Abstract
Phelan–McDermid syndrome (PMS) is a rare, heterogeneous, and complex neurodevelopmental disorder. It is generally caused by a heterozygous microdeletion of contiguous genes located in the distal portion of the long arm of chromosome 22, including the SHANK3 gene. Sequence variants of SHANK3, including frameshift, nonsense mutations, small indels and splice site mutations also result in PMS. Furthermore, haploinsufficiency in SHANK3 has been suggested as the main cause of PMS. SHANK3 is also associated with intellectual disability, autism spectrum disorder and schizophrenia. The phenotype of PMS is variable, and lacks a distinctive phenotypic characteristic, so the clinical diagnosis should be confirmed by genetic analysis. PMS is a multi-system disorder, and clinical care must encompass various specialties and therapists. The role of risperidone, intranasal insulin, insulin growth factor 1, and oxytocin as potential therapeutic options in PMS will be discussed in this review. The diagnosis of PMS is important to provide an appropriate clinical evaluation, treatment, and genetic counseling.
1. Introduction
Phelan–McDermid syndrome (PMS) is a rare, heterogeneous, and complex disorder of neurological development [1,2,3,4,5,6,7,8]. The principal cause of PMS is a heterozygous microdeletion of contiguous genes (including SHANK3, MIM *606230, or just part of this gene), located in the distal portion of the long arm of chromosome 22 [6,8,9,10], of which 70% occur on the paternal chromosome [11]. Other cases result from single nucleotide variants in the SH3 and Multiple Ankyrin Repeat Domains 3 gene (SHANK3) [2,3,6,8,10,12,13,14]. This gene was initially associated with a 4.5-year-old boy manifesting all the features of a 22q13.3 deletion syndrome. Bonaglia and colleagues found in the patient’s karyotype a de novo balanced translocation between chromosomes 12 and 22, with the breakpoint in the 22q13.3 critical region of the 22q distal deletion syndrome [15]. Furthermore, variants in SHANK3 are also associated with intellectual disability in 2% [6,10,14], autism spectrum disorder in 0.5–2%, and schizophrenia in 0.6–2.16% of cases [6,10,14]. SHANK3 is therefore an attractive target for pharmacological intervention in the treatment of these neurodevelopmental disorders with neurological and neuropsychiatric manifestations [6,10,14].
Watt et al. [16] first described PMS in 1985, in a 14-year-old male adolescent with severe intellectual disability, absence of language, minor dysmorphia, and normal muscle tone. In 1988, Herman and colleagues identified a terminal deletion at 22q13.3 in PMS [17], and Phelan and McDermid described the hypotonia associated with the de novo deletion in 22q13.3 in a newborn who later presented with global developmental delay, normal growth, and minor facial dysmorphias [18].
The occurrence rate has been estimated in the range of 2.5–10 per million births, although this is likely to be underestimated (National Organization for Rare Disorders. Phelan–McDermid syndrome), and is genetically detectable by array CGH for a small-size deletion in 22q13.3, or the need for direct or NGS sequencing of SHANK3 to support the specific identification of patients [10,14]. According to the Phelan–McDermid Syndrome International Registry (https://www.pmsf.org/registry/, accessed on 10 November 2021), at least 1200 cases have been reported worldwide [12].
2. Clinical Features
The PMS phenotype can vary widely and lacks a distinctive characteristic phenotype [6]. Clinically, it presents with neonatal hypotonia [2,6,10,19], global delay of development [2,3,8,14,19], absence or severe alteration of language [3,4,6,7,10,14,20], highly variable levels of cognitive functioning with moderate-to-severe intellectual disability in 77% [3,4,5,6,7,8,10,19,20], behavioral disturbances: attention deficit hyperactivity disorder in 36% [2,3,7,8,10], autism spectrum disorder in 84% [4,6,8,14,20], sleep disturbances, stereotyped repetitive movements [5,19], reduced sensitivity to pain, an inability to regulate sweating [8], and seizures of different types [2,3,7,19,20]. Seizures are mostly febrile and do not require medication; however, grand mal seizures, focal seizures, and absence seizures have been reported. No characteristic EEG findings have been described in PMS.
Neuroradiological (for example BRAIN IMAGE) studies in PMS have documented reduced myelination, frontal lobe hypoplasia, corpus callosum thinning or agenesia, ventriculomegaly, and focal cortical atrophy [21].
PET studies in eight children with PMS showed a localized dysfunction of the left temporal polar lobe and significant hypoperfusion of the amygdala compared to 13 children with idiopathic intellectual disability [22].
Growth is normal [5,10], and dysmorphic features are generally mild and include: dolichocephaly [3,5,6,7,8,10,14], long eyelashes [10], midfacial hypoplasia [7], prominent cheeks [10], bulbous nasal tip, pointed chin [10], alterations in ears, fleshy hands, and dysplastic toenails [23]. Additional features include endocrine, immune, ocular, cardiopulmonary, gastrointestinal, renal malformations, and lymphedema abnormalities (see, Table 1), [1,2,3,6,7,8,10,14,19,20,23].

Table 1.
Clinical findings associated with PMS [1,2,6,7,8,10,14,19,20,23].
Natural History
The least-affected clinical skill is motor functioning, with gross motor function being stronger than fine motor function [8,24]. The regression in motor and social skills with a slow and possibly incomplete recovery may occur after acute paroxysmal events [18,24]. The intellectual disability is less striking in younger children than in older children, as relative developmental functioning decreases with increasing age. The most affected developmental skill is language, although receptive language is usually reported to be stronger than expressive language, and social skills essentially present with symptoms in the autism spectrum [18,19].
Most children with PMS seem to acquire basic skills such as walking, but subsequent extension and refinement of these skills is generally very poor. These deficiencies are less striking in younger than older children, a phenomenon known as ‘growing into deficit’. In addition, deficits in adaptive behavior further hamper cognitive development. No improvement of developmental milestones is a common finding in this population, and is important to take into account when evaluating development and treatment effects. At the level of psychopathology, typical characteristics are related to the affective and anxiety domains, with, for example, impulsive, irritable, and demanding behaviors. In post-adolescent patients, psychiatric symptoms such as atypical bipolar disorder appear to be more prominent [19]. This entity is still under-diagnosed in adults who may present with deterioration in cognitive functioning and atypical bipolar disorders with loss of acquired skills [8].
3. Etiopathogenesis
The genetic landscape of PMS is broad and the mechanism in which the deletion occurs can be caused by a variety of rearrangements, including terminal or interstitial deletions, unbalanced translocations, ring chromosomes, and other more complex alterations [3,6,14,25] (Figure 1). Terminal and de novo deletions contribute to the majority of chromosomal anomalies and are reported in approximately 80% of cases. The deletion size can vary from less than 100 kb to 9.2 Mb (4.5 Mb on average), resulting in the loss of the single SHANK3 gene, and ranges from 22q13.2 to 22q13.31-3 [3,14]. Moreover, PMS commonly involves the loss of other genes, such as PARVB (MIM *608121) [8] and SULT4A1 (MIM *608359), and this have been suggested to contribute to phenotype severity [8,26]. Unbalanced translocations or a ring chromosome involving chromosome 22 represent the remaining 20% of cases [3,6,10,14], of which approximately 50% are inherited interchangeably from a balanced carrier parent [9]. Some families with affected siblings have been described, so the origin of the rearrangement may be due to germinal mosaicism in one of the parents [27,28,29].
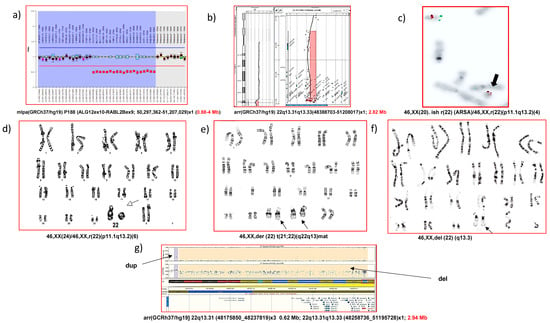
Figure 1.
Different genetic rearrangements present in PMS, established by different cytogenetic/molecular techniques: (a) interstitial deletion by MLPA; (b) terminal deletion by microarray CGH; (c) terminal deletion and ring chromosome by FISH; (d) terminal deletion and ring chromosome by karyotype (GTL bands), (e) unbalanced translocation by karyotype, (f) terminal deletion by karyotype, (g) terminal deletion and other adjacent SNP-array rearrangements.
Interestingly, PMS patients with ring chromosome 22 may develop neurofibromatosis type 2 (NF2) [13], caused by pathogenic variants in NF2, as this gene is located at 22q12.2, adjacent to the PMS deletion region. The risk for NF2 is due to the instability of ring chromosomes during mitosis and follows a two-hit model. The first hit is the loss of NF2 during mitosis; the second hit is a somatic mutation of the remaining NF2 allele [30]. Out of 44 individuals evaluated at the Greenwood Genetic Center or by international collaborators or identified through the PMS International Registry with a ring chromosome 22, 7 (16%) carried a diagnosis of NF2, therefore, this condition is not uncommon [31].
Children with ring chromosome 22 should be monitored for NF2 signs, with baseline and annual ocular, dermal, and neurologic examinations between ages 2 and 10 years with annual audiology screening and brain MRI every two years after age 10 years [32].
SHANKproteins are major scaffolds of the postsynaptic density of excitatory synapses, linking neurotransmitter receptors and ion channels to the actin cytoskeleton and G-protein-coupled signaling pathways, and mutations in SHANK3 gene are associated with autism and intellectual disability [33], as well as psychiatric diseases, such as schizophrenia and bipolar disorder [7]. SHANK3 protein has a critical function in synaptic plasticity by modulating dendrite formation, playing an important role in the development of synapses [3,7,10,14] and the maintenance of long-term potentiation [7]. Defects in SHANK3 protein can also cause a reduction in activated hyperpolarization of cation channels [10,14], which may explain some of the observed phenotypic characteristics in patients [10].
Genotype–phenotype studies are made preferentially in cases with deletions and correlate positively with the number and/or severity of some of the clinical manifestations [3,8], some neurological manifestations including hypotonia [7,34], delayed motor development, deficiencies in social communication related to autism spectrum disorders, aggressive behavior, and intellectual disability [23]. Therefore, individuals with small terminal deletions or with sequence variants in SHANK3 gene may have a less severe phenotype. Those cases with major deletions or truncated nonsense mutations in SHANK3 have been associated with autism spectrum disorders and intellectual disability [3]. In a mouse model of autism spectrum disorders, a truncated mutant form of Shank3 displayed altered plasticity, anxiety-like, motor, social, communications and stereotyped behaviors [35,36,37], which were attributable to reduced synaptic plasticity in the hippocampal–medial prefrontal cortex pathway [37]. Individuals with PMS can exhibit sensitivity to stress, resulting in behavioral deterioration. Using a mouse model, swim stress produces an altered transcriptomic response in pyramidal neurons that impacts genes and pathways involved in synaptic function, signaling, and protein turnover. Several lines of evidence demonstrate that Shank3 expression is regulated by Homer protein homolog 1a (Homer1a), which is part of the Shank3-mGluR-N-methyl-D-aspartate (NMDA) receptor complex and is super-induced and implicated in the stress response. The interaction between stress and genetics and focus attention on activity-dependent changes may contribute to pathogenesis [38].
However, the relationship is not consistent, since patients with deletions of similar size can have variable phenotypes [7]. Individuals with infrequent interstitial deletions, without involvement of the SHANK3 gene, may exhibit an indistinguishable phenotype, raising the possibility that many of the characteristics of PMS may be under the influence of other additional genes located in the 22q13.32 [14] region, or may affect regulatory regions of the SHANK3 gene or have a position effect [2,4].
A deletion that has been identified in PMS contains a region that houses three genes: ACR, (MIM *102480), RABL2B (MIM *605413) and SHANK3, the latter being the strongest candidate for neurobehavioral symptoms [39]. Other genes can potentially influence neurological abnormalities in PMS, such as the MAPK8IP2 gene (OMIM *607755), located approximately 70 Kb proximal to SHANK3, but often deleted together. MAPK8IP2 is highly expressed in the brain at the level of the posterior synaptic space, and studies in mice show that the absence of the MAPK8IP2 protein results in an abnormal dendritic morphology and cognitive and motor deficits. However, it is not yet clear how deletions in SHANK3 and MAPK8IP2 specifically contribute to PMS [7]. The mentioned SULT4A1 gene encodes a cytosolic sulfotransferase highly expressed at postsynaptic sites, which modulates neuronal branching complexity and dendritic spines’ formation, negatively regulates the catalytic activity of Pin1 toward PSD-95, and facilitates NMDAR synaptic expression and function [26].
The genetic heterogeneity of PMS underscores the importance of studying a wide range of alterations [3]. Various case series have been published that have investigated the correlation of the clinical characteristics [3,14], with a wide heterogeneity in the expression and severity of the phenotype [3].
4. Molecular Diagnosis
The diagnosis of PMS is based on confirmed chromosomal and/or molecular analysis. The karyotype is able to identify large deletions and other molecular techniques should be used for the genetic characterization of the deletion (FISH, MLPA, array CGH/SNPs). In the case of variants in SHANK3, either conventional Sanger or next-generation sequencing (NGS) through targeted capture, or whole-exome sequencing (WES), must be performed to sequence the SHANK3 gene. The advantages and disadvantages of the different and main experimental approaches in the diagnosis of PMS are highlighted in Figure 1 and Table 2.
Table 2.
List of advantages and disadvantages of laboratory techniques in the diagnosis of PMS.
The most reasonable sequence or diagnostic algorithm for deletions is CMA (chromosomal microarray). It is necessary to identify the size of the deletion (and the genes deleted), if the deletion is terminal or interstitial, and if there are additional rearrangements. In terminal deletions, a karyotype or FISH will be required to identify if it is the consequence of a translocation or the consequence of a ring. Prenatal diagnosis is possible and is based on cytogenetic and molecular analysis after an invasive amniocentesis test or chorial biopsy. Recently, extended non-invasive prenatal diagnosis has been able to establish variations in copy number in the region affecting PMS and other syndromes as a prenatal screening. However, diagnostic confirmation is still required by invasive techniques, and the application of microarrays CMA, FISH, MLPA, or karyotype could be necessary.
5. Differential Clinical Diagnosis
The differential diagnosis of PMS (Table 3) can be challenging, given the clinical variability in phenotype, and includes: autism spectrum disorders, cerebral palsy, Prader-Willi syndrome (MIM #176270), Angelman syndrome (MIM #105830), velocardiofacial syndrome (MIM #192430), fragile X syndrome (MIM #300624), FG (MIM #305450), Williams–Beuren syndrome (MIM #194050), and Smith–Magenis syndrome (MIM #182290) [12,13,23]. Furthermore, the trichorinophalangeal syndromes (MIM #190350), Clark–Baraitser (MIM #300602) and Sotos (MIM #117550) are also considered differential diagnoses [5].
Table 3.
Differential diagnosis of PMS.
6. Interdisciplinary Clinical Management
The care team should be interdisciplinary and should include neonatologists, pediatricians, neurologists, psychiatrists, endocrinologists, immunologists, cardiologists, gastroenterologists, nephrologists, dentists, as well as speech, respiratory, physical, and occupational therapists who provide follow-up evaluations, exercises and infant massage, and lactation consultants, experts in the evaluation of oral feeding strategies and social workers. All these clinical staff should be coordinated or supervised by a geneticist or a neuropsychiatrist who will provide clinical follow-up and genetic counseling to the family.
More detailed information on developmental characteristics in children is clearly necessary for care and would contribute to improving counselling of parents, identification of specific problems, adequate and individualized support for the persons with PMS [38].
The first line of treatment must address the areas of motor, communication, and language development. Early intervention programs of intensive physical and occupational therapies are highly recommended for motor skills deficits. While evaluating any deterioration of developmental skills, frequent monitoring should be performed to detect and manage medical comorbidities [12,40]. Subsequently, the areas that should be covered are those related to cognitive and behavioral characteristics. These areas should be evaluated and followed to adapt supportive and therapeutic strategies to meet individual needs [8].
An ophthalmological evaluation must be performed to monitor visual development and identify any refractive error or strabismus. Myopia, strabismus, and retinitis pigmentosa have been reported. Dental alterations, such as malocclusion, are frequent and can be serious in some cases, for which reason orthodontics or surgical correction can reduce the risk of dental caries, periodontal disease and relieve pressure on the temporomandibular joint [41].
Patients with PMS can be prone to immune system dysfunction, resulting in recurrent ear and upper respiratory tract infections (complicated by low muscle tone, airway abnormalities, and sputum clearance), asthma, and seasonal and food allergies. Cases of autoimmune hepatitis, atopic dermatitis and recurrent staphylococcal skin infections/cellulitis have been described. Lymphedema has been found in 24% of cases. Clinical management is mainly supportive, but as the underlying pathways responsible for deficits are more clearly understood, there will be the potential to develop more targeted therapies in the future [12].
7. Therapeutic Options
The long-term plan for managing PMS patients should be continuous and interdisciplinary, with support to maximize developmental outcome. Early intervention services should be provided to the family in the first three years. However, life expectancy is uncertain and depends on each individual case [42].
Several pharmacological strategies have been assayed. The use of risperidone (1 mg/day) produced significant improvements in behavioral disturbances in PMS on the global clinical impression scale. It is hypothesized that by blocking dopamine 2 receptors, NMDA transmission is promoted, and glutamatergic dysregulation caused by the loss of SHANK3 is reversed [12].
Intranasal insulin was studied in six PMS patients and resulted in marked short-term improvements in gross and fine motor activities, cognitive functions and educational level. Similarly, long-term positive effects were found for gross and fine motor activities, nonverbal communication, cognitive functions, and autonomy. However, one patient showed changes in balance, extreme sensitivity to touch and general loss of interest, and one patient complained of intermittent nasal bleeding [43]. No adverse effects on glucose levels or HbA1c levels were found [12].
To validate this effect, a randomized, double-blind, placebo-controlled clinical trial was conducted in 25 patients aged 1 to 16 years with a molecularly confirmed 22q13.3 deletion involving the SHANK3 gene. A significant effect was found for cognition and social skills for children older than 3 years, who generally show a decrease in development. Intranasal insulin did not cause serious adverse events. However, clinical trials in larger study populations are required to test the therapeutic effect and safety in PMS [39].
Insulin growth factor 1 (IGF-1) is a small polypeptide that crosses the blood–brain barrier and occurs in higher concentrations during development of the central nervous system, which promotes neuronal maturation and synapse formation. Studies with human neurons created from pluripotential stem cells induced from PMS patients showed that excitatory synapse transmissions can be reversible when treated with IGF-1 in vitro and with overexpression of the SHANK3 protein. IGF-1 also resulted in improvement of NMDA- and AMPA-mediated responses, reducing the decay rate of the NMDA receptor. A control study shows that the administration of IGF-1 in children with PMS improved autistic behavior, probably due to its effect on synapse development and plasticity. This treatment restored the normal density of the dendritic spine in neurons. IGF-1 was also found to work by increasing protein kinase B levels, and therefore direct treatment is another option. Preliminary results from the first clinical study of IGF-1 administration in PMS patients suggest tolerability and significant improvements in both social impairments and restrictive behaviors. No serious adverse effects have been documented and it was determined to be safe in this population. This study is currently in phase 2 and has been expanded to include a larger sample. The cost and availability of the drug are currently restrictive [44].
The acute administration of intra-cerebroventricular oxytocin reversed the deficits of synaptic plasticity in vitro and in vivo in a Shank3 knockout mouse [40]. This was the first study to report that oxytocin may not only reverse this deficit, but also improve behavioral plasticity, suggesting that a reversal effect on synaptic plasticity, specifically long-term potentiation, may be the basis of the improvement in behavior. However, further studies are needed to determine the effect of SHANK3 mutations on the oxytocin system to understand whether the disturbance could explain some of the observed behavioral phenotypes and changes related to plasticity [45]. The variable genetic mechanisms contributing to PMS will impact the search for therapeutic interventions [6].
Using induced pluripotent stem cell technology and transcriptomic studies, some authors have shown in PMS-derived hiPSC neurons have an impact in several developmental pathways, such as altered pre- and post-synaptic signaling and Wnt and ECM signaling [37]. Active compounds were evaluated for efficacy in correcting dysfunctional networks of neurons differentiated from individuals with deleterious point mutations in SHANK3. Among 202 compounds tested, lithium and valproic acid showed the best efficacy in correcting SHANK3 haploinsufficiency-associated phenotypes in cells. Lithium pharmacotherapy was subsequently provided to one patient, and after one year, an encouraging decrease in autism severity was observed [46].
The pharmacological augmentation of mGluR5 activity using 3-cyano-N-(1,3-diphenyl-1H-pyrazol-5-yl)-benzamide as the positive allosteric modulator of these receptors restored mGluR5-dependent signaling (DHPG-induced phosphorylation of ERK1/2) and normalized the frequency of mEPSCs in Shank3 knockdown neurons. These data demonstrate that a deficit in mGluR5-mediated intracellular signaling in Shank3 knockdown neurons can be compensated by 3-cyano-N-(1,3-diphenyl-1H-pyrazol-5-yl)-benzamide; this raises the possibility that pharmacological augmentation of mGluR5 activity represents a possible new therapeutic approach for patients with Shank3 mutations [47,48].
Additionally, treatment strategies have used lithium valproic acid and/or quetiapine, which appears to be a rational pharmacological choice for subjects with atypical bipolar disorder who require a mood-stabilizing pharmacotherapy [49]. Secondly, techniques from the neuropsychological and cognitive behavioral domain, such as goal management and perspective taking training, should be applied to compensate for the impaired orchestration of cognition and emotion. Such a combined treatment strategy is meant to reduce the risk for relapse of a bipolar episode and to reduce hyper-reactivity [19].
Interestingly, the treatment of behavioral abnormalities in PMS patients is complicated by pharmacogenomics issues, as the CYP2D6 enzyme, which metabolizes antidepressants and antipsychotics and is encoded by CYP2D6 gene, which maps to 22q13.2 and is lost in subjects with deletions larger than 8 Mb [50].
For PMS patients with autism spectrum disorders carrying SHANK3 mutations, a rational approach could also be the use of histone deacetylase (HDAC) inhibitors. In fact, a short treatment with a class I HDAC inhibitor such romidepsin has been demonstrated to alleviate social deficits in Shank3-deficient mice, which persisted for ~3 weeks [51].
Further, treating of Shank3-deficient mice with a 4-week ketogenic diet, which can act as an endogenous inhibitor of class I HDAC via the major product β-hydroxybutyrate, elevates the level of histone acetylation in prefrontal cortex neurons. Ketogenic diet treatment lead to the prolonged rescue of social preference deficits in Shank3-deficient mice, elevated the transcription and histone acetylation of Grin2a and Grin2b and restored the diminished N-methyl-D-aspartate (NMDA) receptor synaptic function in prefrontal cortex neurons [52]. HDAC2 transcription was upregulated in these mice, and knockdown of HDAC2 in the prefrontal cortex rescued their social deficits, highlighting an epigenetic mechanism underlying social deficits linked to Shank3 deficiency [51]. The role of valproic acid as a HDAC inhibitor could explain this efficacy in the treatment of some of the symptoms in patients with SHANK3 mutations, as reported in Fragile X syndrome [53].
8. Conclusions
PMS is a complex, heterogeneous, and underdiagnosed entity. Recognizing it is important in order to provide an appropriate evaluation and appropriate treatment option. There are various diagnostic techniques according to the etiological cause. SHANK3 appears to be the critical gene responsible for the PMS phenotype when a heterozygous rearrangement or deletion is detected within the region 22q13.2 to 22q13.31-3, although other genes may contribute to severity or modulate its phenotypic presentation. Preliminary investigations should be focused on the genotype–phenotype correlation, at least in microdeletion cases. A complete medical history and a thorough physical examination are the first step in the evaluation. In addition, an interdisciplinary and individualized therapeutic follow-up is required, as well as imparting adequate genetic counseling according to the etiological cause. The pediatrician must become familiar in order to play a fundamental role in the coordination of care through the different subspecialties involved in this care, such as family support, educators, and therapists.
Author Contributions
F.C.-S., M.C., D.M. and J.N. were responsible for the study concept and design. M.C., D.M., C.E.W., A.C.T., M.A.C., P.G., M.A.L.-R., M.M., E.B. and J.N. performed the supervision. F.C.-S., M.C. and J.N. drafted the manuscript. F.C.-S., D.M., C.E.W., P.G., E.B. and J.N. gave inputs regarding the overall and provided critical revision of the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Support with the literature review.
Acknowledgments
We would like to thank Rosalia Gumina of the University of The Andes for her support with the literature review.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Kniffin, C.L.; McKusick, V.A. #606232 Phelan–McDermid Syndrome, PHMDS. Online Mendelian Inheritance in Man, OMIM®. 2015. Available online: https://www.omim.org/entry/606232 (accessed on 12 December 2021).
- Boccuto, L.; Abenavoli, L.; Cascio, L.; Srikanth, S.; Dupont, B.; Mitz, A.R.; Rogers, R.C.; Phelan, K. Variability in Phelan-McDermid syndrome: The impact of the PNPLA3 p.I148M polymorphism. Clin. Genet. 2018, 94, 590–591. [Google Scholar] [CrossRef]
- Drapeau, E.; Riad, M.; Kajiwara, Y.; Buxbaum, J.D. Behavioral Phenotyping of an Improved Mouse Model of Phelan–McDermid Syndrome with a Complete Deletion of the Shank3 Gene. eNeuro 2018, 5, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurtas, N.; Arrigoni, F.; Errichiello, E.; Zucca, C.; Maghini, C.; D’Angelo, M.G.; Beri, S.; Giorda, R.; Bertuzzo, S.; Delledonne, M.; et al. Chromothripsis and ring chromosome 22: A paradigm of genomic complexity in the Phelan–McDermid syndrome (22q13 deletion syndrome). J. Med. Genet. 2018, 55, 269–277. [Google Scholar] [CrossRef] [PubMed]
- McKelvey, K.D.; Trana, C.J.; Kelsay, J.; Sawyer, J.; Clothier, J. Phelan–McDermid syndrome and cancer predisposition: The value of a karyotype. Am. J. Med. Genet. Part A 2018, 176, 144–145. [Google Scholar] [CrossRef] [PubMed]
- Mitz, A.R.; Philyaw, T.J.; Boccuto, L.; Shcheglovitov, A.; Sarasua, S.M.; Kaufmann, W.E.; Thurm, A. Identification of 22q13 genes most likely to contribute to Phelan McDermid syndrome. Eur. J. Hum. Genet. 2018, 26, 293–302. [Google Scholar] [CrossRef] [Green Version]
- Ziats, C.A.; Grosvenor, L.P.; Sarasua, S.M.; Thurm, A.E.; Swedo, S.E.; Mahfouz, A.; Rennert, O.M.; Ziats, M.N. Functional genomics analysis of Phelan–McDermid syndrome 22q13 region during human neurodevelopment. PLoS ONE 2019, 14, e0213921. [Google Scholar] [CrossRef] [Green Version]
- Zwanenburg, R.J.; Bocca, G.; Ruiter, S.A.J.; Dillingh, J.H.; Flapper, B.C.T.; Heuvel, E.R.V.D.; Van Ravenswaaij-Arts, C.M.A. Is there an effect of intranasal insulin on development and behaviour in Phelan–McDermid syndrome? A randomized, double-blind, placebo-controlled trial. Eur. J. Hum. Genet. 2016, 24, 1696–1701. [Google Scholar] [CrossRef] [Green Version]
- Phelan, K.; McDermid, H. The 22q13.3 Deletion Syndrome (Phelan–McDermid Syndrome). Mol. Syndr. 2011, 2, 186–201. [Google Scholar] [CrossRef] [Green Version]
- Richards, C.; Powis, L.; Moss, J.; Stinton, C.; Nelson, L.; Oliver, C. Prospective study of autism phenomenology and the behavioural phenotype of Phelan–McDermid syndrome: Comparison to fragile X syndrome, Down syndrome and idiopathic autism spectrum disorder. J. Neurodev. Disord. 2017, 9, 37. [Google Scholar] [CrossRef] [Green Version]
- Wilson, H.L.; Wong, A.C.C.; Shaw, S.R.; Tse, W.-Y.; Stapleton, G.A.; Phelan, M.C.; Hu, S.; Marshall, J.; McDermid, H.E. Molecular characterisation of the 22q13 deletion syndrome supports the role of haploinsufficiency of SHANK3/PROSAP2 in the major neurological symptoms. J. Med. Genet. 2003, 40, 575–584. [Google Scholar] [CrossRef] [Green Version]
- Costales, J.L.; Kolevzon, A. Phelan–McDermid Syndrome and SHANK3: Implications for Treatment. Neurotherapeutics 2015, 12, 620–630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Disciglio, V.; Rizzo, C.L.; Mencarelli, M.A.; Mucciolo, M.; Marozza, A.; Di Marco, C.; Massarelli, A.; Canocchi, V.; Baldassarri, M.; Ndoni, E.; et al. Interstitial 22q13 deletions not involving SHANK3 gene: A new contiguous gene syndrome. Am. J. Med. Genet. Part A 2014, 164, 1666–1676. [Google Scholar] [CrossRef] [PubMed]
- Tabet, A.-C.; Rolland, T.; Ducloy, M.; Levy, J.; Buratti, J.; Mathieu, A.; Haye, D.; Perrin, L.; Dupont, C.; Passemard, S.; et al. A framework to identify contributing genes in patients with Phelan–McDermid syndrome. NPJ Genom. Med. 2017, 2, 32. [Google Scholar] [CrossRef] [PubMed]
- Bonaglia, M.C.; Giorda, R.; Borgatti, R.; Felisari, G.; Gagliardi, C.; Selicorni, A.; Zuffardi, O. Disruption of the ProSAP2 Gene in a t(12;22)(q24.1;q13.3) Is Associated with the 22q13.3 Deletion Syndrome. Am. J. Hum. Genet. 2001, 69, 261–268. [Google Scholar] [CrossRef] [Green Version]
- Watt, J.L.; Olson, I.A.; Johnston, A.W.; Ross, H.S.; Couzin, D.A.; Stephen, G.S. A familial pericentric inversion of chromosome 22 with a recombinant subject illustrating a ‘pure’ partial monosomy syndrome. J. Med. Genet. 1985, 22, 283–287. [Google Scholar] [CrossRef] [Green Version]
- Herman, G.E.; Greenberg, F.; Ledbetter, D.H.; Optiz, J.M.; Reynolds, J.F. Multiple congenital anomaly/mental retardation (MCA/MR) syndrome with Goldenhar complex due to a terminal del(22q). Am. J. Med. Genet. 1988, 29, 909–915. [Google Scholar] [CrossRef]
- Phelan, M.C.; Rogers, R.C.; Stevenson, R.E. A de Novo Terminal Deletion of 22q. Am. J. Hum. Genet. 1988, 43, A118. [Google Scholar]
- Egger, J.I.M.; Zwanenburg, R.J.; Ravenswaaij-Arts, C.M.A.; Kleefstra, T.; Verhoeven, W.M.A. Neuropsychological phenotype and psychopathology in seven adult patients with Phelan–McDermid syndrome: Implications for treatment strategy. Genes Brain Behav. 2016, 15, 395–404. [Google Scholar] [CrossRef]
- Reierson, G.; Bernstein, J.; Froehlich-Santino, W.; Urban, A.; Purmann, C.; Berquist, S.; Jordan, J.; O’Hara, R.; Hallmayer, J. Characterizing regression in Phelan McDermid Syndrome (22q13 deletion syndrome). J. Psychiatr. Res. 2017, 91, 139–144. [Google Scholar] [CrossRef]
- Tabolacci, E.; Zollino, M.; Lecce, R.; Sangiorgi, E.; Gurrieri, F.; Leuzzi, V.; Opitz, J.M.; Neri, G. Two Brothers with 22q13 Deletion Syndrome and Features Suggestive of the Clark–Baraitser Syndrome. Clin. Dysmorphol. 2005, 14, 127–132. [Google Scholar] [CrossRef]
- Philippe, A.; Boddaert, N.; Vaivre-Douret, L.; Robel, L.; Danon-Boileau, L.; Malan, V.; de Blois, M.-C.; Heron, D.; Colleaux, L.; Golse, B.; et al. Neurobehavioral Profile and Brain Imaging Study of the 22q13.3 Deletion Syndrome in Childhood. Pediatrics 2008, 122, e376–e382. [Google Scholar] [CrossRef]
- De Rubeis, S.; Siper, P.M.; Durkin, A.; Weissman, J.; Muratet, F.; Halpern, D.; Trelles, M.D.P.; Frank, Y.; Lozano, R.; Wang, A.T.; et al. Delineation of the genetic and clinical spectrum of Phelan–McDermid syndrome caused by SHANK3 point mutations. Mol. Autism 2018, 9, 31. [Google Scholar] [CrossRef] [PubMed]
- Soorya, L.; Kolevzon, A.; Zweifach, J.; Lim, T.; Dobry, Y.; Schwartz, L.; Frank, Y.; Wang, A.T.; Cai, G.; Parkhomenko, E.; et al. Prospective investigation of autism and genotype-phenotype correlations in 22q13 deletion syndrome and SHANK3 deficiency. Mol. Autism 2013, 4, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, H.L.; Crolla, J.A.; Walker, D.; Artifoni, L.; Dallapiccola, B.; Takano, T.; Vasudevan, P.; Huang, S.; Maloney, V.; Yobb, T.; et al. Interstitial 22q13 deletions: Genes other than SHANK3 have major effects on cognitive and language development. Eur. J. Hum. Genet. 2008, 16, 1301–1310. [Google Scholar] [CrossRef] [PubMed]
- Culotta, L.; Scalmani, P.; Vinci, E.; Terragni, B.; Sessa, A.; Broccoli, V.; Mantegazza, M.; Verpelli, C. SULT4A1 modulates synaptic development and function by promoting the formation of PSD-95/NMDAR complex. J. Neurosci. 2020, 40, 7013–7026. [Google Scholar] [CrossRef] [PubMed]
- Durand, C.M.; Betancur, C.; Boeckers, T.M.; Bockmann, J.; Chaste, P.; Fauchereau, F.; Nygren, G.; Rastam, M.; Gillberg, I.C.; Anckarsäter, H.; et al. Mutations in the gene encoding the synaptic scaffolding protein SHANK3 are associated with autism spectrum disorders. Nat. Genet. 2006, 39, 25–27. [Google Scholar] [CrossRef] [Green Version]
- Gauthier, J.; Champagne, N.; Lafrenière, R.G.; Xiong, L.; Spiegelman, D.; Brustein, E.; Lapointe, M.; Peng, H.; Côté, M.; Noreau, A.; et al. De novo mutations in the gene encoding the synaptic scaffolding protein SHANK3 in patients ascertained for schizophrenia. Proc. Natl. Acad. Sci. USA 2010, 107, 7863–7868. [Google Scholar] [CrossRef] [Green Version]
- Nemirovsky, S.I.; Córdoba, M.; Zaiat, J.J.; Completa, S.P.; Vega, P.A.; González-Morón, D.; Medina, N.M.; Fabbro, M.; Romero, S.; Brun, B.; et al. Whole Genome Sequencing Reveals a De Novo SHANK3 Mutation in Familial Autism Spectrum Disorder. PLoS ONE 2015, 10, e0116358. [Google Scholar] [CrossRef]
- Zirn, B.; Arning, L.; Bartels, I.; Shoukier, M.; Hoffjan, S.; Neubauer, B.; Hahn, A. Ring chromosome 22 and neurofibromatosis type II: Proof of two-hit model for the loss of the NF2 gene in the development of meningioma. Clin. Genet. 2010, 81, 82–87. [Google Scholar] [CrossRef]
- Ziats, C.A.; Jain, L.; McLarney, B.; Vandenboom, E.; DuPont, B.R.; Rogers, C.; Sarasua, S.; Nevado, J.; Cordisco, E.L.; Phelan, K.; et al. Neurofibromatosis type 2 in Phelan–McDermid syndrome: Institutional experience and review of the literature. Eur. J. Med Genet. 2020, 63, 104042. [Google Scholar] [CrossRef]
- Lyons-Warren, A.M.; Cheung, S.W.; Lloyd Holder, J., Jr. Clinical Reasoning: A common cause for Phelan–McDermid syndrome and neurofibromatosis type 2. Neurology 2017, 89, e205–e209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woike, D.; Wang, E.; Tibbe, D.; Nia, F.H.; Failla, A.V.; Kibæk, M.; Overgård, T.M.; Larsen, M.J.; Fagerberg, C.R.; Barsukov, I.; et al. Mutations affecting the N-terminal domains of SHANK3 point to different pathomechanisms in neurodevelopmental disorders. Sci. Rep. 2022, 12, 902. [Google Scholar] [CrossRef] [PubMed]
- Sarasua, S.M.; Dwivedi, A.; Boccuto, L.; Rollins, J.D.; Chen, C.-F.; Rogers, R.C.; Phelan, K.; DuPont, B.R.; Collins, J.S. Association between deletion size and important phenotypes expands the genomic region of interest in Phelan–McDermid syndrome (22q13 deletion syndrome). J. Med. Genet. 2011, 48, 761–766. [Google Scholar] [CrossRef] [PubMed]
- Moutin, E.; Sakkaki, S.; Compan, V.; Bouquier, N.; Giona, F.; Areias, J.; Goyet, E.; Hemonnot-Girard, A.-L.; Seube, V.; Glasson, B.; et al. Restoring glutamate receptosome dynamics at synapses rescues autism-like deficits in Shank3-deficient mice. Mol. Psychiatry 2021, 26, 7596–7609. [Google Scholar] [CrossRef]
- Ma, K.; Qin, L.; Matas, E.; Duffney, L.J.; Liu, A.; Yan, Z. Histone deacetylase inhibitor MS-275 restores social and synaptic function in a Shank3-deficient mouse model of autism. Neuropsychopharmacology 2018, 43, 1779–1788. [Google Scholar] [CrossRef]
- Breen, M.S.; Browne, A.; Hoffman, G.E.; Stathopoulos, S.; Brennand, K.; Buxbaum, J.D.; Drapeau, E. Transcriptional signatures of participant-derived neural progenitor cells and neurons implicate altered Wnt signaling in Phelan–McDermid syndrome and autism. Mol. Autism 2020, 11, 53. [Google Scholar] [CrossRef]
- Lin, R.; Learman, L.N.; Bangash, M.A.; Melnikova, T.; Leyder, E.; Reddy, S.C.; Naidoo, N.; Park, J.M.; Savonenko, A.; Worley, P.F. Homer1a regulates Shank3 expression and underlies behavioral vulnerability to stress in a model of Phelan–McDermid syndrome. Cell Rep. 2021, 37, 110014. [Google Scholar] [CrossRef]
- Zwanenburg, R.J.; Ruiter, S.A.; Heuvel, E.R.V.D.; Flapper, B.C.; Van Ravenswaaij-Arts, C.M. Developmental phenotype in Phelan–McDermid (22q13.3 deletion) syndrome: A systematic and prospective study in 34 children. J. Neurodev. Disord. 2016, 8, 16. [Google Scholar] [CrossRef] [Green Version]
- Harony-Nicolas, H.; De Rubeis, S.; Kolevzon, A.; Buxbaum, J.D. Phelan McDermid Syndrome. J. Child Neurol. 2015, 30, 1861–1870. [Google Scholar] [CrossRef] [Green Version]
- Kolevzon, A.; Angarita, B.; Bush, L.; Wang, A.T.; Frank, Y.; Yang, A.; Rapaport, R.; Saland, J.; Srivastava, S.; Farrell, C.; et al. Phelan–McDermid syndrome: A review of the literature and practice parameters for medical assessment and monitoring. J. Neurodev. Disord. 2014, 6, 39. [Google Scholar] [CrossRef]
- Omansky, G.L.; Abdulhayoglu, E.; Zhurbilo, B.; Leary, O.G.; Elisa, A.; Bella, Z. Phelan–McDermid Syndrome. Neonatal Netw. 2017, 36, 98–102. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, H.; Kern, W.; Giese, R.; Hallschmid, M.; Enders, A. Intranasal insulin to improve developmental delay in children with 22q13 deletion syndrome: An exploratory clinical trial. J. Med. Genet. 2008, 46, 217–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shcheglovitov, A.; Shcheglovitova, O.; Yazawa, M.; Portmann, T.; Shu, R.; Sebastiano, V.; Krawisz, A.; Froehlich, W.; Bernstein, J.A.; Hallmayer, J.F.; et al. SHANK3 and IGF1 restore synaptic deficits in neurons from 22q13 deletion syndrome patients. Nature 2013, 503, 267–271. [Google Scholar] [CrossRef] [Green Version]
- Rajamani, K.T.; Wagner, S.; Grinevich, V.; Harony-Nicolas, H. Oxytocin as a Modulator of Synaptic Plasticity: Implications for Neurodevelopmental Disorders. Front. Synaptic Neurosci. 2018, 10, 17. [Google Scholar] [CrossRef] [Green Version]
- Darville, H.; Poulet, A.; Rodet-Amsellem, F.; Chatrousse, L.; Pernelle, J.; Boissart, C.; Héron, D.; Nava, C.; Perrier, A.; Jarrige, M.; et al. Human Pluripotent Stem Cell-derived Cortical Neurons for High Throughput Medication Screening in Autism: A Proof of Concept Study in SHANK3 Haploinsufficiency Syndrome. EBioMedicine 2016, 9, 293–305. [Google Scholar] [CrossRef] [Green Version]
- Verpelli, C.; Dvoretskova, E.; Vicidomini, C.; Rossi, F.; Chiappalone, M.; Schoen, M.; Di Stefano, B.; Mantegazza, R.; Broccoli, V.; Böckers, T.M.; et al. Importance of Shank3 Protein in Regulating Metabotropic Glutamate Receptor 5 (mGluR5) Expression and Signaling at Synapses. J. Biol. Chem. 2011, 286, 34839–34850. [Google Scholar] [CrossRef] [Green Version]
- Vicidomini, C.; Ponzoni, L.; Lim, D.; Schmeisser, M.; Reim, D.; Morello, N.; Orelanna, D.; Tozzi, A.; Durante, V.; Scalmani, P.; et al. Pharmacological enhancement of mGlu5 receptors rescues behavioral deficits in SHANK3 knock-out mice. Mol. Psychiatry 2016, 22, 689–702. [Google Scholar] [CrossRef] [Green Version]
- Serret, S.; Thümmler, S.; Dor, E.; Vesperini, S.; Santos, A.; Askenazy, F. Lithium as a rescue therapy for regression and catatonia features in two SHANK3 patients with autism spectrum disorder: Case reports. BMC Psychiatry 2015, 15, 107. [Google Scholar] [CrossRef] [Green Version]
- Dyar, B.; Meaddough, E.; Sarasua, S.; Rogers, C.; Phelan, K.; Boccuto, L. Genetic Findings as the Potential Basis of Personalized Pharmacotherapy in Phelan–McDermid Syndrome. Genes 2021, 12, 1192. [Google Scholar] [CrossRef]
- Qin, L.; Ma, K.; Wang, Z.-J.; Hu, Z.; Matas, E.; Wei, J.; Yan, Z. Social deficits in Shank3-deficient mouse models of autism are rescued by histone deacetylase (HDAC) inhibition. Nat. Neurosci. 2018, 21, 564–575. [Google Scholar] [CrossRef]
- Qin, L.; Ma, K.; Yan, Z. Rescue of histone hypoacetylation and social deficits by ketogenic diet in a Shank3 mouse model of autism. Neuropsychopharmacology 2021, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Torrioli, M.; Vernacotola, S.; Setini, C.; Bevilacqua, F.; Martinelli, D.; Snape, M.; Hutchison, J.A.; Di Raimo, F.R.; Tabolacci, E.; Neri, G. Treatment with valproic acid ameliorates ADHD symptoms in fragile X syndrome boys. Am. J. Med. Genet. Part A 2010, 152A, 1420–1427. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).