
Molecular Epidemiology of Mycobacterium tuberculosis Complex Strains in Urban and Slum Settings of Nairobi, Kenya


 , ,
, ,  ,
, 
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Design and Setting
2.2. Laboratory Procedures
2.2.1. Sputum Decontamination, Mycobacterial Culture, and DNA Extraction
2.2.2. Whole-Genome Sequencing
2.3. Data Analysis
2.3.1. Phylogenetic Reconstruction and WGS-Based Cluster Inference
2.3.2. Molecular Drug Resistance Prediction
2.3.3. Statistics
2.4. Ethical Considerations
3. Results
3.1. Study Population
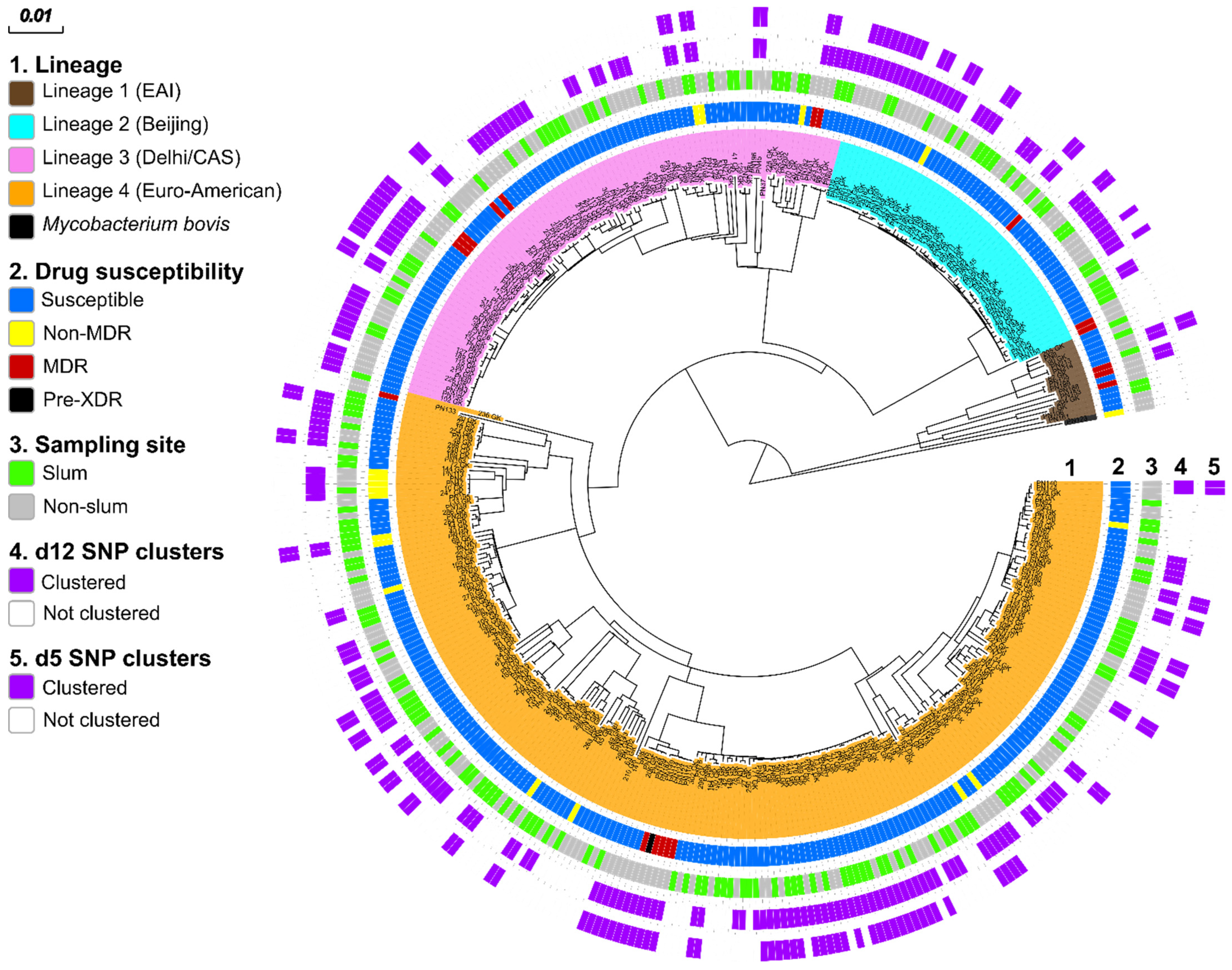
3.2. MTBC Population Structure
3.3. Genome-Based Drug Resistance Prediction
3.4. Genome-Based Cluster Analysis
3.5. Transmission of MDR MTBC Strains
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- WHO. Global Tuberculosis Report 2021; World Health Organization: Geneva, Switzerland, 2021; p. 232. [Google Scholar]
- Glaziou, P.; Sismanidis, C.; Floyd, K.; Raviglione, M. Global epidemiology of tuberculosis. Cold Spring Harb. Perspect. Med. 2014, 5, a017798. [Google Scholar] [CrossRef] [PubMed]
- Kipruto, H.; Mung’atu, J.; Ogila, K.; Adem, A.; Mwalili, S.; Masini, E.; Kibuchi, E. The epidemiology of tuberculosis in Kenya, a high TB HIV burden country (2000-2013). Int. J. Public Health Epidemiol. Res. 2015, 1, 002–013. [Google Scholar]
- Kwan, C.K.; Ernst, J.D. HIV and tuberculosis: A deadly human syndemic. Clin. Microbiol. Rev. 2011, 24, 351–376. [Google Scholar] [CrossRef] [PubMed]
- Campbell, P.J.; Morlock, G.P.; Sikes, R.D.; Dalton, T.L.; Metchock, B.; Starks, A.M.; Hooks, D.P.; Cowan, L.S.; Plikaytis, B.B.; Posey, J.E. Molecular detection of mutations associated with first- and second-line drug resistance compared with conventional drug susceptibility testing of Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2011, 55, 2032–2041. [Google Scholar] [CrossRef]
- Cilloni, L.; Fu, H.; Vesga, J.F.; Dowdy, D.; Pretorius, C.; Ahmedov, S.; Nair, S.A.; Mosneaga, A.; Masini, E.; Sahu, S.; et al. The potential impact of the COVID-19 pandemic on the tuberculosis epidemic a modelling analysis. EClinicalMedicine 2020, 28, 100603. [Google Scholar] [CrossRef]
- McQuaid, C.F.; Vassall, A.; Cohen, T.; Fiekert, K.; White, R.G. The impact of COVID-19 on TB: A review of the data. Int. J. Tuberc. Lung Dis. 2021, 25, 436–446. [Google Scholar] [CrossRef]
- Singhal, R.; Myneedu, V.P. Microscopy as a diagnostic tool in pulmonary tuberculosis. Int J. Mycobacteriol. 2015, 4, 1–6. [Google Scholar] [CrossRef]
- Shuaib, Y.A.; Khalil, E.A.G.; Schaible, U.E.; Wieler, L.H.; Bakheit, M.A.M.; Mohamed-Noor, S.E.; Abdalla, M.A.; Homolka, S.; Andres, S.; Hillemann, D.; et al. Smear Microscopy for Diagnosis of Pulmonary Tuberculosis in Eastern Sudan. Tuberc. Res. Treat. 2018, 2018, 8038137. [Google Scholar] [CrossRef]
- Merker, M.; Blin, C.; Mona, S.; Duforet-Frebourg, N.; Lecher, S.; Willery, E.; Blum, M.G.; Rusch-Gerdes, S.; Mokrousov, I.; Aleksic, E.; et al. Evolutionary history and global spread of the Mycobacterium tuberculosis Beijing lineage. Nat. Genet. 2015, 47, 242–249. [Google Scholar] [CrossRef]
- Merker, M.; Barbier, M.; Cox, H.; Rasigade, J.P.; Feuerriegel, S.; Kohl, T.A.; Diel, R.; Borrell, S.; Gagneux, S.; Nikolayevskyy, V.; et al. Compensatory evolution drives multidrug-resistant tuberculosis in Central Asia. Elife 2018, 7. [Google Scholar] [CrossRef]
- Shuaib, Y.A.; Khalil, E.A.G.; Wieler, L.H.; Schaible, U.E.; Bakheit, M.A.; Mohamed-Noor, S.E.; Abdalla, M.A.; Kerubo, G.; Andres, S.; Hillemann, D.; et al. Mycobacterium tuberculosis Complex Lineage 3 as Causative Agent of Pulmonary Tuberculosis, Eastern Sudan. Emerg. Infect. Dis. 2020, 26, 427–436. [Google Scholar] [CrossRef] [PubMed]
- Kompala, T.; Shenoi, S.V.; Friedland, G. Transmission of tuberculosis in resource-limited settings. Curr. HIV/AIDS Rep. 2013, 10, 264–272. [Google Scholar] [CrossRef] [PubMed]
- Enos, M.; Sitienei, J.; Ong’ang’o, J.; Mungai, B.; Kamene, M.; Wambugu, J.; Kipruto, H.; Manduku, V.; Mburu, J.; Nyaboke, D.; et al. Kenya tuberculosis prevalence survey 2016: Challenges and opportunities of ending TB in Kenya. PLoS ONE 2018, 13, e0209098. [Google Scholar] [CrossRef] [PubMed]
- Wiens, K.E.; Woyczynski, L.P.; Ledesma, J.R.; Ross, J.M.; Zenteno-Cuevas, R.; Goodridge, A.; Ullah, I.; Mathema, B.; Djoba Siawaya, J.F.; Biehl, M.H.; et al. Global variation in bacterial strains that cause tuberculosis disease: A systematic review and meta-analysis. BMC Med. 2018, 16, 196. [Google Scholar] [CrossRef]
- Middelkoop, K.; Mathema, B.; Myer, L.; Shashkina, E.; Whitelaw, A.; Kaplan, G.; Kreiswirth, B.; Wood, R.; Bekker, L.G. Transmission of tuberculosis in a South African community with a high prevalence of HIV infection. J. Infect. Dis. 2015, 211, 53–61. [Google Scholar] [CrossRef]
- Ganoza, C.A.; Ricaldi, J.N.; Chauca, J.; Rojas, G.; Munayco, C.; Agapito, J.; Palomino, J.C.; Guerra, H. Novel hypertonic saline-sodium hydroxide (HS-SH) method for decontamination and concentration of sputum samples for Mycobacterium tuberculosis microscopy and culture. J. Med. Microbiol. 2008, 57, 1094–1098. [Google Scholar] [CrossRef][Green Version]
- Basavaraj, A.; Chandanwale, A.; Patil, A.; Kadam, D.; Joshi, S.; Gupte, N.; McIntire, K.; Jain, D.; Dalal, H.; Badave, R.; et al. Tuberculosis Risk among Medical Trainees, Pune, India. Emerg. Infect. Dis. 2016, 22, 541–543. [Google Scholar] [CrossRef]
- GLI. Mycobacteriology Laboratory Manual; WHO: Geneva, Swizerland, 2014. [Google Scholar]
- Somerville, W.; Thibert, L.; Schwartzman, K.; Behr, M.A. Extraction of Mycobacterium tuberculosis DNA: A question of containment. J. Clin. Microbiol. 2005, 43, 2996–2997. [Google Scholar] [CrossRef]
- Baym, M.; Kryazhimskiy, S.; Lieberman, T.D.; Chung, H.; Desai, M.M.; Kishony, R. Inexpensive multiplexed library preparation for megabase-sized genomes. PLoS ONE 2015, 10, e0128036. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef]
- Kohl, T.A.; Utpatel, C.; Schleusener, V.; De Filippo, M.R.; Beckert, P.; Cirillo, D.M.; Niemann, S. MTBseq: A comprehensive pipeline for whole genome sequence analysis of Mycobacterium tuberculosis complex isolates. PeerJ 2018, 6, e5895. [Google Scholar] [CrossRef] [PubMed]
- Feuerriegel, S.; Schleusener, V.; Beckert, P.; Kohl, T.A.; Miotto, P.; Cirillo, D.M.; Cabibbe, A.M.; Niemann, S.; Fellenberg, K. PhyResSE: A Web Tool Delineating Mycobacterium tuberculosis Antibiotic Resistance and Lineage from Whole-Genome Sequencing Data. J. Clin. Microbiol. 2015, 53, 1908–1914. [Google Scholar] [CrossRef] [PubMed]
- Posada, D. jModelTest: Phylogenetic model averaging. Mol. Biol. Evol. 2008, 25, 1253–1256. [Google Scholar] [CrossRef] [PubMed]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2—approximately maximum-likelihood trees for large alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef]
- He, Z.; Zhang, H.; Gao, S.; Lercher, M.J.; Chen, W.H.; Hu, S. Evolview v2: An online visualization and management tool for customized and annotated phylogenetic trees. Nucleic Acids Res. 2016, 44, W236–W241. [Google Scholar] [CrossRef]
- Walker, T.M.; Ip, C.L.C.; Harrell, R.H.; Evans, J.T.; Kapatai, G.; Dedicoat, M.J.; Eyre, D.W.; Wilson, D.J.; Hawkey, P.M.; Crook, D.W.; et al. Whole-genome sequencing to delineate Mycobacterium tuberculosis outbreaks: A retrospective observational study. Lancet Infect. Dis. 2013, 13, 137–146. [Google Scholar] [CrossRef]
- Meehan, C.J.; Moris, P.; Kohl, T.A.; Pecerska, J.; Akter, S.; Merker, M.; Utpatel, C.; Beckert, P.; Gehre, F.; Lempens, P.; et al. The relationship between transmission time and clustering methods in Mycobacterium tuberculosis epidemiology. EBioMedicine 2018, 37, 410–416. [Google Scholar] [CrossRef]
- MARCH. DLTLD Guidelines on Management of Leprosy and Tuberculosis; Ministry of Public Health and Sanitation: Nairobi, Kenya, 2009; p. 108.
- Coll, F.; McNerney, R.; Guerra-Assuncao, J.A.; Glynn, J.R.; Perdigao, J.; Viveiros, M.; Portugal, I.; Pain, A.; Martin, N.; Clark, T.G. A robust SNP barcode for typing Mycobacterium tuberculosis complex strains. Nat. Commun. 2014, 5, 4812. [Google Scholar] [CrossRef]
- Niemann, S.; Merker, M.; Kohl, T.; Supply, P. Impact of Genetic Diversity on the Biology of Mycobacterium tuberculosis Complex Strains. Microbiol. Spectr. 2016, 4, 1–18. [Google Scholar] [CrossRef]
- Nuru, A.; Mamo, G.; Worku, A.; Admasu, A.; Medhin, G.; Pieper, R.; Ameni, G. Genetic Diversity of Mycobacterium tuberculosis Complex Isolated from Tuberculosis Patients in Bahir Dar City and Its Surroundings, Northwest Ethiopia. Biomed. Res. Int. 2015, 2015, 174732. [Google Scholar] [CrossRef]
- Mbugi, E.V.; Katale, B.Z.; Siame, K.K.; Keyyu, J.D.; Kendall, S.L.; Dockrell, H.M.; Streicher, E.M.; Michel, A.L.; Rweyemamu, M.M.; Warren, R.M.; et al. Genetic diversity of Mycobacterium tuberculosis isolated from tuberculosis patients in the Serengeti ecosystem in Tanzania. Tuberculosis 2015, 95, 170–178. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Asiimwe, B.B.; Ghebremichael, S.; Kallenius, G.; Koivula, T.; Joloba, M.L. Mycobacterium tuberculosis spoligotypes and drug susceptibility pattern of isolates from tuberculosis patients in peri-urban Kampala, Uganda. BMC Infect. Dis. 2008, 8, 101. [Google Scholar] [CrossRef] [PubMed]
- Ejo, M.; Torrea, G.; Uwizeye, C.; Kassa, M.; Girma, Y.; Bekele, T.; Ademe, Y.; Diro, E.; Gehre, F.; Rigouts, L.; et al. Genetic diversity of the Mycobacterium tuberculosis complex s.strains from newly diagnosed tuberculosis patients in Northwest Ethiopia reveals a predominance of East-African-Indian and Euro-American lineages. Int. J. Infect. Dis. 2021, 103, 72–80. [Google Scholar] [CrossRef] [PubMed]
- Stucki, D.; Brites, D.; Jeljeli, L.; Coscolla, M.; Liu, Q.; Trauner, A.; Fenner, L.; Rutaihwa, L.; Borrell, S.; Luo, T.; et al. Mycobacterium tuberculosis lineage 4 comprises globally distributed and geographically restricted sublineages. Nat. Genet. 2016, 48, 1535–1543. [Google Scholar] [CrossRef]
- Bazira, J.; Asiimwe, B.B.; Joloba, M.L.; Bwanga, F.; Matee, M.I. Mycobacterium tuberculosis spoligotypes and drug susceptibility pattern of isolates from tuberculosis patients in South-Western Uganda. BMC Infect. Dis. 2011, 11, 81. [Google Scholar] [CrossRef]
- Githui, W.A.; Jordaan, A.M.; Juma, E.S.; Kinyanjui, P.; Karimi, F.G.; Kimwomi, J.; Meme, H.; Mumbi, P.; Streicher, E.M.; Warren, R.; et al. Identification of MDR-TB BeijingW and other Mycobacterium tuberculosis genotypes in Nairobi, Kenya. Int J. Tuberc. Lung Dis. 2004, 8, 352–360. [Google Scholar]
- Yimer, S.A.; Norheim, G.; Namouchi, A.; Zegeye, E.D.; Kinander, W.; Tonjum, T.; Bekele, S.; Mannsaker, T.; Bjune, G.; Aseffa, A.; et al. Mycobacterium tuberculosis lineage 7 strains are associated with prolonged patient delay in seeking treatment for pulmonary tuberculosis in Amhara Region, Ethiopia. J. Clin. Microbiol. 2015, 53, 1301–1309. [Google Scholar] [CrossRef]
- Rutaihwa, L.K.; Menardo, F.; Stucki, D.; Gygli, S.M.; Ley, S.D.; Malla, B.; Feldmann, J.; Borrell, S.; Beisel, C.; Middelkoop, K.; et al. Multiple Introductions of Mycobacterium tuberculosis Lineage 2–Beijing Into Africa Over Centuries. Front. Ecol. Evol. 2019, 7, 112. [Google Scholar] [CrossRef]
- Mekonnen, A.; Merker, M.; Collins, J.M.; Addise, D.; Aseffa, A.; Petros, B.; Ameni, G.; Niemann, S. Molecular epidemiology and drug resistance patterns of Mycobacterium tuberculosis complex isolates from university students and the local community in Eastern Ethiopia. PLoS ONE 2018, 13, e0198054. [Google Scholar] [CrossRef]
- Cain, K.P.; Marano, N.; Kamene, M.; Sitienei, J.; Mukherjee, S.; Galev, A.; Burton, J.; Nasibov, O.; Kioko, J.; De Cock, K.M. The movement of multidrug-resistant tuberculosis across borders in East Africa needs a regional and global solution. PLoS Med. 2015, 12, e1001791. [Google Scholar] [CrossRef]
- Ogaro, T.D.; Githui, W.; Kikuvi, G.; Okari, J.; Wangui, E.; Asiko, V. Anti-tuberculosis drug resistance in Nairobi, Kenya. African J. Health Sci. 2012, 20, 21–27. [Google Scholar]
- Obonyo, L.; Amukoye, E.; Zipporah, P. First line anti-tuberculosis drug resistance among human immunodeficiency virus infected patients attending Maryland Comprehensive Care Centre, Mathare 4a, Nairobi, Kenya. Int. J. Sci. Basic Appl. Res. 2014, 15, 661–668. [Google Scholar]
- Kateregga, N.; Atuheire, C.; Ntambi, S.; Ocaka, D.; Ndukui, G.J.; Wampande, E. Prevalence of multidrug resistant Mycobacterium tuberculosis and risk factors among youth attending MDR-TB unit in Mulago hospital. J. Bacteriol. Mycol. 2018, 5, 1088. [Google Scholar]
- Hoza, A.S.; Mfinanga, S.G.M.; Konig, B. Anti-TB drug resistance in Tanga, Tanzania: A cross sectional facility-base prevalence among pulmonary TB patients. Asian Pac. J. Trop. Med. 2015, 8, 907–913. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Variable | No. | Cohort A | Cohort B | X2 | p-Value |
|---|---|---|---|---|---|
| Lineage | 7.70 | 0.103 | |||
| L1 (EAI) | 14 | 9 (5.0%) | 5 (2.5%) | ||
| L2 (Beijing) | 56 | 30 (16.6%) | 26 (12.7%) | ||
| L3 (Delhi/CAS) | 99 | 52 (28.7%) | 47 (23.0%) | ||
| L4 (Euro-American) | 215 | 89 (49.2%) | 126 (61.8%) | ||
| M. bovis | 1 | 1 (0.5%) | 0 (0.00%) | ||
| DST | 25.3 | <0.001 a | |||
| Susceptible | 348 | 151 (83.4%) | 197 (96.6%) | ||
| MDR | 20 | 20 (11.1%) | 0 (0.00%) | ||
| Non-MDR | 17 | 10 (5.5%) | 7 (3.4%) | ||
| d12 | 3.08 | 0.079 | |||
| Clustered | 218 | 111 (61.3%) | 107 (52.5%) | ||
| Not clustered | 167 | 70 (38.7%) | 97 (47.5%) | ||
| d5 | 14.2 | <0.001 a | |||
| Clustered | 117 | 72 (39.8%) | 45 (22.1%) | ||
| Not clustered | 268 | 109 (60.2%) | 159 (77.9%) | ||
| TB history | 0.11 | 0.747 | |||
| New | 326 | 152 (84.0%) | 174 (85.3%) | ||
| Retreatment | 56 | 28 (15.5%) | 28 (15.5%) | ||
| NA * | 3 | 1 (0.5%) | 2 (1.0%) | ||
| Age group | 0.11 | 0.917 | |||
| ≤30 | 166 | 63 (34.8%) | 103 (50.5%) | ||
| >30 | 161 | 62 (34.3%) | 99 (48.5%) | ||
| NA * | 58 | 56 (30.9%) | 2 (1.0%) | ||
| Sex | 4.97 | 0.026 a | |||
| Male | 246 | 105 (58.0%) | 141 (69.1%) | ||
| Female | 136 | 75 (41.4%) | 61 (29.9%) | ||
| NA * | 3 | 1 (0.6%) | 2 (1.0%) | ||
| Sampling area | 178.2 | <0.001 a | |||
| Urban | 215 | 166 (91.7%) | 49 (24.0%) | ||
| Slum | 170 | 15 (8.3%) | 155 (76.0%) | ||
| Total | 385 | 181 (47.0%) | 204 (53.0%) | ─ | ─ |
| Drug | Gene | Mutation | Codon Change | Total |
|---|---|---|---|---|
| INH | katG | Ser315Thr | agc/aCc | 21 |
| Ser315Arg | agc/aGa | 2 | ||
| fabG1-inhA | −15 c/t | C→T | 8 | |
| inhA | Leu203Leu | ctg/ctA | 2 | |
| Ile194Thr | atc/aCc | 2 | ||
| RIF | rpoB | Gln432Pro | Caa/cCa | 2 |
| His 445Tyr | Cac/Tac | 5 | ||
| His 445Arg | Cac/cGc | 1 | ||
| His445Asp | Cac/Gac | 1 | ||
| Ser450Leu | Tcg/tTg | 11 | ||
| Leu452Pro | Ctg/cCg | 1 | ||
| STR | rpsL | Lys43Arg | Aag/aGg | 9 |
| gidB | Ala138Val | Gcg/gTg | 1 | |
| rrs | 513 a/c | A→C | 3 | |
| EMB | embB | Met306Ile | Atg/atA | 7 |
| Met306Val | Atg/Gtg | 2 | ||
| Asp328Gly | Gat/gGt | 1 | ||
| Asp354Ala | Gac/Aac | 1 | ||
| PZA | pncA | Gln10Pro | cag/cCg | 1 |
| His57Asp | cac/Gac | 1 | ||
| Asp63Gly | gac/gGc | 2 | ||
| Lsy96Thr | aag/aCg | 6 | ||
| Thr135Pro | acc/Ccc | 1 | ||
| Gln141_ | cag/tag | 1 | ||
| FQs | gyrA | Asp94Gly | gac/Cac | 1 |
| Variables | No. | No. of Wild Type | No. of Resistant (%) | OR (95% CI) | p-Value |
|---|---|---|---|---|---|
| Gender | |||||
| Male | 246 | 226 | 20 (8.10) | Ref | |
| Female | 136 | 120 | 16 (11.8) | 1.5 (0.75–3.01) | 0.247 |
| NA * | 3 | 2 | 1 (33.3) | ─ | ─ |
| Age | |||||
| ≤30 | 166 | 148 | 18 (10.8) | Ref | |
| >30 | 161 | 149 | 12 (5.70) | 1.5 (0.70–3.24) | 0.291 |
| NA * | 58 | 51 | 7 (12.1) | ─ | ─ |
| Study area | |||||
| Slum | 170 | 163 | 7 (4.1) | Ref | |
| Urban | 215 | 185 | 30 (14.0) | 3.8 (1.62–8.83) | 0.002 |
| TB history | |||||
| New | 326 | 296 | 30 (9.2) | Ref | |
| Retreatment | 56 | 50 | 6 (10.7) | 1.2 (0.47–2.99) | 0.721 |
| NA * | 3 | 2 | 1 (33.3) | ─ | ─ |
| Lineage | |||||
| L2 (Beijing) | 56 | 52 | 4 (7.1) | Ref | |
| L1 (EAI) | 14 | 11 | 3 (21.4) | 3.5 (0.69–18.1) | 0.129 |
| L3 (Delhi/CAS) | 99 | 89 | 10 (10.1) | 1.5 (0.44–4.89) | 0.539 |
| L4 (Euro-American) | 215 | 196 | 19 (8.8) | 1.3 (0.41–3.86) | 0.868 |
| M. bovis * | 1 | 0 | 1 (100) | ─ | ─ |
| Total | 385 | 348 | 37 (9.6) | ─ | ─ |
| Variable | No. | d12 | d5 | ||||||
|---|---|---|---|---|---|---|---|---|---|
| NC | C (%) | OR (95% CI) | p-Value | NC | C (%) | OR (95% CI) | p-Value | ||
| Lineage | |||||||||
| L3 (Delhi/CAS) | 99 | 45 | 54 (54.5%) | Ref | 78 | 21 (21.2%) | Ref | ||
| L2 (Beijing) | 56 | 19 | 37 (66.1%) | 1.6 (0.82–3.20) | 0.163 | 36 | 20 (35.7%) | 2.1 (1.00–4.28 | 0.051 |
| L4 (Euro-American) | 215 | 92 | 123 (57.2%) | 1.1 (0.69–1.80) | 0.658 | 141 | 74 (34.4%) | 1.9 (1.12–3.41) | 0.019 |
| L1 (EAI) * | 14 | 10 | 4 (28.6%) | ─ | ─ | 12 | 2 (14.3%) | ─ | ─ |
| M. bovis * | 1 | 0 | 0 (0.00%) | ─ | ─ | 0 | 0 (0.00%) | ─ | ─ |
| Total | 385 | 166 | 218 (56.6) | ─ | ─ | 267 | 117 (30.5) | ─ | ─ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kerubo, G.; Ndungu, P.; Shuaib, Y.A.; Amukoye, E.; Revathi, G.; Homolka, S.; Kariuki, S.; Merker, M.; Niemann, S. Molecular Epidemiology of Mycobacterium tuberculosis Complex Strains in Urban and Slum Settings of Nairobi, Kenya. Genes 2022, 13, 475. https://doi.org/10.3390/genes13030475
Kerubo G, Ndungu P, Shuaib YA, Amukoye E, Revathi G, Homolka S, Kariuki S, Merker M, Niemann S. Molecular Epidemiology of Mycobacterium tuberculosis Complex Strains in Urban and Slum Settings of Nairobi, Kenya. Genes. 2022; 13(3):475. https://doi.org/10.3390/genes13030475
Chicago/Turabian StyleKerubo, Glennah, Perpetua Ndungu, Yassir Adam Shuaib, Evans Amukoye, Gunturu Revathi, Susanne Homolka, Samuel Kariuki, Matthias Merker, and Stefan Niemann. 2022. "Molecular Epidemiology of Mycobacterium tuberculosis Complex Strains in Urban and Slum Settings of Nairobi, Kenya" Genes 13, no. 3: 475. https://doi.org/10.3390/genes13030475
APA StyleKerubo, G., Ndungu, P., Shuaib, Y. A., Amukoye, E., Revathi, G., Homolka, S., Kariuki, S., Merker, M., & Niemann, S. (2022). Molecular Epidemiology of Mycobacterium tuberculosis Complex Strains in Urban and Slum Settings of Nairobi, Kenya. Genes, 13(3), 475. https://doi.org/10.3390/genes13030475