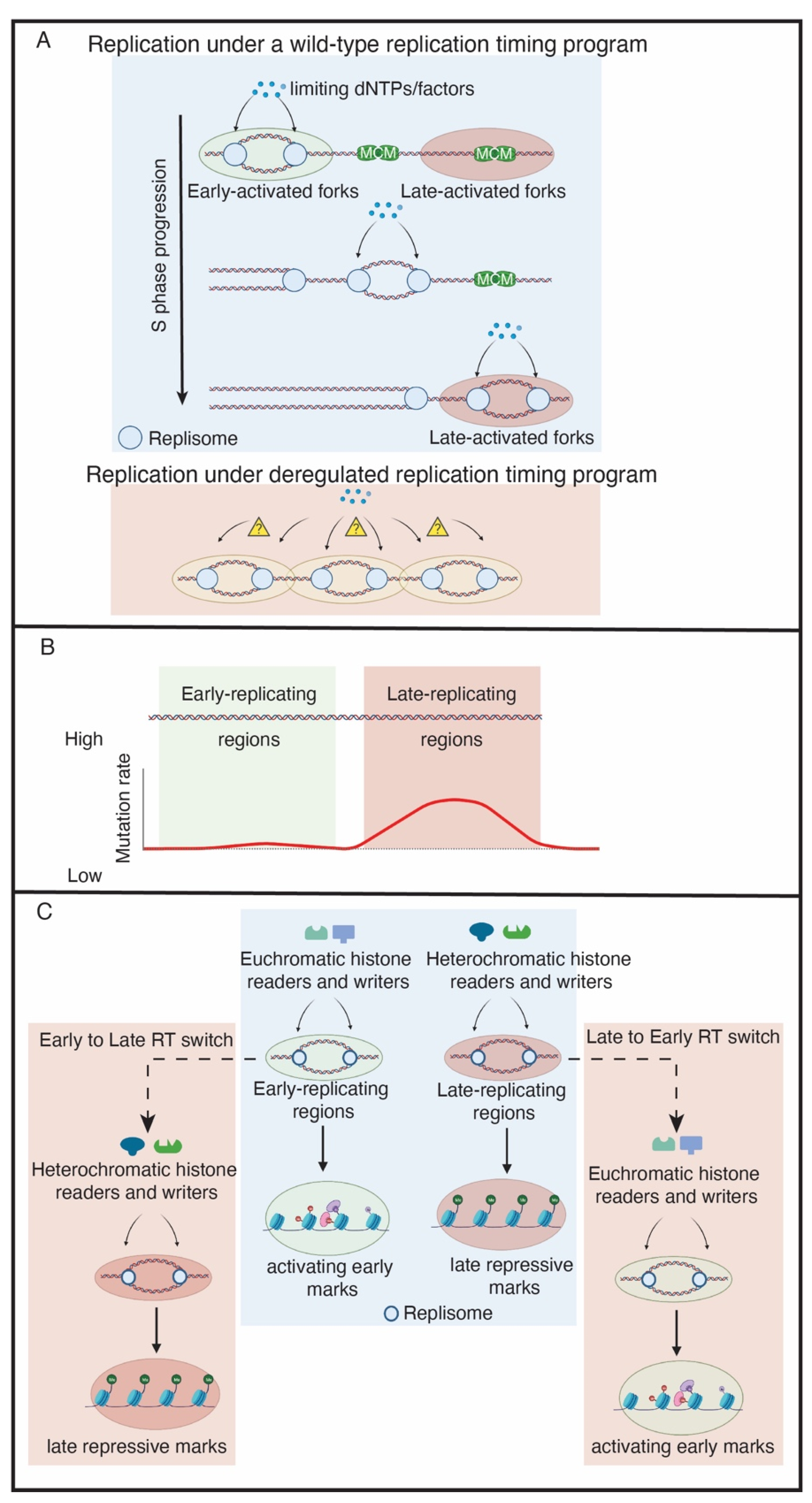
{kind=link}
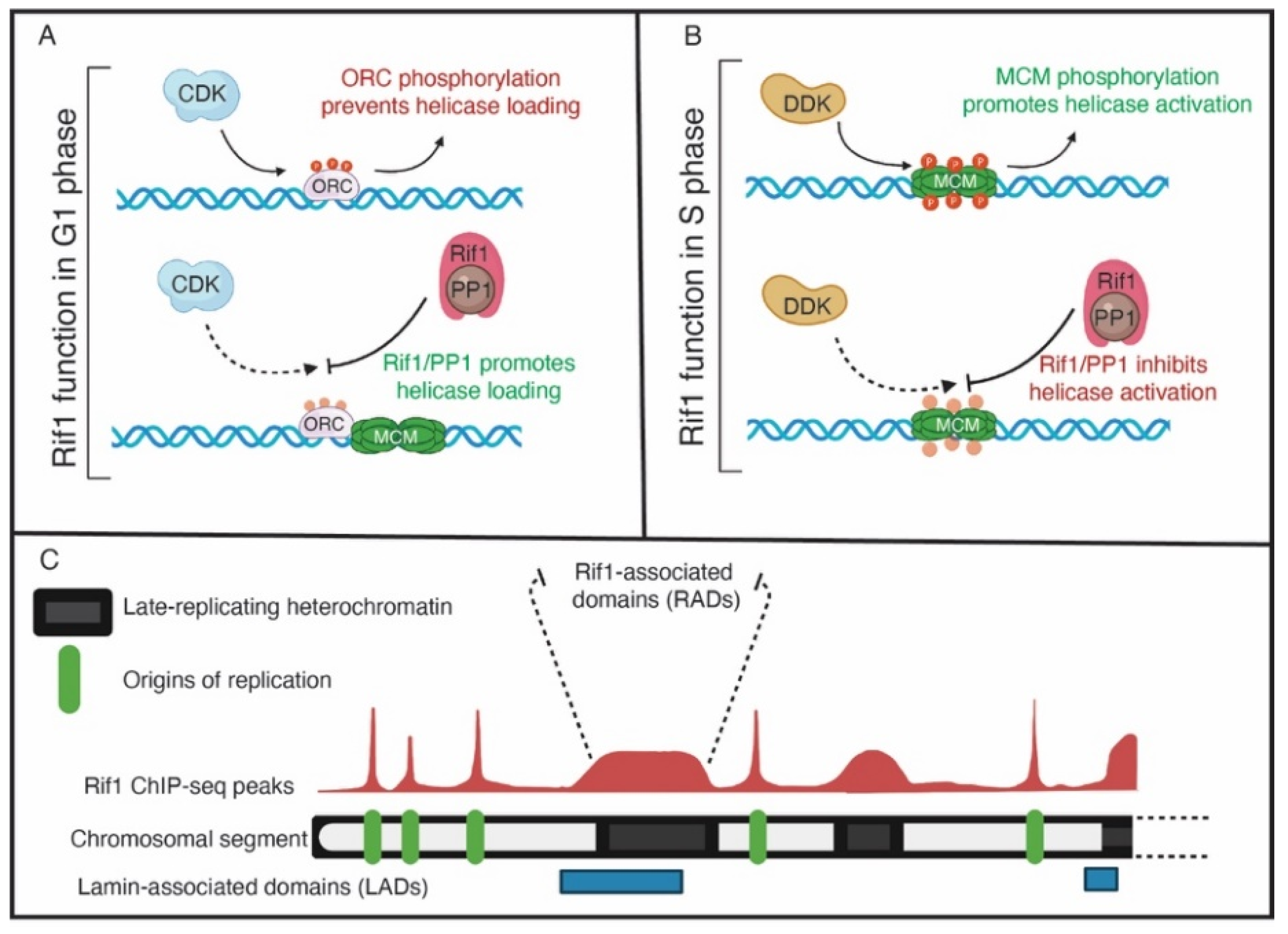
{kind=link}
1. Introduction
2. The Biological Function of Replication Timing
3. Rif1 Is a PP1 Specificity Factor That Regulates RT across Species
4. Rif1 Activity Is Regulated during Development
5. Rif1 Dynamically Associates with Chromatin through the Cell Cycle and Development
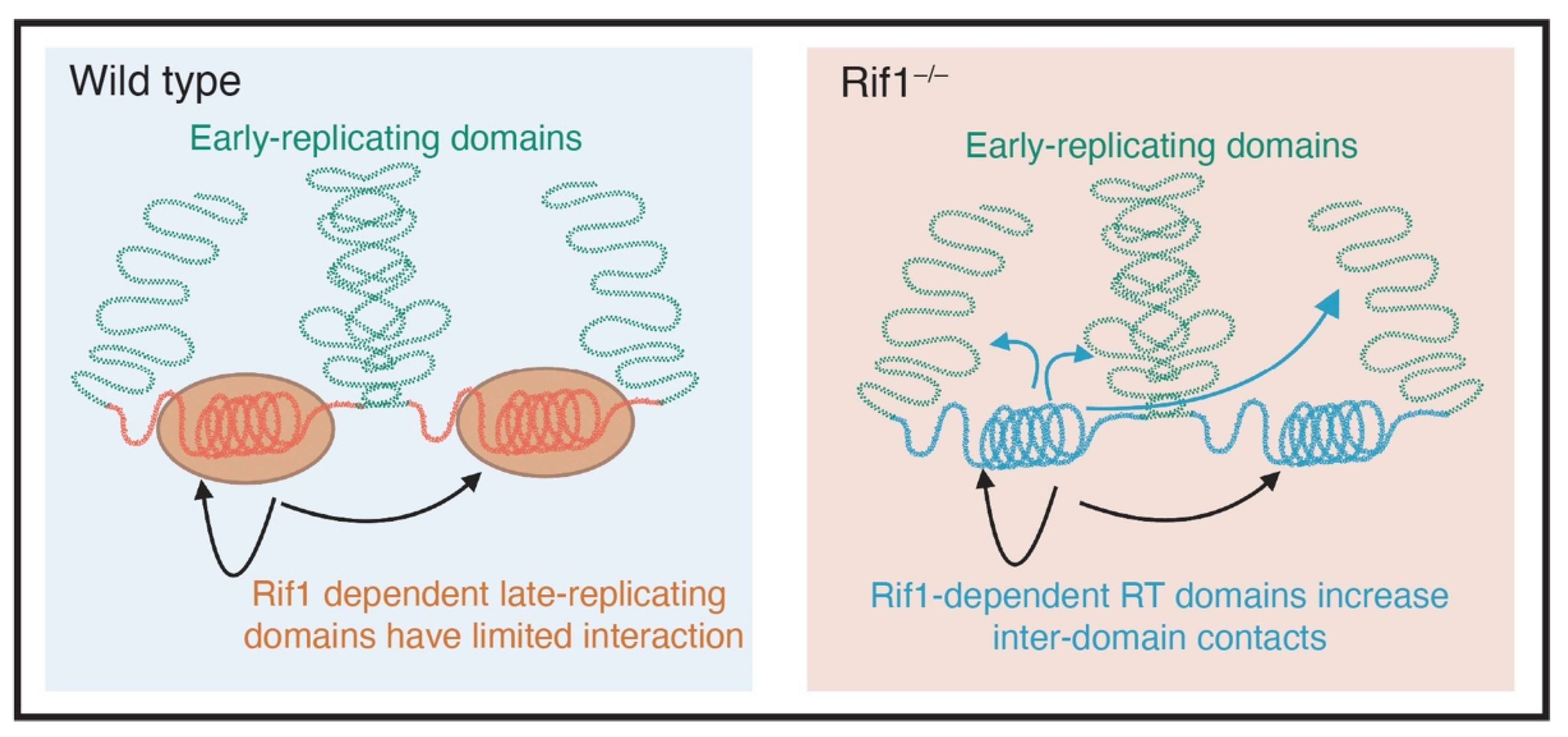
6. Rif1 and Nuclear Organization
7. Future Directions and Outstanding Questions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Leonard, A.C.; Mechali, M. DNA replication origins. Cold Spring Harb. Perspect. Med. 2013, 5, a010116. [Google Scholar] [CrossRef]
- Bell, S.P.; Labib, K. Chromosome duplication in Saccharomyces cerevisiae. Genetics 2016, 203, 1027–1067. [Google Scholar] [CrossRef]
- Diffley, J.F.X. Regulation of Early Events in Chromosome Replication. Curr. Biol. 2004, 14, R778–R786. [Google Scholar] [CrossRef] [PubMed]
- Sheu, Y.-J.; Stillman, B. Cdc7-Dbf4 phosphorylates MCM proteins via a docking site-mediated mechanism to promote S phase progression. Mol. Cell 2006, 24, 101–113. [Google Scholar] [CrossRef] [PubMed]
- Bell, S.P.; Dutta, A. DNA Replication in Eukaryotic Cells. Annu. Rev. Biochem. 2003, 71, 333–374. [Google Scholar] [CrossRef]
- Siddiqui, K.; On, K.F.; Diffley, J.F.X. Regulating DNA replication in Eukarya. Cold Spring Harb. Perspect. Biol. 2013, 5, a012930. [Google Scholar] [CrossRef] [PubMed]
- Moyer, S.E.; Lewis, P.W.; Botchan, M.R. Isolation of the Cdc45/Mcm2-7/GINS (CMG) complex, a candidate for the eukaryotic DNA replication fork helicase. Proc. Natl. Acad. Sci. USA 2006, 103, 10236–10241. [Google Scholar] [CrossRef] [PubMed]
- Douglas, M.E.; Ali, F.A.; Costa, A.; Diffley, J.F.X. The mechanism of eukaryotic CMG helicase activation. Nature 2018, 555, 265–268. [Google Scholar] [CrossRef] [PubMed]
- Remus, D.; Diffley, J.F. Eukaryotic DNA replication control: Lock and load, then fire. Curr. Opin. Cell Biol. 2009, 21, 771–777. [Google Scholar] [CrossRef] [PubMed]
- Fragkos, M.; Ganier, O.; Coulombe, P.; Méchali, M. DNA replication origin activation in space and time. Nat. Rev. Mol. Cell Biol. 2015, 16, 360–374. [Google Scholar] [CrossRef]
- Rhind, N.; Yang, S.C.H.; Bechhoefer, J. Reconciling stochastic origin firing with defined replication timing. Chromosom. Res. 2010, 18, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Mantiero, D.; MacKenzie, A.; Donaldson, A.; Zegerman, P. Limiting replication initiation factors execute the temporal programme of origin firing in budding yeast. EMBO J. 2011, 30, 4805–4814. [Google Scholar] [CrossRef] [PubMed]
- Collart, C.; Allen, G.E.; Bradshaw, C.R.; Smith, J.C.; Zegerman, P. Titration of four replication factors is essential for the Xenopus laevis midblastula transition. Science 2013, 341, 893–896. [Google Scholar] [CrossRef] [PubMed]
- Demczuk, A.; Gauthier, M.G.; Veras, I.; Kosiyatrakul, S.; Schildkraut, C.L.; Busslinger, M.; Bechhoefer, J.; Norio, P. Regulation of DNA Replication within the Immunoglobulin Heavy-Chain Locus During B Cell Commitment. PLoS Biol. 2012, 10, e1001360. [Google Scholar] [CrossRef] [PubMed]
- Dijkwel, P.A.; Wang, S.; Hamlin, J.L. Initiation Sites Are Distributed at Frequent Intervals in the Chinese Hamster Dihydrofolate Reductase Origin of Replication but Are Used with Very Different Efficiencies. Mol. Cell. Biol. 2002, 22, 3053–3065. [Google Scholar] [CrossRef]
- Rhind, N.; Gilbert, D.M. DNA replication timing. Cold Spring Harb. Perspect. Biol. 2013, 5, a010132. [Google Scholar] [CrossRef]
- Wang, W.; Klein, K.N.; Proesmans, K.; Yang, H.; Marchal, C.; Zhu, X.; Borrman, T.; Hastie, A.; Weng, Z.; Bechhoefer, J.; et al. Genome-wide mapping of human DNA replication by optical replication mapping supports a stochastic model of eukaryotic replication. Mol. Cell 2021, 81, 2975–2988.e6. [Google Scholar] [CrossRef]
- Gindin, Y.; Valenzuela, M.S.; Aladjem, M.I.; Meltzer, P.S.; Bilke, S. A chromatin structure-based model accurately predicts DNA replication timing in human cells. Mol. Syst. Biol. 2014, 10, 722. [Google Scholar] [CrossRef]
- Marchal, C.; Sasaki, T.; Vera, D.; Wilson, K.; Sima, J.; Rivera-Mulia, J.C.; Trevilla-García, C.; Nogues, C.; Nafie, E.; Gilbert, D.M. Genome-wide analysis of replication timing by next-generation sequencing with E/L Repli-seq. Nat. Protoc. 2018, 13, 819–839. [Google Scholar] [CrossRef]
- Koren, A.; Massey, D.J.; Bracci, A.N. TIGER: Inferring DNA replication timing from whole-genome sequence data. Bioinformatics 2021, 37, 4001–4005. [Google Scholar] [CrossRef]
- Zhang, J.; Xu, F.; Hashimshony, T.; Keshet, I.; Cedar, H. Establishment of transcriptional competence in early and late S phase. Nature 2002, 420, 198–202. [Google Scholar] [CrossRef] [PubMed]
- Klein, K.N.; Zhao, P.A.; Lyu, X.; Sasaki, T.; Bartlett, D.A.; Singh, A.M.; Tasan, I.; Zhang, M.; Watts, L.P.; Hiraga, S.I.; et al. Replication timing maintains the global epigenetic state in human cells. Science 2021, 372, 371–378. [Google Scholar] [CrossRef] [PubMed]
- Sima, J.; Gilbert, D.M. Complex correlations: Replication timing and mutational landscapes during cancer and genome evolution. Curr. Opin. Genet. Dev. 2014, 25, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Jia, P.; Jin, H.; Meador, C.B.; Xia, J.; Ohashi, K.; Liu, L.; Pirazzoli, V.; Dahlman, K.B.; Politi, K.; Michor, F.; et al. Next-generation sequencing of paired tyrosine kinase inhibitor-sensitive and -resistant EGFR mutant lung cancer cell lines identifies spectrum of DNA changes associated with drug resistance. Genome Res. 2013, 23, 1434–1445. [Google Scholar] [CrossRef] [PubMed]
- Stamatoyannopoulos, J.A.; Adzhubei, I.; Thurman, R.E.; Kryukov, G.V.; Mirkin, S.M.; Sunyaev, S.R. Human mutation rate associated with DNA replication timing. Nat. Genet. 2009, 41, 393–395. [Google Scholar] [CrossRef]
- Gilbert, D.M. Replication timing and transcriptional control: Beyond cause and effect. Curr. Opin. Cell Biol. 2002, 14, 377–383. [Google Scholar] [CrossRef]
- Schwaiger, M.; Schübeler, D. A question of timing: Emerging links between transcription and replication. Curr. Opin. Genet. Dev. 2006, 16, 177–183. [Google Scholar] [CrossRef]
- Dileep, V.; Gilbert, D.M. Single-cell replication profiling to measure stochastic variation in mammalian replication timing. Nat. Commun. 2018, 9, 427. [Google Scholar] [CrossRef] [PubMed]
- Hiratani, I.; Takahashi, S. DNA Replication Timing Enters the Single-Cell Era. Genes 2019, 10, 221. [Google Scholar] [CrossRef] [PubMed]
- Hardy, C.F.J.; Sussel, L.; Shore, D. A RAP1-interacting protein involved in transcriptional silencing and telomere length regulation. Genes Dev. 1992, 6, 801–814. [Google Scholar] [CrossRef] [PubMed]
- Lian, H.Y.H.-Y.; Robertson, E.D.; Hiraga, S.I.; Alvino, G.M.; Collingwood, D.; McCune, H.J.; Sridhar, A.; Brewer, B.J.; Raghuraman, M.K.; Donaldson, A.D. The effect of Ku on telomere replication time is mediated by telomere length but is independent of histone tail acetylation. Mol. Biol. Cell 2011, 22, 1753–1765. [Google Scholar] [CrossRef]
- Hayano, M.; Kanoh, Y.; Matsumoto, S.; Renard-Guillet, C.; Shirahige, K.; Masai, H. Rif1 is a global regulator of timing of replication origin firing in fission yeast. Genes Dev. 2012, 26, 137–150. [Google Scholar] [CrossRef] [PubMed]
- Cornacchia, D.; Dileep, V.; Quivy, J.P.; Foti, R.; Tili, F.; Santarella-Mellwig, R.; Antony, C.; Almouzni, G.; Gilbert, D.M.; Buonomo, S.B.C. Mouse Rif1 is a key regulator of the replication-timing programme in mammalian cells. EMBO J. 2012, 31, 3678–3690. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, S.; Ishii, A.; Kanoh, Y.; Oda, M.; Nishito, Y.; Masai, H. Rif1 regulates the replication timing domains on the human genome. EMBO J. 2012, 31, 3667–3677. [Google Scholar] [CrossRef] [PubMed]
- Seller, C.A.; O’Farrell, P.H. Rif1 prolongs the embryonic S phase at the Drosophila mid-blastula transition. PLoS Biol. 2018, 16, e2005687. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, R.L.; Das, S.; Hill, C.A.; Duronio, R.J.; Nordman, J.T. Rif1 Functions in a Tissue-Specific Manner to Control Replication Timing through Its PP1-Binding Motif. Genetics 2020, 215, 75–87. [Google Scholar] [CrossRef] [PubMed]
- Buonomo, S.B.C.; Wu, Y.; Ferguson, D.; De Lange, T. Mammalian Rif1 contributes to replication stress survival and homology-directed repair. J. Cell Biol. 2009, 187, 385–398. [Google Scholar] [CrossRef] [PubMed]
- Castaño, I.; Pan, S.J.; Zupancic, M.; Hennequin, C.; Dujon, B.; Cormack, B.P. Telomere length control and transcriptional regulation of subtelomeric adhesins in Candida glabrata. Mol. Microbiol. 2005, 55, 1246–1258. [Google Scholar] [CrossRef]
- Kanoh, J.; Ishikawa, F. spRap1 and spRif1, recruited to telomeres by Taz1, are essential for telomere function in fission yeast. Curr. Biol. 2001, 11, 1624–1630. [Google Scholar] [CrossRef]
- Levy, D.L.; Blackburn, E.H. Counting of Rif1p and Rif2p on Saccharomyces cerevisiae Telomeres Regulates Telomere Length. Mol. Cell. Biol. 2004, 24, 10857–10867. [Google Scholar] [CrossRef]
- Silverman, J.; Takai, H.; Buonomo, S.B.C.; Eisenhaber, F.; De Lange, T. Human Rif1, ortholog of a yeast telomeric protein, is regulated by ATM and 53BP1 and functions in the S-phase checkpoint. Genes Dev. 2004, 18, 2108–2119. [Google Scholar] [CrossRef]
- Xu, L.; Blackburn, E.H. Human Rif1 protein binds aberrant telomeres and aligns along anaphase midzone microtubules. J. Cell Biol. 2004, 167, 819–830. [Google Scholar] [CrossRef] [PubMed]
- Chapman, J.R.; Barral, P.; Vannier, J.B.; Borel, V.; Steger, M.; Tomas-Loba, A.; Sartori, A.A.; Adams, I.R.; Batista, F.D.; Boulton, S.J. RIF1 Is Essential for 53BP1-Dependent Nonhomologous End Joining and Suppression of DNA Double-Strand Break Resection. Mol. Cell 2013, 49, 858–871. [Google Scholar] [CrossRef]
- Di Virgilio, M.; Callen, E.; Yamane, A.; Zhang, W.; Jankovic, M.; Gitlin, A.D.; Feldhahn, N.; Resch, W.; Oliveira, T.Y.; Chait, B.T.; et al. Rif1 prevents resection of DNA breaks and promotes immunoglobulin class switching. Science 2013, 339, 711–715. [Google Scholar] [CrossRef]
- Escribano-Díaz, C.; Orthwein, A.; Fradet-Turcotte, A.; Xing, M.; Young, J.T.F.; Tkáč, J.; Cook, M.A.; Rosebrock, A.P.; Munro, M.; Canny, M.D.; et al. A Cell Cycle-Dependent Regulatory Circuit Composed of 53BP1-RIF1 and BRCA1-CtIP Controls DNA Repair Pathway Choice. Mol. Cell 2013, 49, 872–883. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, M.; Lottersberger, F.; Buonomo, S.B. 53BP1 Regulates DSB Repair Using. Science 2013, 339, 700–704. [Google Scholar] [CrossRef] [PubMed]
- Mirman, Z.; Lottersberger, F.; Takai, H.; Kibe, T.; Gong, Y.; Takai, K.; Bianchi, A.; Zimmermann, M.; Durocher, D.; de Lange, T. 53BP1/Rif1/Shieldin counteract DSB resection through CST/Polα-dependent fill-in. Nature 2018, 560, 112. [Google Scholar] [CrossRef] [PubMed]
- Noordermeer, S.M.; Adam, S.; Setiaputra, D.; Barazas, M.; Pettitt, S.J.; Ling, A.K.; Olivieri, M.; Álvarez-Quilón, A.; Moatti, N.; Zimmermann, M.; et al. The Shieldin complex mediates 53BP1-dependent DNA repair. Nature 2018, 560, 117. [Google Scholar] [CrossRef]
- Hengeveld, R.C.C.; de Boer, H.R.; Schoonen, P.M.; de Vries, E.G.E.; Lens, S.M.A.; van Vugt, M.A.T.M. Rif1 Is Required for Resolution of Ultrafine DNA Bridges in Anaphase to Ensure Genomic Stability. Dev. Cell 2015, 34, 466–474. [Google Scholar] [CrossRef]
- Mukherjee, C.; Tripathi, V.; Manolika, E.M.; Margriet Heijink, A.; Ricci, G.; Merzouk, S.; Rudolf De Boer, H.; Demmers, J.; Van Vugt, M.A.T.M.; Chaudhuri, A.R. RIF1 promotes replication fork protection and efficient restart to maintain genome stability. Nat. Commun. 2019, 10, 3287. [Google Scholar] [CrossRef]
- Jares, P.; Donaldson, A.; Blow, J.J. The Cdc7/Dbf4 protein kinase: Target of the S phase checkpoint? EMBO Rep. 2000, 1, 319. [Google Scholar] [CrossRef] [PubMed]
- Takeda, T.; Ogino, K.; Tatebayashi, K.; Ikeda, H.; Arai, K.I.; Masai, H. Regulation of Initiation of S Phase, Replication Checkpoint Signaling, and Maintenance of Mitotic Chromosome Structures during S Phase by Hsk1 Kinase in the Fission Yeast. Mol. Biol. Cell 2001, 12, 1257. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sreesankar, E.; Senthilkumar, R.; Bharathi, V.; Mishra, R.K.; Mishra, K. Functional diversification of yeast telomere associated protein, Rif1, in higher eukaryotes. BMC Genom. 2012, 13, 255. [Google Scholar] [CrossRef] [PubMed]
- Sukackaite, R.; Cornacchia, D.; Jensen, M.R.; Mas, P.J.; Blackledge, M.; Enervald, E.; Duan, G.; Auchynnikava, T.; Köhn, M.; Hart, D.J.; et al. Mouse Rif1 is a regulatory subunit of protein phosphatase 1 (PP1). Sci. Rep. 2017, 7, 2119. [Google Scholar] [CrossRef]
- Hiraga, S.; Ly, T.; Garzón, J.; Hořejší, Z.; Ohkubo, Y.; Endo, A.; Obuse, C.; Boulton, S.J.; Lamond, A.I.; Donaldson, A.D. Human RIF1 and protein phosphatase 1 stimulate DNA replication origin licensing but suppress origin activation. EMBO Rep. 2017, 18, 403–419. [Google Scholar] [CrossRef] [PubMed]
- Mattarocci, S.; Shyian, M.; Lemmens, L.; Damay, P.; Altintas, D.M.; Shi, T.; Bartholomew, C.R.; Thomä, N.H.; Hardy, C.F.J.; Shore, D. Rif1 Controls DNA replication timing in yeast through the PP1 Phosphatase Glc7. Cell Rep. 2014, 7, 62–69. [Google Scholar] [CrossRef] [PubMed]
- Davé, A.; Cooley, C.; Garg, M.; Bianchi, A. Protein Phosphatase 1 Recruitment by Rif1 Regulates DNA Replication Origin Firing by Counteracting DDK Activity. Cell Rep. 2014, 7, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Hiraga, S.I.; Alvino, G.M.; Chang, F.; Lian, H.Y.; Sridhar, A.; Kubota, T.; Brewer, B.J.; Weinreich, M.; Raghuraman, M.K.; Donaldson, A.D. Rif1 controls DNA replication by directing Protein Phosphatase 1 to reverse Cdc7- mediated phosphorylation of the MCM complex. Genes Dev. 2014, 28, 372–383. [Google Scholar] [CrossRef]
- Alver, R.C.; Chadha, G.S.; Gillespie, P.J.; Blow, J.J. Reversal of DDK-Mediated MCM Phosphorylation by Rif1-PP1 Regulates Replication Initiation and Replisome Stability Independently of ATR/Chk1. Cell Rep. 2017, 18, 2508–2520. [Google Scholar] [CrossRef]
- Deegan, T.D.; Yeeles, J.T.; Diffley, J.F. Phosphopeptide binding by Sld3 links Dbf4-dependent kinase to MCM replicative helicase activation. EMBO J. 2016, 35, 961–973. [Google Scholar] [CrossRef]
- Tanaka, S.; Nakato, R.; Katou, Y.; Shirahige, K.; Araki, H. Origin Association of Sld3, Sld7, and Cdc45 Proteins Is a Key Step for Determination of Origin-Firing Timing. Curr. Biol. 2011, 21, 2055–2063. [Google Scholar] [CrossRef] [PubMed]
- De Jesús-Kim, L.; Friedman, L.J.; Lõoke, M.; Ramsoomair, C.K.; Gelles, J.; Bell, S.P. Ddk regulates replication initiation by controlling the multiplicity of cdc45-gins binding to mcm2-7. Elife 2021, 10, e65471. [Google Scholar] [CrossRef]
- Peace, J.M.; Ter-Zakarian, A.; Aparicio, O.M. Rif1 regulates initiation timing of late replication origins throughout the S. cerevisiae genome. PLoS ONE 2014, 9, e98501. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Caballero, M.; Kolesnikova, T.; Zhimulev, I.; Koren, A.; Nordman, J. Replication timing analysis in polyploid cells reveals rif1 uses multiple mechanisms to promote underreplication in drosophila. Genetics 2021, 219, iyab147. [Google Scholar] [CrossRef] [PubMed]
- Foti, R.; Gnan, S.; Cornacchia, D.; Dileep, V.; Bulut-Karslioglu, A.; Diehl, S.; Buness, A.; Klein, F.A.; Huber, W.; Johnstone, E.; et al. Nuclear Architecture Organized by Rif1 Underpins the Replication-Timing Program. Mol. Cell 2016, 61, 260–273. [Google Scholar] [CrossRef] [PubMed]
- Farrell, J.A.; O’Farrell, P.H. From egg to gastrula: How the cell cycle is remodeled during the Drosophila mid-blastula transition. Annu. Rev. Genet. 2014, 48, 269–294. [Google Scholar] [CrossRef] [PubMed]
- Blumenthal, A.B.; Kriegstein, H.J.; Hogness, D.S. The Units of DNA Replication in Drosophila melanogaster Chromosomes. Cold Spring Harb. Symp. Quant. Biol. 1974, 38, 205–223. [Google Scholar] [CrossRef] [PubMed]
- Shermoen, A.W.; McCleland, M.L.; O’Farrell, P.H. Developmental Control of Late Replication and S Phase Length. Curr. Biol. 2010, 20, 2067–2077. [Google Scholar] [CrossRef]
- Moiseeva, T.N.; Qian, C.; Sugitani, N.; Osmanbeyoglu, H.U.; Bakkenist, C.J. WEE1 kinase inhibitor AZD1775 induces CDK1 kinase-dependent origin firing in unperturbed G1- and S-phase cells. Proc. Natl. Acad. Sci. USA 2019, 116, 23891–23893. [Google Scholar] [CrossRef] [PubMed]
- Ciardo, D.; Haccard, O.; Narassimprakash, H.; Cornu, D.; Chiara Guerrera, I.; Goldar, A.; Marheineke, K. Polo-like kinase 1 (Plk1) regulates DNA replication origin firing and interacts with Rif1 in Xenopus. Nucleic Acids Res. 2021, 49, 9851–9869. [Google Scholar] [CrossRef]
- Edgar, B.A.; Orr-Weaver, T.L. Endoreplication cell cycles: More for less. Cell 2001, 105, 297–306. [Google Scholar] [CrossRef]
- Nordman, J.; Orr-Weaver, T.L. Regulation of DNA replication during development. Development 2012, 139, 455–464. [Google Scholar] [CrossRef]
- Munden, A.; Rong, Z.; Sun, A.; Gangula, R.; Mallal, S.; Nordman, J.T. Rif1 inhibits replication fork progression and controls DNA copy number in Drosophila. Elife 2018, 7, e39140. [Google Scholar] [CrossRef]
- Hannibal, R.L.; Baker, J.C. Selective Amplification of the Genome Surrounding Key Placental Genes in Trophoblast Giant Cells. Curr. Biol. 2016, 26, 230–236. [Google Scholar] [CrossRef]
- Nordman, J.T.; Kozhevnikova, E.N.; Verrijzer, C.P.; Pindyurin, A.V.; Andreyeva, E.N.; Shloma, V.V.; Zhimulev, I.F.; Orr-Weaver, T.L. DNA copy-number control through inhibition of replication fork progression. Cell Rep. 2014, 9, 841–849. [Google Scholar] [CrossRef] [PubMed]
- Hiraga, S.; Monerawela, C.; Katou, Y.; Shaw, S.; Clark, K.R.; Shirahige, K.; Donaldson, A.D. Budding yeast Rif1 binds to replication origins and protects DNA at blocked replication forks. EMBO Rep. 2018, 19, e46222. [Google Scholar] [CrossRef]
- Kanoh, Y.; Matsumoto, S.; Fukatsu, R.; Kakusho, N.; Kono, N.; Renard-Guillet, C.; Masuda, K.; Iida, K.; Nagasawa, K.; Shirahige, K.; et al. Rif1 binds to G quadruplexes and suppresses replication over long distances. Nat. Struct. Mol. Biol. 2015, 22, 889–897. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.D.; Smith, D.L.; DeRisi, J.L.; Blackburn, E.H. Telomeric Protein Distributions and Remodeling Through the Cell Cycle in Saccharomyces cerevisiae. Mol. Biol. Cell 2003, 14, 556. [Google Scholar] [CrossRef]
- Dimitrova, D.S.; Gilbert, D.M. The spatial position and replication timing of chromosomal domains are both established in early G1 phase. Mol. Cell 1999, 4, 983–993. [Google Scholar] [CrossRef]
- Wu, R.; Terry, A.V.; Singh, P.B.; Gilbert, D.M. Differential Subnuclear Localization and Replication Timing of Histone H3 Lysine 9 Methylation States. Mol. Biol. Cell 2005, 16, 2872. [Google Scholar] [CrossRef] [PubMed]
- Ebrahimi, H.; Robertson, E.D.; Taddei, A.; Gasser, S.M.; Donaldson, A.D.; Hiraga, S.I. Early initiation of a replication origin tethered at the nuclear periphery. J. Cell Sci. 2010, 123, 1015–1019. [Google Scholar] [CrossRef]
- Lieberman-Aiden, E.; Van Berkum, N.L.; Williams, L.; Imakaev, M.; Ragoczy, T.; Telling, A.; Amit, I.; Lajoie, B.R.; Sabo, P.J.; Dorschner, M.O.; et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 2009, 326, 289–293. [Google Scholar] [CrossRef] [PubMed]
- Ryba, T.; Hiratani, I.; Lu, J.; Itoh, M.; Kulik, M.; Zhang, J.; Schulz, T.C.; Robins, A.J.; Dalton, S.; Gilbert, D.M. Evolutionarily conserved replication timing profiles predict long-range chromatin interactions and distinguish closely related cell types. Genome Res. 2010, 20, 761–770. [Google Scholar] [CrossRef] [PubMed]
- Nora, E.P.; Lajoie, B.R.; Schulz, E.G.; Giorgetti, L.; Okamoto, I.; Servant, N.; Piolot, T.; Van Berkum, N.L.; Meisig, J.; Sedat, J.; et al. Spatial partitioning of the regulatory landscape of the X-inactivation center. Nature 2012, 485, 381. [Google Scholar] [CrossRef]
- Dixon, J.R.; Selvaraj, S.; Yue, F.; Kim, A.; Li, Y.; Shen, Y.; Hu, M.; Liu, J.S.; Ren, B. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 2012, 485, 376–380. [Google Scholar] [CrossRef] [PubMed]
- Pope, B.D.; Ryba, T.; Dileep, V.; Yue, F.; Wu, W.; Denas, O.; Vera, D.L.; Wang, Y.; Hansen, R.S.; Canfield, T.K.; et al. Topologically associating domains are stable units of replication-timing regulation. Nature 2014, 515, 402–405. [Google Scholar] [CrossRef] [PubMed]
- Gnan, S.; Flyamer, I.M.; Klein, K.N.; Castelli, E.; Rapp, A.; Maiser, A.; Chen, N.; Weber, P.; Enervald, E.; Cardoso, M.C.; et al. Nuclear organisation and replication timing are coupled through RIF1–PP1 interaction. Nat. Commun. 2021, 12, 2910. [Google Scholar] [CrossRef] [PubMed]
- Ogiyama, Y.; Schuettengruber, B.; Papadopoulos, G.L.; Chang, J.M.; Cavalli, G. Polycomb-Dependent Chromatin Looping Contributes to Gene Silencing during Drosophila Development. Mol. Cell 2018, 71, 73–88.e5. [Google Scholar] [CrossRef]
- Kumar, S.; Yoo, H.Y.; Kumagai, A.; Shevchenko, A.; Shevchenko, A.; Dunphy, W.G. Role for Rif1 in the checkpoint response to damaged DNA in Xenopus egg extracts. Cell Cycle 2012, 11, 1183–1194. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Richards, L.; Das, S.; Nordman, J.T. Rif1-Dependent Control of Replication Timing. Genes 2022, 13, 550. https://doi.org/10.3390/genes13030550
Richards L, Das S, Nordman JT. Rif1-Dependent Control of Replication Timing. Genes. 2022; 13(3):550. https://doi.org/10.3390/genes13030550
Chicago/Turabian StyleRichards, Logan, Souradip Das, and Jared T. Nordman. 2022. "Rif1-Dependent Control of Replication Timing" Genes 13, no. 3: 550. https://doi.org/10.3390/genes13030550
APA StyleRichards, L., Das, S., & Nordman, J. T. (2022). Rif1-Dependent Control of Replication Timing. Genes, 13(3), 550. https://doi.org/10.3390/genes13030550