
Population Structure of a Worldwide Collection of Tropical Japonica Rice Indicates Limited Geographic Differentiation and Shows Promising Genetic Variability Associated with New Plant Type

 , ,
, ,  , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials and Experimentation
2.2. Phenotyping
2.3. Genotyping
2.4. Diversity Analysis
2.5. Determination of Population Structure
3. Results
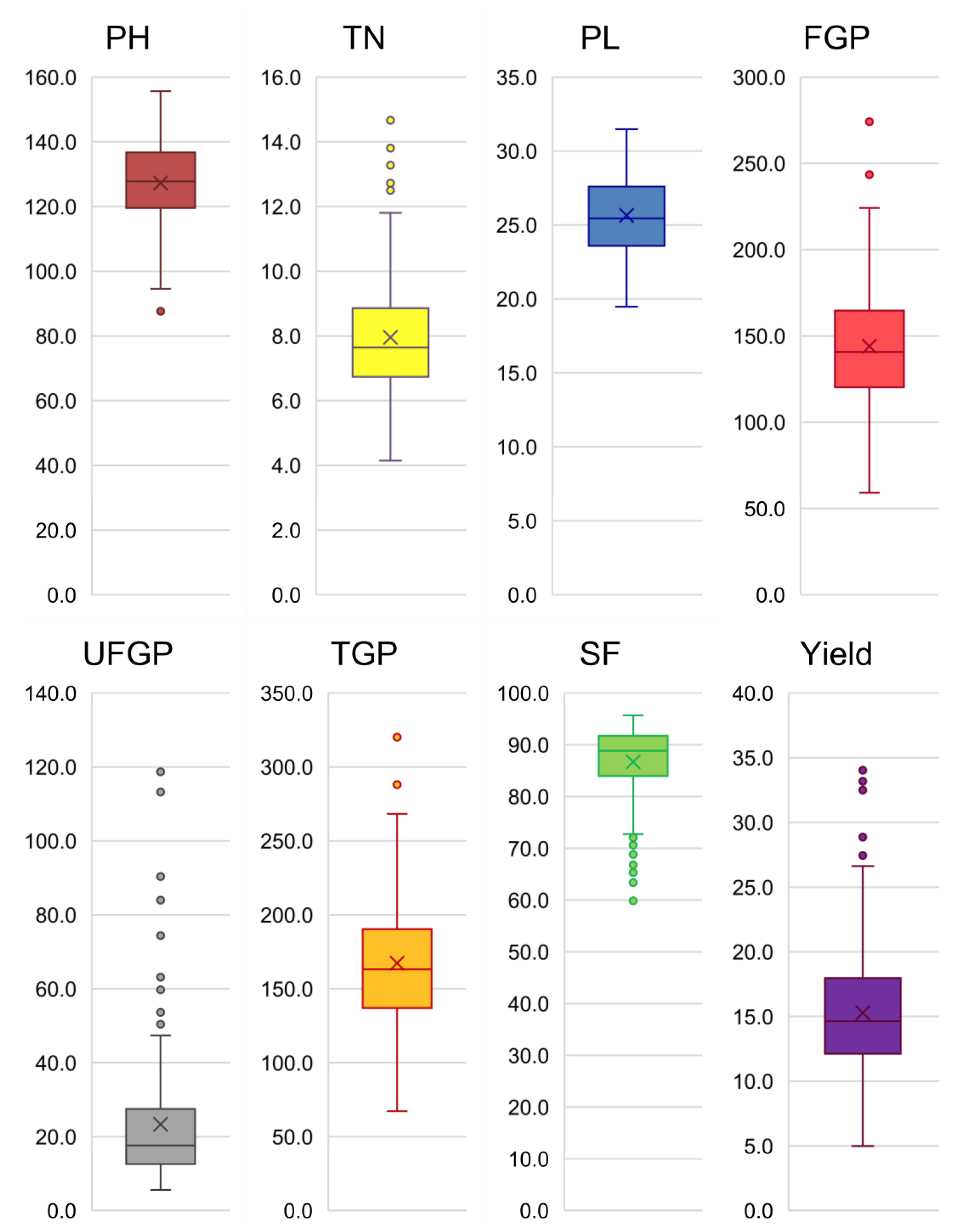
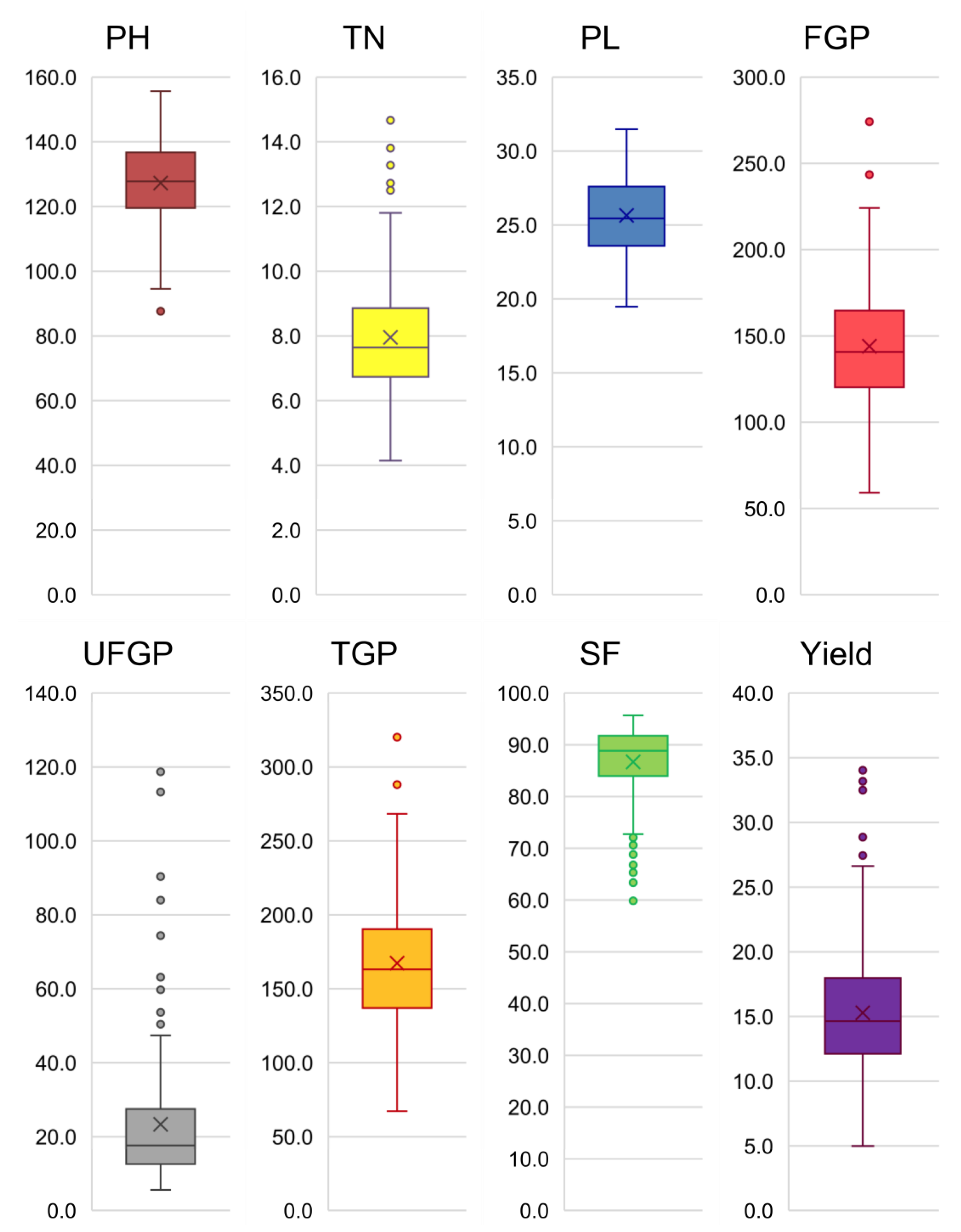
3.1. Agro-Morphological Variability
3.2. Phenotypic Diversity of Tropical Japonica Lines
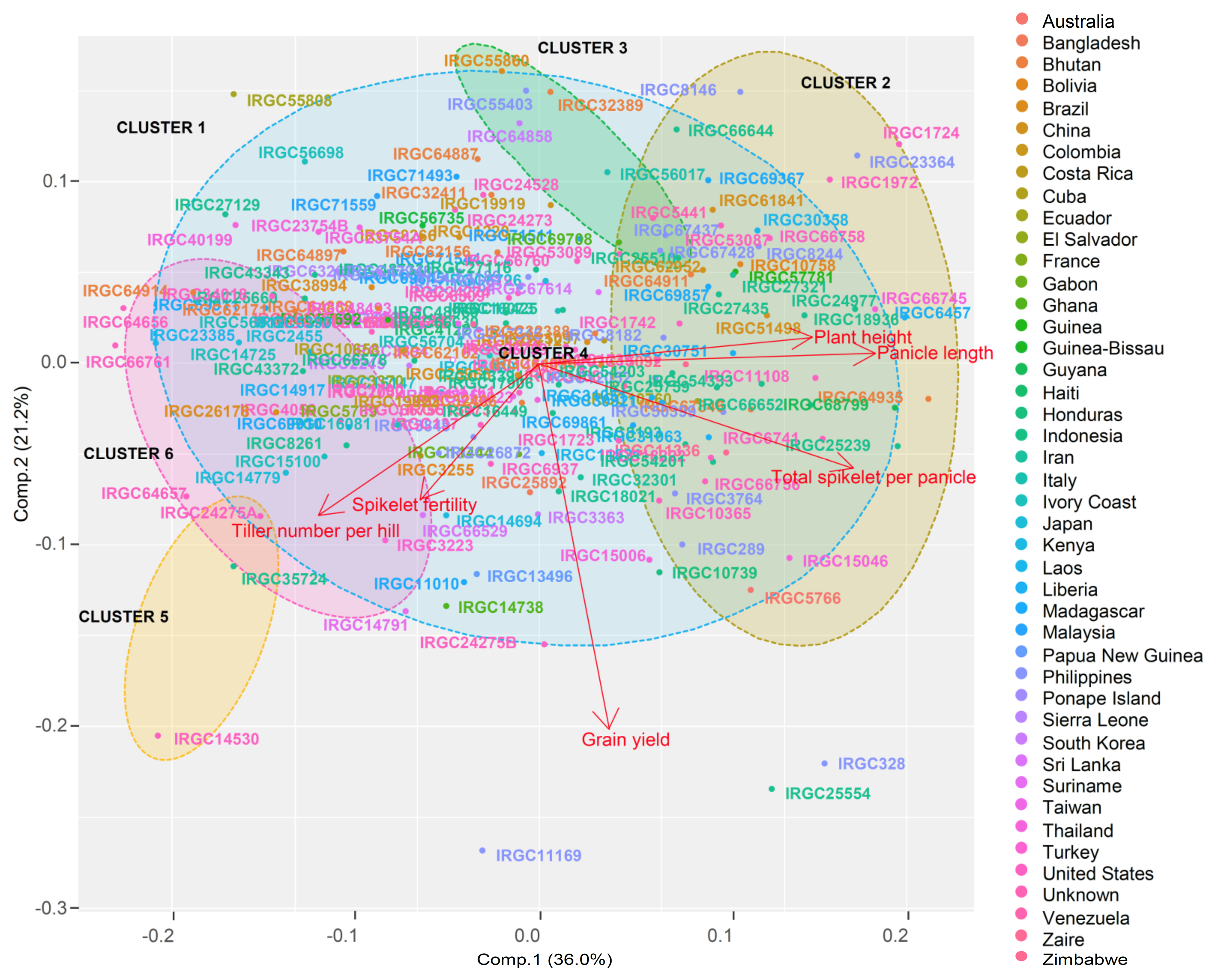
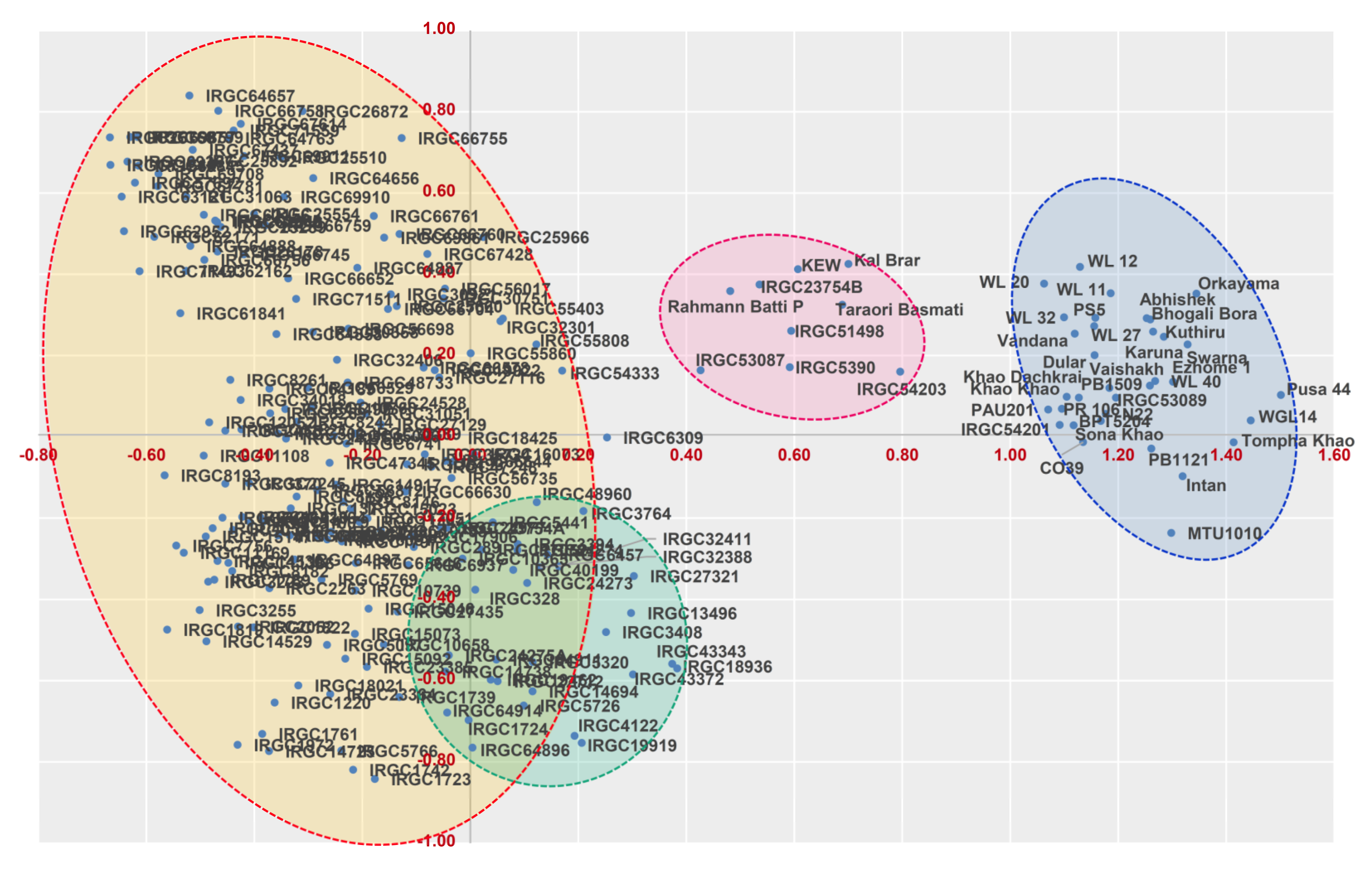
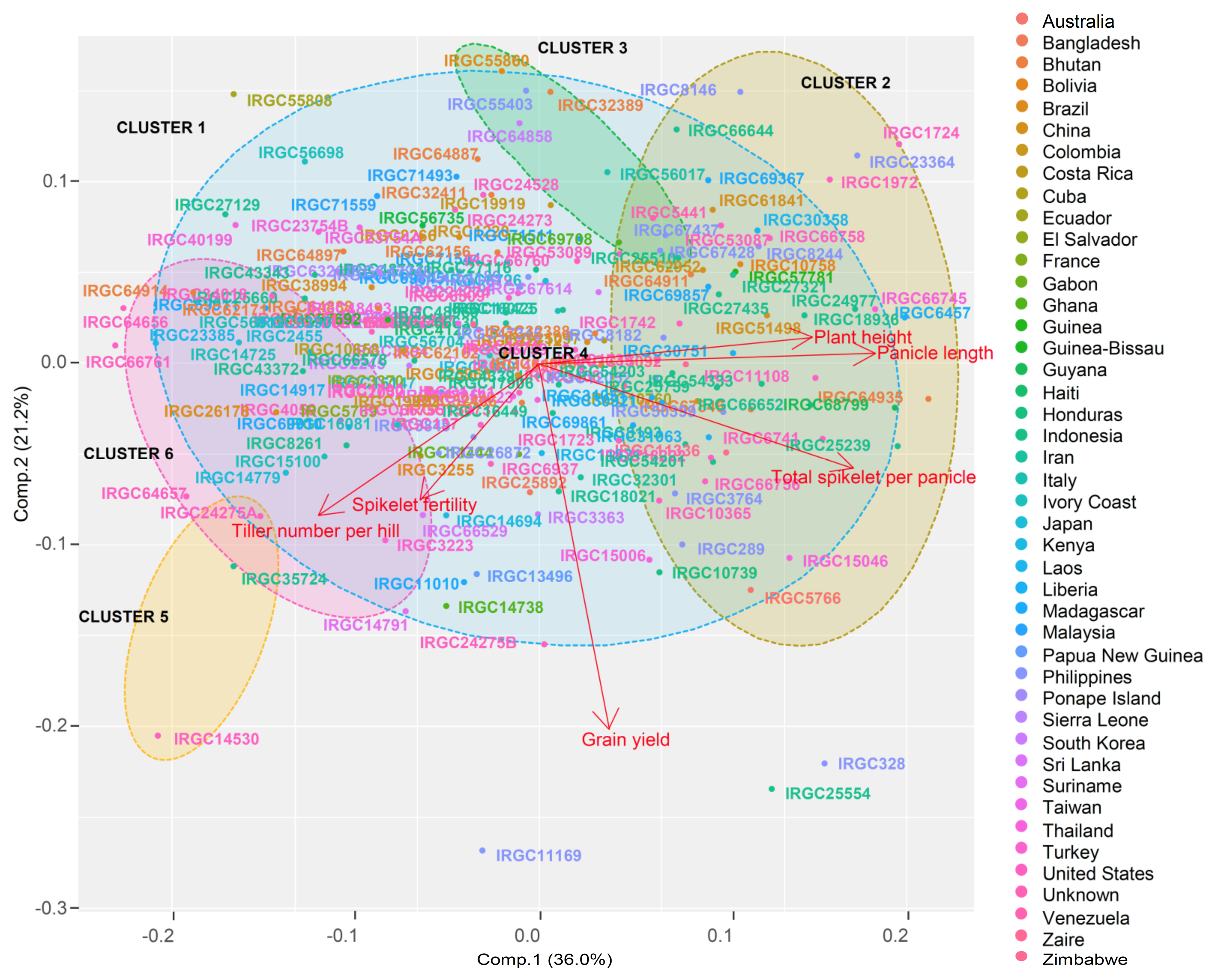
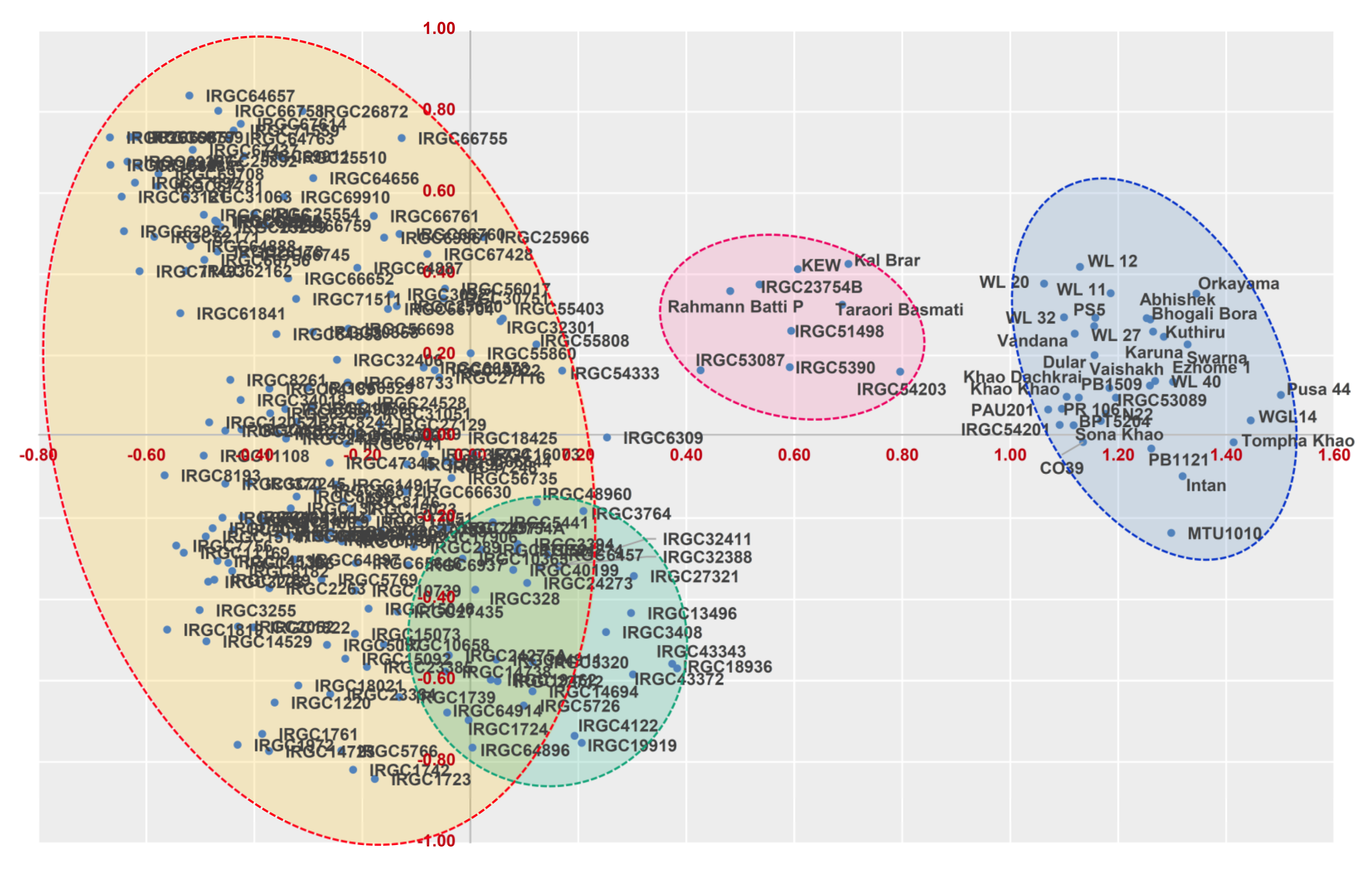
3.3. Marker-Based Genetic Distance and Grouping
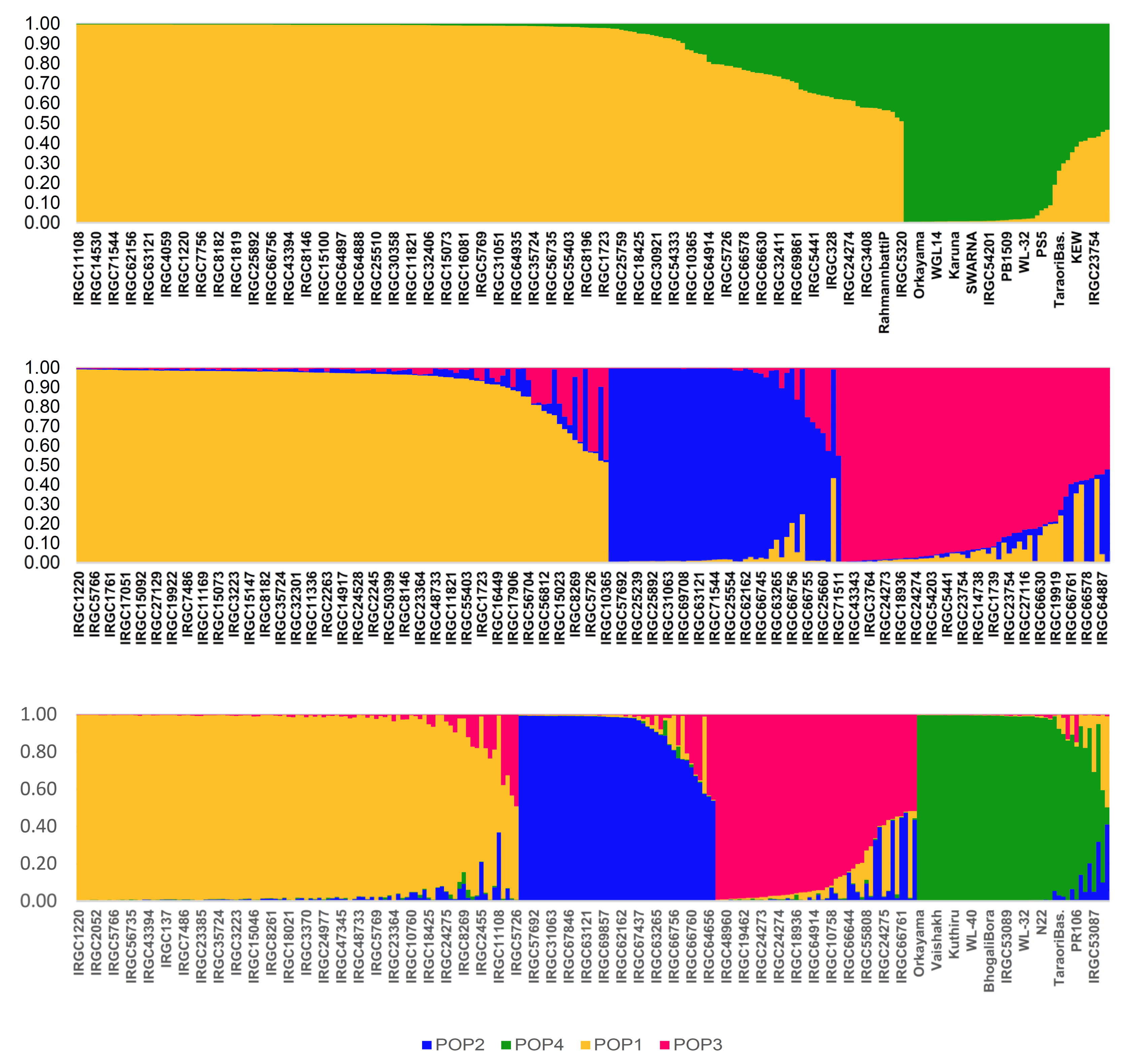
3.4. Population Structure
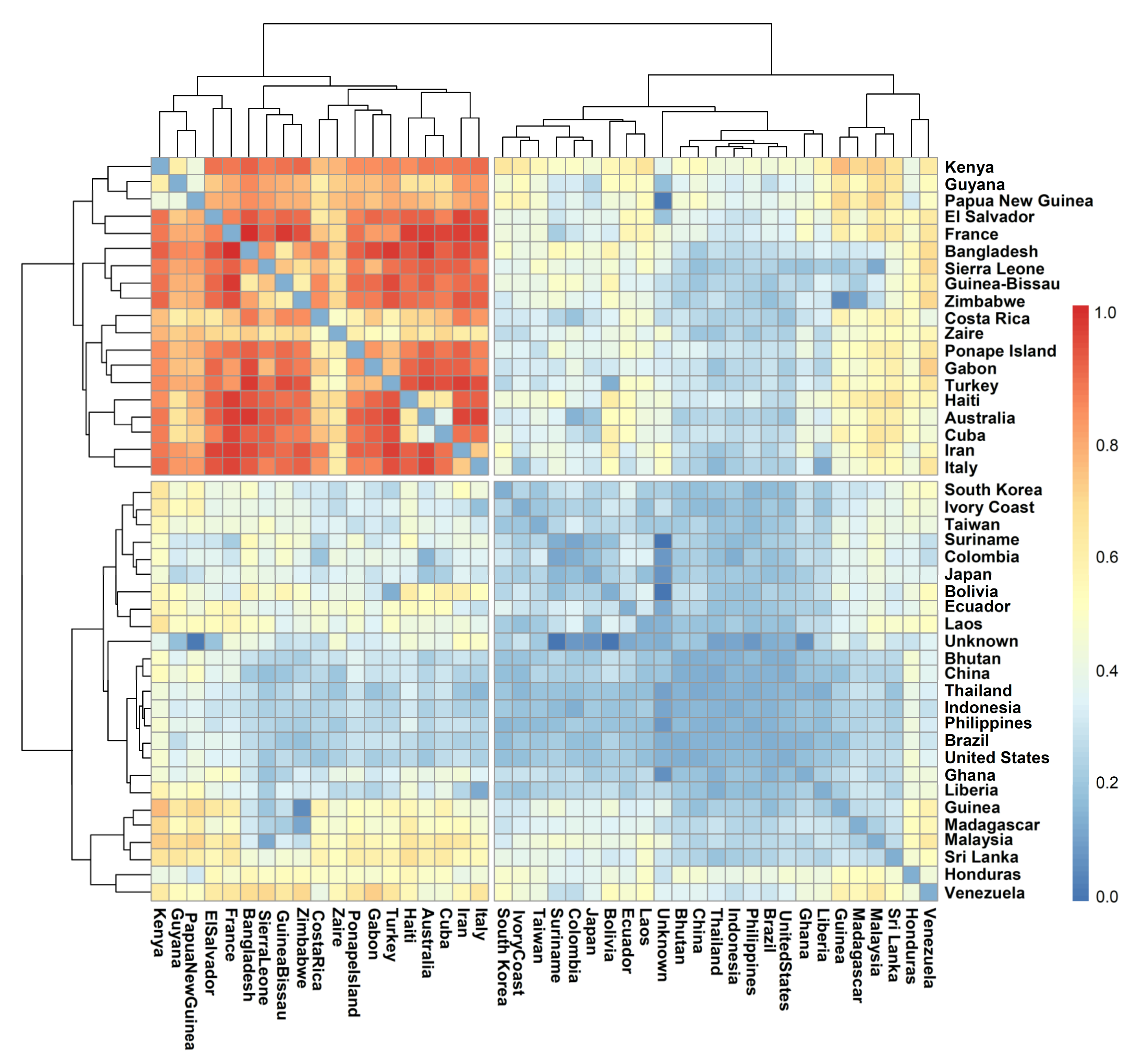
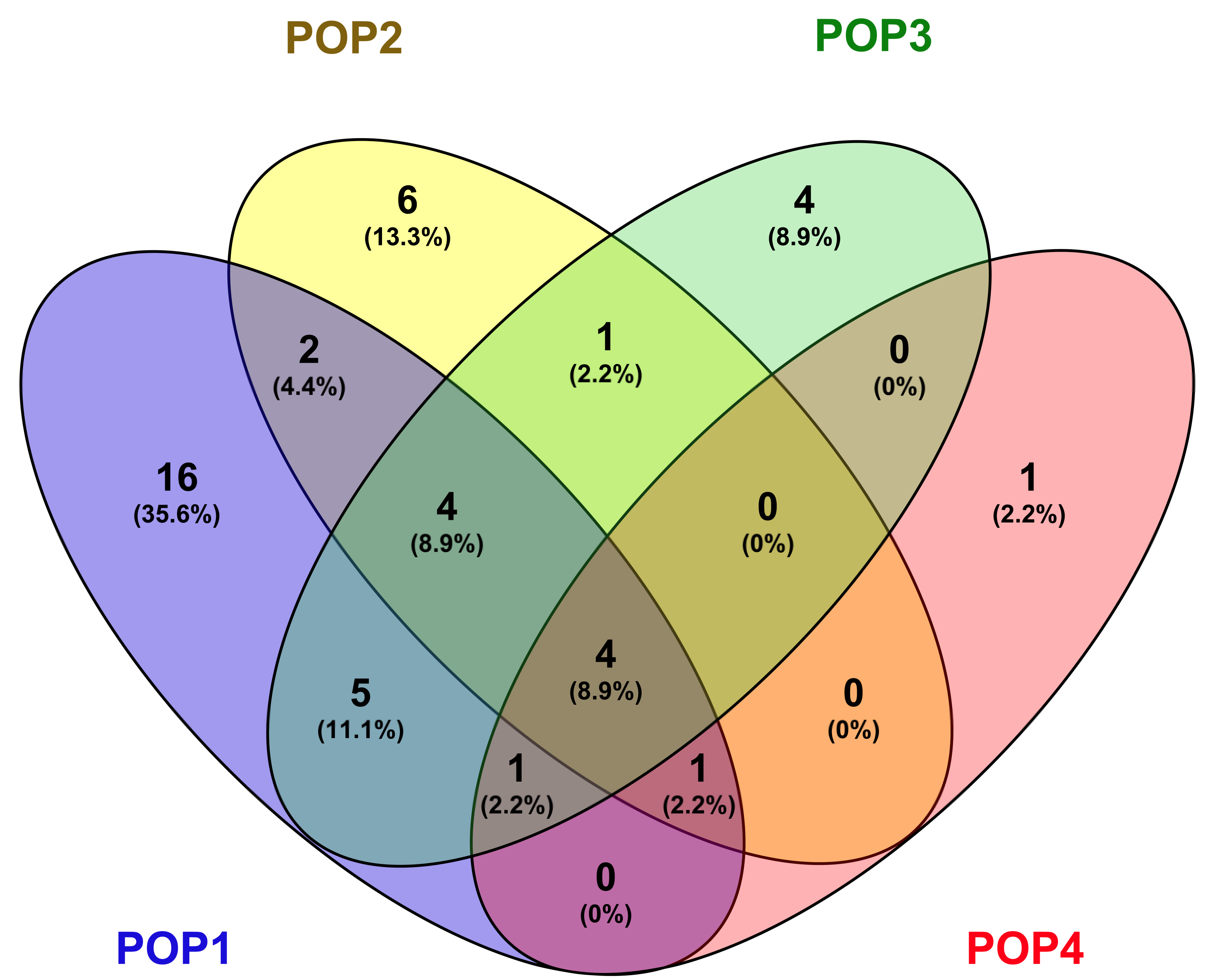
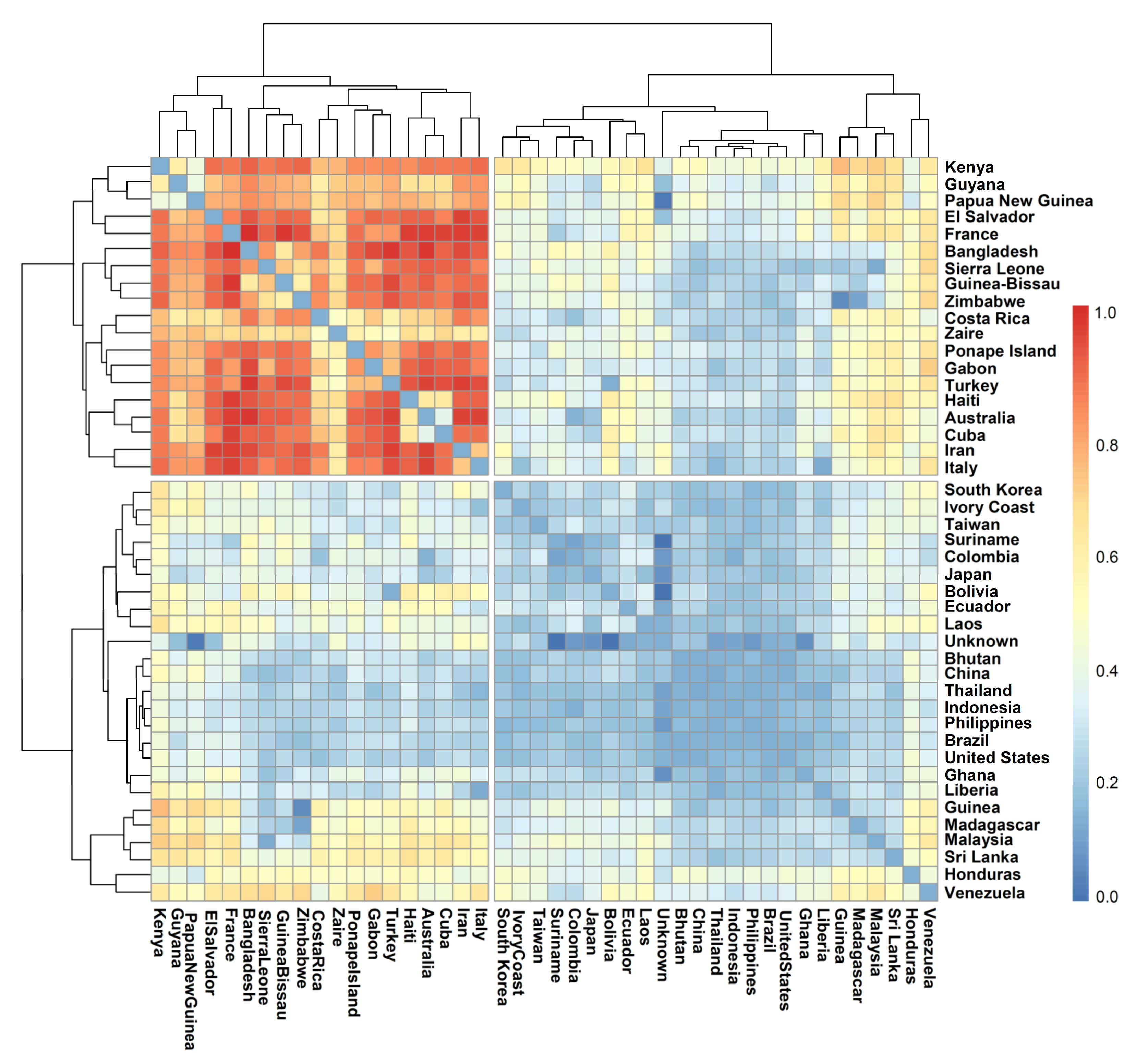
3.5. Population Distribution and Differentiation Based on Geographic Origin
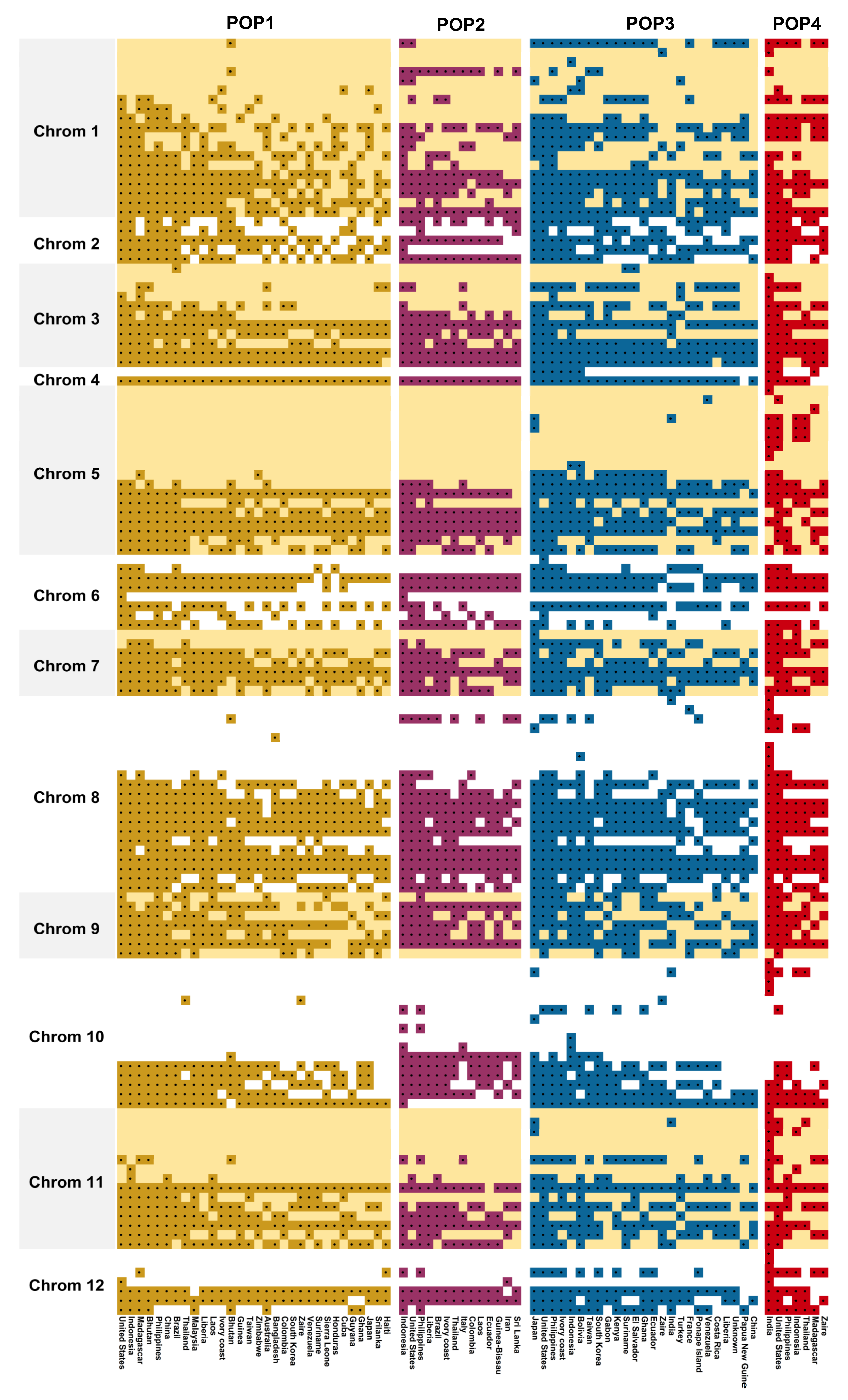
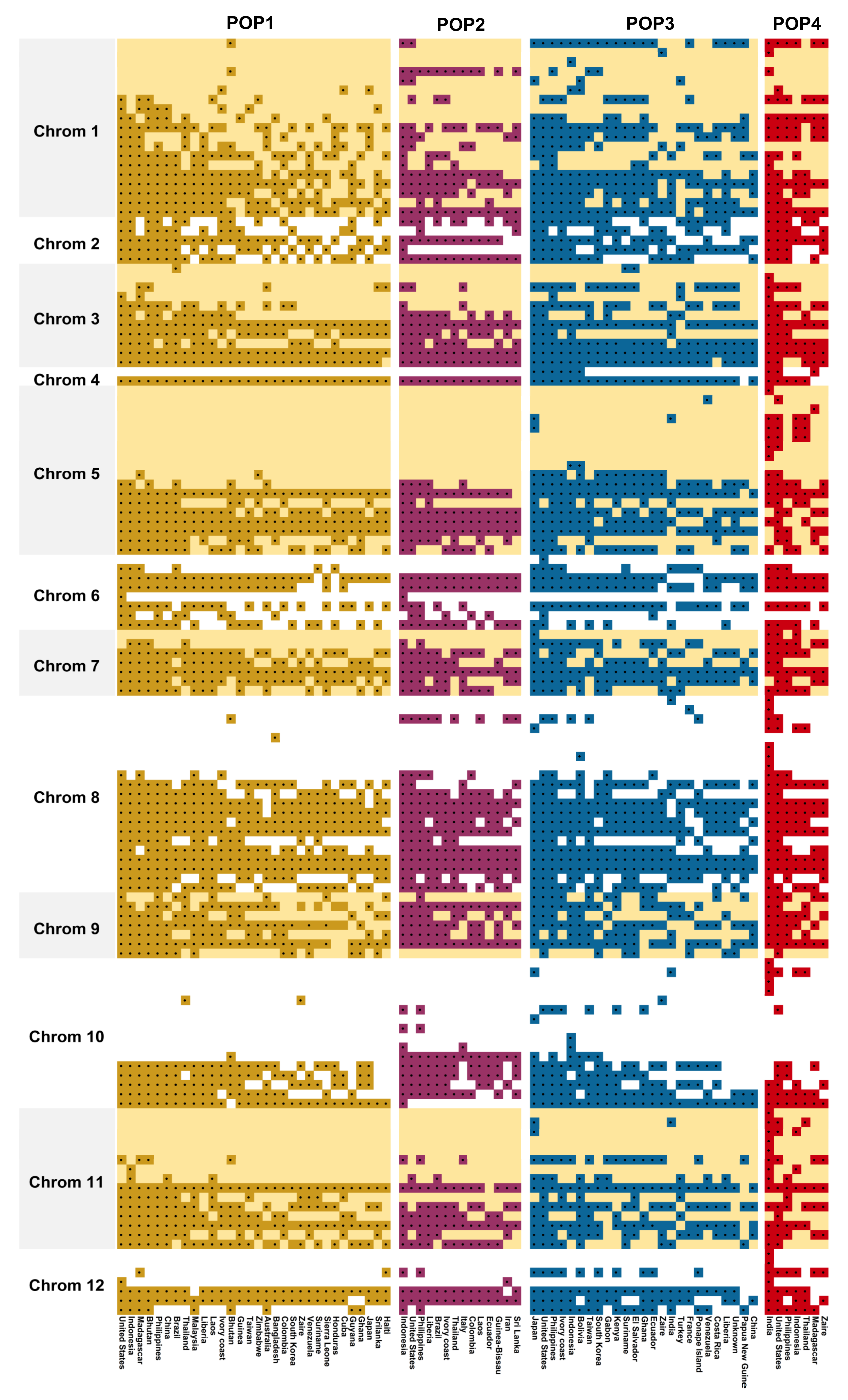
3.6. Chromosome-Wise Allele Distribution among the Subpopulations
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Singh, V.J.; Vinod, K.K.; Gopalakrishnan, S.; Singh, A.K. Rice Adaptation to Climate Change: Opportunities and Priorities in Molecular Breeding. In Molecular Breeding for Rice Abiotic Stress Tolerance and Nutritional Quality; John Wiley & Sons: Hoboken, NJ, USA, 2021; pp. 1–25. [Google Scholar]
- Nachimuthu, V.V.; Raveendran, M.; Sudhakar, D.; Rajeswari, S.; Balaji, A.; Pandian, G.P.; Karthika, G.; Manonmani, S.; Suji, K.K.; Robin, S. Analysis of population structure and genetic diversity in rice germplasm using SSR markers: An initiative towards association mapping of agronomic traits in Oryza sativa. Rice 2015, 8, 30. [Google Scholar] [CrossRef] [Green Version]
- Singh, V.J.; Gampala, S.; Chakraborti, S.K.; Singh, A.K. Molecular characterization and genetic diversity analysis of rice varieties and landraces based on SSR markers. Ecoscan 2015, 9, 363–368. [Google Scholar]
- Bandumula, N. Rice production in Asia: Key to global food security. Proc. Natl. Acad. Sci. India Sect. B Biol. Sci. 2018, 88, 1323–1328. [Google Scholar] [CrossRef]
- Kumar, A.; Bhowmick, P.K.; Singh, V.J.; Malik, M.; Gupta, A.K.; Seth, R.; Nagarajan, M.; Krishnan, S.G.; Singh, A.K. Marker-assisted identification of restorer gene(s) in iso-cytoplasmic restorer lines of WA cytoplasm in rice and assessment of their fertility restoration potential across environments. Physiol. Mol. Biol. Plants 2017, 23, 891–909. [Google Scholar] [CrossRef]
- Kumar, A.; Singh, V.J.; Krishnan, S.G.; Vinod, K.K.; Bhowmick, P.K.; Nagarajan, M.; Ellur, R.K.; Bollinedi, H.; Singh, A.K. WA-CMS-based iso-cytoplasmic restorers derived from commercial rice hybrids reveal distinct population structure and genetic divergence towards restorer diversification. 3Biotech 2019, 9, 299. [Google Scholar] [CrossRef]
- Shidenur, S.; Singh, V.J.; Vinod, K.K.; Krishnan, S.G.; Ghritlahre, S.K.; Bollinedi, H.; Ellur, R.K.; Dixit, B.K.; Singh, B.; Nagarajan, M.; et al. Molecular detection of WA-CMS restorers from tropical japonica derived lines, their evaluation for fertility restoration and adaptation. Plant Breed. 2019, 138, 553–567. [Google Scholar] [CrossRef]
- Khush, G.S. Breaking the yield frontier of rice. GeoJournal 1995, 35, 329–332. [Google Scholar] [CrossRef]
- Shidenur, S.; Singh, V.J.; Vinod, K.K.; Krishnan, S.G.; Ghritlahre, S.K.; Bollinedi, H.; Dixit, B.K.; Ellur, R.K.; Nagarajan, M.; Singh, A.K.; et al. Enhanced grain yield in rice hybrids through complementation of fertility restoration by Rf3 and Rf4 genes as revealed by multi-location evaluation of tropical japonica derived rice (Oryza sativa L.) hybrids. Plant Breed. 2020, 139, 743–753. [Google Scholar] [CrossRef]
- Phillips, R.L.; Vasil, I.K. DNA-Based Markers in Plants; Kluwer Academic Publishers: Dordrecht, The Netherlands, 2001; Volume 497. [Google Scholar]
- Varshney, R.K.; Graner, A.; Sorrells, M.E. Genic microsatellite markers in plants: Features and applications. Trends Biotechnol. 2005, 23, 48–55. [Google Scholar] [CrossRef]
- Garris, A.J.; Tai, T.H.; Coburn, J.; Kresovich, S.; McCouch, S. Genetic structure and diversity in Oryza sativa L. Genetics 2005, 169, 1631–1638. [Google Scholar] [CrossRef] [Green Version]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2020; Available online: https://www.R-project.org/ (accessed on 22 February 2022).
- Chen, X.; Cho, Y.; McCouch, S. Sequence divergence of rice microsatellites in Oryza and other plant species. Mol. Genet. Genomics 2002, 268, 331–343. [Google Scholar] [CrossRef] [PubMed]
- Prabhu, K.V.; Somers, D.J.; Rakow, G.; Gugel, R.K. Molecular markers linked to white rust resistance in mustard Brassica juncea. Theor. Appl. Genet. 1998, 97, 865–870. [Google Scholar] [CrossRef]
- Liu, K.; Muse, S.V. PowerMarker V3.25: Integrated analysis environment for genetic marker data. Bioinformatics 2005, 9, 2128–2129. [Google Scholar] [CrossRef] [Green Version]
- Botstein, D.; White, R.L.; Skolnick, M.; Davis, R.W. Construction of genetic linkage map in man using restriction fragment length polymorphisms. Am. J. Hum. Genet. 1980, 32, 314–331. [Google Scholar]
- Rogers, J.S. Measures of Genetic Similarity and Genetic Distance. In Studies in Genetics VII; University of Texas Publication 7213: Austin, TX, USA, 1972; pp. 145–153. [Google Scholar]
- Yeh, F.C.; Yand, R.C.; Boyle, T.J.B. POPGENE, the User-Friendly Shareware for Population Genetic Analysis (Ver.1.32); University of Alberta Press: Edmonton, AB, Canada, 2000. [Google Scholar]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Earl, D.A.; von Holdt, B.M. STRUCTURE HARVESTER: A website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Res. 2012, 4, 359–361. [Google Scholar] [CrossRef]
- Excoffier, L.; Laval, G.; Schneider, S. Arlequin ver. 3.0: An integrated software package for population genetics data analysis. Evol. Bioinform. 2005, 1, 47–50. [Google Scholar] [CrossRef] [Green Version]
- Weir, B.S.; Cockerham, C.C. Estimating F-statistics for the analysis of population structure. Evolution 1984, 38, 1358–1370. [Google Scholar]
- Thomson, M.J.; Septiningsih, E.M.; Suwardjo, F.; Santoso, T.J.; Silitonga, T.S.; McCouch, S.R. Genetic diversity analysis of traditional and improved Indonesian rice (Oryza sativa L.) germplasm using microsatellite markers. Theor. Appl. Genet. 2007, 114, 559–568. [Google Scholar] [CrossRef]
- Gampala, S.; Singh, V.J.; Chakraborti, S.K. Analysis of variability and genetic parameter for grain quality attributes in high yielding rice (Oryza sativa L.) genotypes. Ecoscan 2015, 9, 411–414. [Google Scholar]
- Wang, C.H.; Zheng, X.M.; Xu, Q.; Yuan, X.P.; Huang, L.; Zhou, H.F.; Wei, X.H.; Ge, S. Genetic diversity and classification of Oryza sativa with emphasis on Chinese rice germplasm. Heredity 2014, 112, 489–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khush, G.S.; Peng, S. Improving yield potential by modifying plant type. In IRRI Discussion Papers 287587; International Rice Research Institute (IRRI): Los Baños, Philippines, 1998. [Google Scholar] [CrossRef]
- Kim, B. Classifying Oryza sativa accessions into indica and japonica using logistic regression model with phenotypic data. Peer J. 2019, 7, 7259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, S.; Cassman, K.G.; Virmani, S.S.; Sheehy, J.; Khush, G.S. Yield potential trends of tropical rice since the release of IR8 and the challenge of increasing rice yield potential. Crop Sci. 1999, 39, 1552–1559. [Google Scholar] [CrossRef] [Green Version]
- Virk, P.S.; Khush, G.S.; Peng, S. Breeding to enhance yield potential of rice at IRRI: The ideotype approach. Int. Rice Res. Note 2004, 29, 5–9. [Google Scholar]
- Bollinedi, H.; Yadav, A.K.; Vinod, K.K.; Gopala Krishnan, S.; Bhowmick, P.K.; Nagarajan, M.; Neeraja, C.N.; Ellur, R.K.; Singh, A.K. Genome-wide association study reveals novel marker-trait associations (MTAs) governing the localization of Fe and Zn in the rice grain. Front. Genet. 2020, 11, 213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, A.; Singh, V.J.; Bhowmick, P.K.; Vinod, K.K.; Seth, R.; Nagarajan, M.; Ellur, R.K.; Bollinedi, H.; Krishnan, S.G.; Singh, A.K. Assessing the performance of hybrids developed using iso-cytoplasmic restorers and identification of promising combiners in rice. Indian J. Genet. 2019, 79, 523–535. [Google Scholar] [CrossRef]
- Burman, D.; Maji, B.; Singh, S.; Mandal, S.; Sarangi, S.K.; Bandyopadhyay, B.K.; Bal, A.R.; Sharma, D.K.; Krishnamurthy, S.L.; Singh, H.N.; et al. Participatory evaluation guides the development and selection of farmers’ preferred rice varieties for salt-and flood-affected coastal deltas of South and Southeast Asia. Field Crops Res. 2018, 220, 67–77. [Google Scholar] [CrossRef]
- Wang, Y.; Lu, J.; Ren, T.; Hussain, S.; Guo, C.; Wang, S.; Cong, R.; Li, X. Effects of nitrogen and tiller type on grain yield and physiological responses in rice. AoB Plants 2017, 9, plx012. [Google Scholar] [CrossRef]
- Jyothi, B.; Divya, B.; Rao, L.S.; Bhavani, P.L.; Revathi, P.; Rao, P.R.; Rachana, B.; Padmavathi, G.; Kumar, J.A.; Gireesh, C.; et al. New plant type trait characterization and development of core set among indica and tropical japonica genotypes of rice. Plant Genet. Resour. 2018, 16, 504–512. [Google Scholar] [CrossRef]
- Messmer, M.M.; Melchinger, A.E.; Herrmann, R.G.; Boppenmaier, J. Relationships among early European maize inbreds: II. Comparison of pedigree and RFLP data. Crop Sci. 1993, 33, 944–950. [Google Scholar] [CrossRef]
- Gross, B.L.; Zhao, Z. Archaeological and genetic insights into the origins of domesticated rice. Proc. Natl. Acad. Sci. USA 2014, 111, 6190–6197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silva, F.; Stevens, C.J.; Weisskopf, A.; Castillo, C.; Qin, L.; Bevan, A.; Fuller, D.Q. Modelling the geographical origin of rice cultivation in Asia using the rice archaeological database. PLoS ONE 2015, 10, e0137024. [Google Scholar] [CrossRef] [PubMed]
- Anandan, A.; Anumlla, M.; Pradhan, S.K.; Ali, J. Population structure, diversity and trait association analysis in rice (Oryza sativa L.) germplasm for early seedling vigor (ESV) using trait linked SSR markers. PLoS ONE 2016, 11, e0152406. [Google Scholar] [CrossRef] [Green Version]
- DeWoody, J.A.; Honeycutt, R.L.; Skow, L.C. Microsatellite markers in white-tailed deer. J. Hered. 1995, 86, 317–319. [Google Scholar] [CrossRef]
- Hashimoto, Z.; Mori, N.; Kawamura, M.; Ishii, T.; Yoshida, S.; Ikegami, M.; Takumi, S.; Nakamura, C. Genetic diversity and phylogeny of Japanese sake-brewing rice as revealed by AFLP and nuclear and chloroplast SSR markers. Theor. Appl. Genet. 2004, 109, 1586–1596. [Google Scholar] [CrossRef]
- Siwach, P.; Jain, S.; Saini, N.; Chowdhury, V.K.; Jain, R.K. Allelic diversity among Basmati and non-Basmati long-grain indica rice varieties using microsatellite markers. J. Plant Biochem. Biotechnol. 2004, 13, 25–32. [Google Scholar] [CrossRef]
- Shah, S.M.; Naveed, S.A.; Arif, M. Genetic diversity in basmati and non-basmati rice varieties based on microsatellite markers. Pak. J. Bot. 2013, 45, 423–431. [Google Scholar]
- Behera, L.; Mohanty, S.; Pradhan, S.K.; Singh, S.; Singh, O.N.; Sahu, R.; Sahu, S.C.; Dash, S.K.; Mohapatra, T. Assessment of genetic diversity of rainfed lowland rice genotypes using microsatellite markers. Indian J. Genet. 2013, 73, 142–152. [Google Scholar] [CrossRef]
- Ravi, M.; Geethanjali, S.; Sameeyafarheen, F.; Maheswaran, M. Molecular marker based genetic diversity analysis in rice (Oryza sativa L.) using RAPD and SSR markers. Euphytica 2003, 133, 243–252. [Google Scholar] [CrossRef]
- Sruthi, K.; Divya, B.; Senguttuvel, P.; Revathi, P.; Kemparaju, K.B.; Koteswararao, P.; Sundaram, R.M.; Singh, V.J.; Kumar, E.R.; Bhowmick, P.K.; et al. Evaluation of genetic diversity of parental lines for development of heterotic groups in hybrid rice (Oryza sativa L.). J Plant Biochem. Biotechnol. 2020, 29, 236–252. [Google Scholar] [CrossRef]
- Oka, H.I. Functions and genetic base of reproductive barriers. In Origin of Cultivated Rice; Oka, H.I., Ed.; Elsevier Science; Scientific Societies Press: Tokyo, Japan; Amsterdam, The Netherlands, 1988; Volume 156, pp. 181–209. [Google Scholar]
- Kato, S. The affinity of rice varieties as shown by fertility of hybrid plants. Bull. Sci. Fac. Agric. Kyushu Univ. 1998, 3, 132–147. [Google Scholar]
- Ikehashi, H.; Araki, H. Varietal screening of compatibility types revealed in F1 fertility of distant crosses in rice. Jap. J. Breed. 1984, 34, 304–313. [Google Scholar] [CrossRef] [Green Version]
- Kumar, R.V.; Virmani, S.S. Wide compatibility in rice (Oryza sativa L.). Euphytica 1992, 64, 71–80. [Google Scholar] [CrossRef]
- Glaszmann, J.C. Isozymes and classification of Asian rice varieties. Appl Genet. 1987, 74, 21–30. [Google Scholar] [CrossRef]
- Parsons, B.J.; Newbury, H.J.; Jackson, M.T. The genetic structure and conservation of aus, aman and boro rices from Bangladesh. Genet. Res. Crop Evol. 1999, 46, 587–598. [Google Scholar] [CrossRef]
- Ni, J.; Colowit, P.M.; Mackill, D.J. Evaluation of genetic diversity in rice subspecies using microsatellite markers. Crop Sci. 2002, 42, 601–607. [Google Scholar] [CrossRef] [Green Version]
- Upadhyay, P.; Neeraj, C.N.; Kole, C.; Singh, V.K. Population structure and genetic diversity in popular rice varieties of India as evidenced from SSR analysis. Biochem. Genet. 2012, 50, 70–83. [Google Scholar] [CrossRef]
- Salgotra, R.K.; Gupta, B.B.; Javaid, A.B.; Sandeep, S. Genetic Diversity and Population Structure of Basmati Rice (Oryza sativa L.) germplasm collected from North Western Himalayas using trait linked SSR Markers. PLoS ONE 2015, 10, e0131858. [Google Scholar] [CrossRef] [Green Version]
- Babu, B.K.; Meena, V.; Agarwal, V.; Agrawal, P.K. Population structure and genetic diversity analysis of Indian and exotic rice (Oryza sativa L.) accessions using SSR markers. Mol. Biol. Rep. 2014, 41, 4328–4339. [Google Scholar] [CrossRef]
- Sun, C.Q.; Wang, X.K.; Yoshimura, A.; Doi, K. Genetic differentiation for nuclear mitochondrial and chloroplast genomes in common wild rice (O. rufipogon Griff.) and cultivated rice (O. sativa L.). Theor. Appl. Genet. 2002, 104, 1335–1345. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Source | Df | Mean Sum of Squares | |||||||
|---|---|---|---|---|---|---|---|---|---|
| PH | TLN | PL | FG | UFG | TG | SF | YLD | ||
| Genotype (G) | 199 | 1000.2 ** | 21.1 ** | 43.9 ** | 6929.0 ** | 1784.7 ** | 10007.0 ** | 305.2 ** | 142.4 ** |
| Year (Y) | 2 | 106.0 | 1.3 | 0.8 | 481.0 | 150.2 | 111.0 | 136.1 | 33.5 |
| G × Y | 398 | 481.5 ** | 12.7 ** | 14.5 ** | 2946.0 ** | 569.7 ** | 3900.0 ** | 131.1 ** | 42.1 ** |
| Rep (Year) | 3 | 2410.3 | 34.3 | 219.8 | 11745.0 | 932.9 | 19070.0 | 62.1 | 311.8 |
| Residual | 597 | 144.4 | 2.15 | 20.8 | 160.0 | 34.4 | 201.0 | 9.5 | 3.3 |
| CV (%) | 15.8 | 37.5 | 18.7 | 32.8 | 96.2 | 33.3 | 11.5 | 41.4 | |
| LSD (0.05) | 2.2 | 0.3 | 0.8 | 2.3 | 3.4 | 2.6 | 0.6 | 0.3 | |
| Mean | 127.3 | 8.0 | 25.6 | 144.0 | 23.4 | 167.4 | 86.7 | 15.3 | |
| Minimum | 87.7 | 4.2 | 19.5 | 59.1 | 5.6 | 67.2 | 59.8 | 5.0 | |
| Maximum | 155.7 | 14.8 | 31.5 | 274.1 | 118.7 | 323.8 | 95.7 | 34.1 | |
| Statistics | PC1 | PC2 | PC3 | PC4 | PC5 | PC6 | |
|---|---|---|---|---|---|---|---|
| Standard deviation | 1.47 | 1.12 | 1.06 | 0.89 | 0.66 | 0.46 | |
| Proportion of Variance | 0.36 | 0.21 | 0.19 | 0.13 | 0.07 | 0.04 | |
| Cumulative Proportion (%) | 36.0 | 57.2 | 75.8 | 89.1 | 96.5 | 100.0 | |
| Eigenvalues | 2.16 | 1.27 | 1.12 | 0.79 | 0.44 | 0.21 | |
| Trait contribution (%) | PH | 21.17 | 0.32 | 0.04 | 54.39 | 23.86 | 0.21 |
| TLN | 13.80 | 12.08 | 35.36 | 11.99 | 0.35 | 26.43 | |
| PL | 31.79 | 0.05 | 0.43 | 3.28 | 62.65 | 1.81 | |
| TG | 27.92 | 5.99 | 0.35 | 25.69 | 12.12 | 27.93 | |
| SF | 3.92 | 9.91 | 63.69 | 4.61 | 0.94 | 16.95 | |
| YLD | 1.41 | 71.66 | 0.13 | 0.05 | 0.08 | 26.67 | |
| Cluster | Membership % | Centroid ± Standard Error | |||||
|---|---|---|---|---|---|---|---|
| PH | TLN | PL | TG | SF | YLD | ||
| 1 | 86.5 | 127.9 ± 1.0 | 7.8 ± 0.1 | 25.9 ± 0.2 | 165.0 ± 2.6 | 88.6 ± 0.35 | 15.5 ± 0.4 |
| 2 | 7.5 | 137.3 ± 2.7 | 7.8 ± 0.5 | 28.1 ± 0.5 | 246.3 ± 10.7 | 73.4 ± 2.34 | 16.1 ± 1.1 |
| 3 | 2.5 | 142.4 ± 3.6 | 8.5 ± 0.4 | 26.0 ± 1.0 | 121.2 ± 12.0 | 70.9 ± 2.20 | 8.2 ± 0.6 |
| 4 | 0.5 | 102.0 ± 0.0 | 10.8 ± 0.0 | 24.8 ± 0.0 | 238.8 ± 0.0 | 58.9 ± 0.0 | 20.1 ± 0.0 |
| 5 | 1.5 | 95.3 ± 5.2 | 11.4 ± 0.2 | 23.8 ± 1.1 | 136.7 ± 7.8 | 87.7 ± 1.18 | 25.0 ± 3.9 |
| 6 | 1.5 | 131.9 ± 8.3 | 15.5 ± 0.9 | 24.7 ± 1.0 | 109.1 ± 23.1 | 91.8 ± 1.31 | 14.6 ± 2.9 |
| Chrom | Loci | Allele Frequency | AFm | He | PIC | Ae | Rare Alleles | ||
|---|---|---|---|---|---|---|---|---|---|
| Total | Average | Class | |||||||
| 1 | 6 | 19 | 3.17 | 2, 3, 6 | 0.689 | 0.423 | 0.366 | 1.863 | 1 |
| 2 | 2 | 5 | 2.50 | 2, 3 | 0.603 | 0.519 | 0.432 | 2.114 | - |
| 3 | 5 | 11 | 2.20 | 2, 3 | 0.761 | 0.294 | 0.237 | 1.528 | - |
| 4 | 1 | 2 | 2.00 | 2 | 0.931 | 0.128 | 0.120 | 1.147 | - |
| 5 | 5 | 18 | 3.60 | 2, 3, 4, 6 | 0.705 | 0.405 | 0.353 | 1.848 | 2 |
| 6 | 3 | 8 | 2.67 | 2, 4 | 0.772 | 0.298 | 0.250 | 1.579 | 1 |
| 7 | 3 | 7 | 2.33 | 2, 3 | 0.615 | 0.455 | 0.380 | 2.002 | - |
| 8 | 7 | 21 | 3.00 | 2, 3, 4 | 0.669 | 0.400 | 0.338 | 1.888 | 2 |
| 9 | 3 | 7 | 2.33 | 2, 3 | 0.693 | 0.432 | 0.360 | 1.786 | - |
| 10 | 3 | 16 | 5.33 | 2, 3, 11 | 0.566 | 0.511 | 0.436 | 2.261 | 5 |
| 11 | 4 | 15 | 3.75 | 3, 4, 5 | 0.714 | 0.402 | 0.345 | 1.734 | - |
| 12 | 2 | 7 | 3.50 | 3, 5 | 0.772 | 0.361 | 0.312 | 1.565 | 2 |
| K | Reps | Mean LnP(K) | SD LnP(K) | Ln’(K) | |Ln”(K)| | Delta K |
|---|---|---|---|---|---|---|
| 1 | 3 | −12876.9 | 0.2 | - | - | - |
| 2 | 3 | −10938.2 | 2.2 | 1938.7 | 1334.5 | 603.3 |
| 3 | 3 | −10334.1 | 27.0 | 604.2 | 35.8 | 1.3 |
| 4 | 3 | −9765.7 | 3.6 | 568.3 | 301.7 | 83.3 |
| 5 | 3 | −9499.1 | 13.7 | 266.6 | 46.6 | 3.4 |
| 6 | 3 | −9279.1 | 62.9 | 220.0 | 26.9 | 0.4 |
| 7 | 3 | −9086.0 | 31.6 | 193.1 | 91.2 | 2.9 |
| 8 | 3 | −8984.0 | 63.5 | 102.0 | - | - |
| Particulars | Source of Variation | Df | SS | Variance | Percentage of Variation | |
|---|---|---|---|---|---|---|
| Country of origin | Among populations | 25 | 1164.52 | 46.58 | 1.93 | 0.19 |
| Within populations | 210 | 2817.14 | 13.41 | 5.26 | 0.52 | |
| Individuals within Populations | 236 | 684.00 | 2.90 | 2.90 | 0.29 | |
| Total | 471 | 4665.67 | 10.09 | 1.00 | ||
| Fixation Index (FST) | 0.19 | |||||
| Sub-species | Among populations | 3 | 493.07 | 164.36 | 3.55 | 0.28 |
| Within populations | 232 | 3491.13 | 15.05 | 6.12 | 0.49 | |
| Individuals within Populations | 236 | 664.00 | 2.81 | 2.81 | 0.23 | |
| Total | 471 | 4648.20 | 12.48 | 1.00 | ||
| Fixation Index (FST) | 0.28 | |||||
| Populations | Among populations | 3 | 478.25 | 159.42 | 1.30 | 0.13 |
| Within populations | 232 | 3505.95 | 15.11 | 6.15 | 0.60 | |
| Individuals within Populations | 236 | 664.00 | 2.81 | 2.81 | 0.27 | |
| Total | 471 | 4648.20 | 10.26 | 1.00 | ||
| Fixation Index (FST) | 0.127 | |||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Singh, V.J.; Bhowmick, P.K.; Vinod, K.K.; Krishnan, S.G.; Nandakumar, S.; Kumar, A.; Kumar, M.; Shekhawat, S.; Dixit, B.K.; Malik, A.; et al. Population Structure of a Worldwide Collection of Tropical Japonica Rice Indicates Limited Geographic Differentiation and Shows Promising Genetic Variability Associated with New Plant Type. Genes 2022, 13, 484. https://doi.org/10.3390/genes13030484
Singh VJ, Bhowmick PK, Vinod KK, Krishnan SG, Nandakumar S, Kumar A, Kumar M, Shekhawat S, Dixit BK, Malik A, et al. Population Structure of a Worldwide Collection of Tropical Japonica Rice Indicates Limited Geographic Differentiation and Shows Promising Genetic Variability Associated with New Plant Type. Genes. 2022; 13(3):484. https://doi.org/10.3390/genes13030484
Chicago/Turabian StyleSingh, Vikram Jeet, Prolay Kumar Bhowmick, Kunnummal Kurungara Vinod, Subbaiyan Gopala Krishnan, Shekharappa Nandakumar, Amit Kumar, Manoj Kumar, Sonu Shekhawat, Brijesh Kumar Dixit, Ankit Malik, and et al. 2022. "Population Structure of a Worldwide Collection of Tropical Japonica Rice Indicates Limited Geographic Differentiation and Shows Promising Genetic Variability Associated with New Plant Type" Genes 13, no. 3: 484. https://doi.org/10.3390/genes13030484
APA StyleSingh, V. J., Bhowmick, P. K., Vinod, K. K., Krishnan, S. G., Nandakumar, S., Kumar, A., Kumar, M., Shekhawat, S., Dixit, B. K., Malik, A., Ellur, R. K., Bollinedi, H., Nagarajan, M., & Singh, A. K. (2022). Population Structure of a Worldwide Collection of Tropical Japonica Rice Indicates Limited Geographic Differentiation and Shows Promising Genetic Variability Associated with New Plant Type. Genes, 13(3), 484. https://doi.org/10.3390/genes13030484