
Complex Autism Spectrum Disorder with Epilepsy, Strabismus and Self-Injurious Behaviors in a Patient with a De Novo Heterozygous POLR2A Variant

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patient Recruitment
2.2. Psychometric Analysis of ASD
2.3. Whole Genome Sequencing Pipeline and Variant Analysis
2.4. Dynamic Molecular Simulations
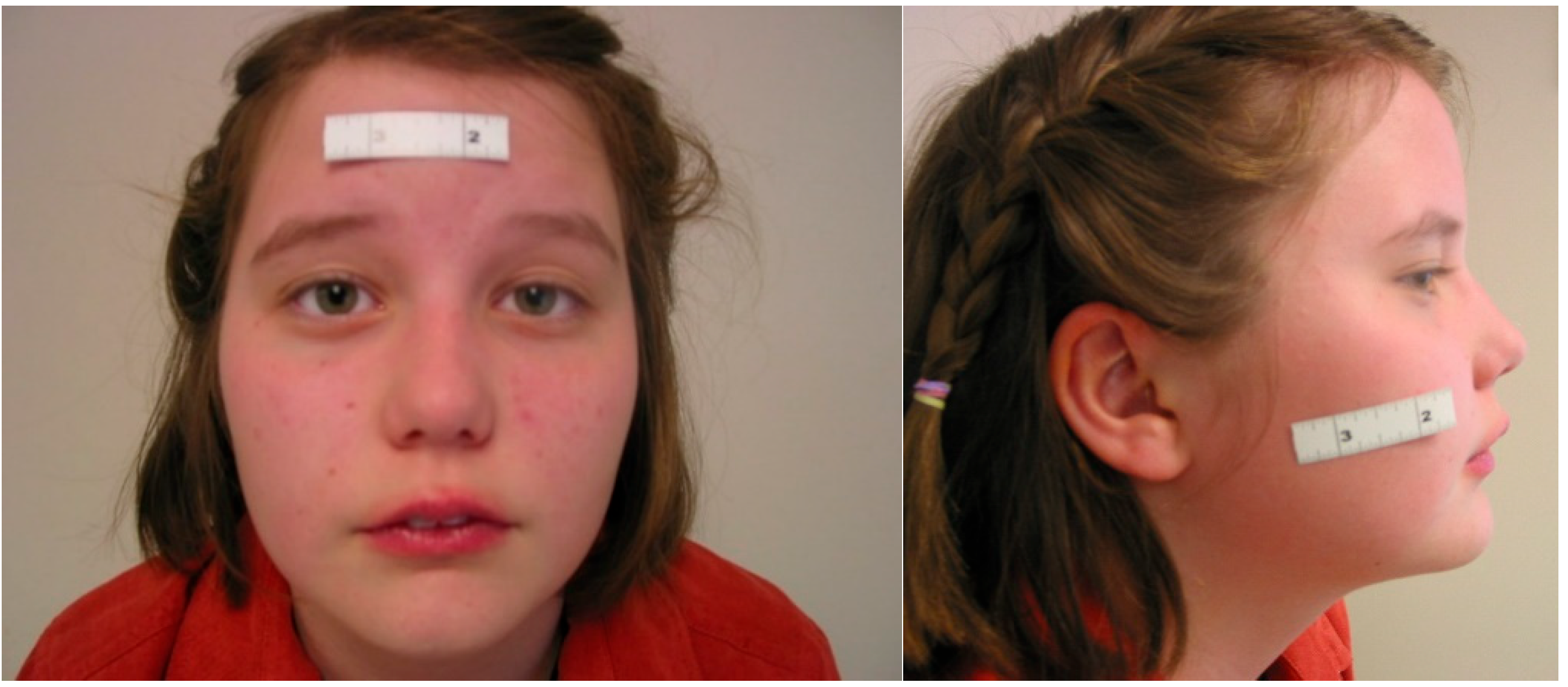
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, 5th ed.; American Psychiatric Association: Arlington, VA, USA, 2013. [Google Scholar] [CrossRef]
- Public Health Agency of Canada. Autism Spectrum Disorder among Children and Youth in Canada 2018. 29 March 2018. Available online: https://www.canada.ca/en/public-health/services/publications/diseases-conditions/autism-spectrum-disorder-children-youth-canada-2018.html (accessed on 24 May 2021).
- Tick, B.; Bolton, P.; Happé, F.; Rutter, M.; Rijsdijk, F. Heritability of autism spectrum disorders: A meta-analysis of twin studies. J. Child Psychol. Psychiatry 2016, 57, 585–595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilbert, J.; Man, H.-Y. Fundamental Elements in Autism: From Neurogenesis and Neurite Growth to Synaptic Plasticity. Front. Cell. Neurosci. 2017, 11, 359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guang, S.; Pang, N.; Deng, X.; Yang, L.; He, F.; Wu, L.; Chen, C.; Yin, F.; Peng, J. Synaptopathology Involved in Autism Spectrum Disorder. Front. Cell. Neurosci. 2018, 12, 470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rylaarsdam, L.E.; Guemez-Gamboa, A. Genetic Causes and Modifiers of Autism Spectrum Disorder. Front. Cell. Neurosci. 2019, 13, 385. [Google Scholar] [CrossRef]
- Sestan, N.; State, M.W. Lost in Translation: Traversing the Complex Path from Genomics to Therapeutics in Autism Spectrum Disorder. Neuron 2018, 100, 406–423. [Google Scholar] [CrossRef] [Green Version]
- Plummer, J.T.; Gordon, A.J.; Levitt, P. The Genetic Intersection of Neurodevelopmental Disorders and Shared Medical Comorbidities–Relations that Translate from Bench to Bedside. Front. Psychiatry 2016, 7, 142. [Google Scholar] [CrossRef] [Green Version]
- Ayhan, F.; Konopka, G. Regulatory genes and pathways disrupted in autism spectrum disorders. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2019, 89, 57–64. [Google Scholar] [CrossRef]
- Gazestani, V.H.; Pramparo, T.; Nalabolu, S.; Kellman, B.P.; Murray, S.; Lopez, L.; Pierce, K.; Courchesne, E.; Lewis, N.E. Transcriptional organization of autism spectrum disorder and its connection to ASD risk genes and phenotypic variation. bioRxiv 2018, 435917. [Google Scholar] [CrossRef]
- Hahn, S. Structure and mechanism of the RNA polymerase II transcription machinery. Nat. Struct. Mol. Biol. 2004, 11, 394–403. [Google Scholar] [CrossRef] [Green Version]
- Liou, S.-H.; Singh, S.K.; Singer, R.H.; Coleman, R.A.; Liu, W.-L. Structure of the p53/RNA polymerase II assembly. Commun. Biol. 2021, 4, 1–12. [Google Scholar] [CrossRef]
- Kaplan, C.D.; Jin, H.; Zhang, I.L.; Belyanin, A. Dissection of Pol II Trigger Loop Function and Pol II Activity–Dependent Control of Start Site Selection In Vivo. PLoS Genet. 2012, 8, e1002627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, D.; Bushnell, D.A.; Westover, K.; Kaplan, C.; Kornberg, R.D. Structural Basis of Transcription: Role of the Trigger Loop in Substrate Specificity and Catalysis. Cell 2006, 127, 941–954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haijes, H.; Koster, M.J.; Rehmann, H.; Li, D.; Hakonarson, H.; Cappuccio, G.; Hancarova, M.; Lehalle, D.; Reardon, W.; Schaefer, G.B.; et al. De Novo Heterozygous POLR2A Variants Cause a Neurodevelopmental Syndrome with Profound Infantile-Onset Hypotonia. Am. J. Hum. Genet. 2019, 105, 283–301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hansen, A.W.; Arora, P.; Khayat, M.M.; Smith, L.J.; Lewis, A.M.; Rossetti, L.Z.; Jayaseelan, J.; Cristian, I.; Haynes, D.; DiTroia, S.; et al. Germline mutation in POLR2A: A heterogeneous, multi-systemic developmental disorder characterized by transcriptional dysregulation. Hum. Genet. Genom. Adv. 2021, 2, 100014. [Google Scholar] [CrossRef] [PubMed]
- Callaghan, D.B.; Rogic, S.; Tan, P.P.C.; Calli, K.; Qiao, Y.; Baldwin, R.; Jacobson, M.; Belmadani, M.; Holmes, N.; Yu, C.; et al. Whole genome sequencing and variant discovery in the ASPIRE autism spectrum disorder cohort. Clin. Genet. 2019, 96, 199–206. [Google Scholar] [CrossRef]
- Dhaliwal, J.; Qiao, Y.; Calli, K.; Martell, S.; Race, S.; Chijiwa, C.; Glodjo, A.; Jones, S.; Rajcan-Separovic, E.; Scherer, S.; et al. Contribution of Multiple Inherited Variants to Autism Spectrum Disorder (ASD) in a Family with 3 Affected Siblings. Genes 2021, 12, 1053. [Google Scholar] [CrossRef]
- Ng, P.C.; Henikoff, S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003, 31, 3812–3814. [Google Scholar] [CrossRef] [Green Version]
- Adzhubei, I.; Jordan, D.M.; Sunyaev, S.R. Predicting Functional Effect of Human Missense Mutations Using PolyPhen-2. Curr. Protoc. Hum. Genet 2013, 7. [Google Scholar] [CrossRef] [Green Version]
- Steinhaus, R.; Proft, S.; Schuelke, M.; Cooper, D.N.; Schwarz, J.M.; Seelow, D. MutationTaster2021. Nucleic Acids Res. 2021, 49, W446–W451. [Google Scholar] [CrossRef]
- Rogers, M.F.; Shihab, H.A.; Mort, M.E.; Cooper, D.N.; Gaunt, T.; Campbell, C. FATHMM-XF: Accurate prediction of pathogenic point mutations via extended features. Bioinformatics 2018, 34, 511–513. [Google Scholar] [CrossRef] [Green Version]
- Rentzsch, P.; Witten, D.; Cooper, G.M.; Shendure, J.; Kircher, M. CADD: Predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2019, 47, D886–D894. [Google Scholar] [CrossRef] [PubMed]
- Davydov, E.V.; Goode, D.; Sirota, M.; Cooper, G.M.; Sidow, A.; Batzoglou, S. Identifying a High Fraction of the Human Genome to be under Selective Constraint Using GERP++. PLoS Comput. Biol. 2010, 6, e1001025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grantham, R. Amino Acid Difference Formula to Help Explain Protein Evolution. Science 1974, 185, 862–864. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Schrodinger. The PyMOL Molecular Graphics System, Version 2.5.2. November 2015. Available online: https://pymol.org/2/ (accessed on 22 November 2015).
- Kuriata, A.; Gierut, A.M.; Oleniecki, T.; Ciemny, M.P.; Kolinski, A.; Kurcinski, M.; Kmiecik, S. CABS-flex 2.0: A web server for fast simulations of flexibility of protein structures. Nucleic Acids Res. 2018, 46, W338–W343. [Google Scholar] [CrossRef] [Green Version]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alfoldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef]
- Accogli, A.; Geraldo, A.F.; Piccolo, G.; Riva, A.; Scala, M.; Balagura, G.; Salpietro, V.; Madia, F.; Maghnie, M.; Zara, F.; et al. Diagnostic Approach to Macrocephaly in Children. Front. Pediatr. 2022, 9, 794069. [Google Scholar] [CrossRef]

{kind=link}
| Tool | Score | Prediction |
|---|---|---|
| SIFT | 0.01 | Deleterious |
| Polyphen-2 (HDIV) | 0.998 | Probably damaging |
| MutationTaster | 0.999889 | Damaging |
| FATHMM | −1.09 | Tolerated |
| MutationAssessor | 0.94726 | Functional |
| CADD PHRED | 29.6 | Deleterious |
| GERP ++ RS | 5.93 | Conserved |
| Grantham | 64 | Similar |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Evans, D.R.; Qiao, Y.; Trost, B.; Calli, K.; Martell, S.; Jones, S.J.M.; Scherer, S.W.; Lewis, M.E.S. Complex Autism Spectrum Disorder with Epilepsy, Strabismus and Self-Injurious Behaviors in a Patient with a De Novo Heterozygous POLR2A Variant. Genes 2022, 13, 470. https://doi.org/10.3390/genes13030470
Evans DR, Qiao Y, Trost B, Calli K, Martell S, Jones SJM, Scherer SW, Lewis MES. Complex Autism Spectrum Disorder with Epilepsy, Strabismus and Self-Injurious Behaviors in a Patient with a De Novo Heterozygous POLR2A Variant. Genes. 2022; 13(3):470. https://doi.org/10.3390/genes13030470
Chicago/Turabian StyleEvans, Daniel R., Ying Qiao, Brett Trost, Kristina Calli, Sally Martell, Steven J. M. Jones, Stephen W. Scherer, and M. E. Suzanne Lewis. 2022. "Complex Autism Spectrum Disorder with Epilepsy, Strabismus and Self-Injurious Behaviors in a Patient with a De Novo Heterozygous POLR2A Variant" Genes 13, no. 3: 470. https://doi.org/10.3390/genes13030470
APA StyleEvans, D. R., Qiao, Y., Trost, B., Calli, K., Martell, S., Jones, S. J. M., Scherer, S. W., & Lewis, M. E. S. (2022). Complex Autism Spectrum Disorder with Epilepsy, Strabismus and Self-Injurious Behaviors in a Patient with a De Novo Heterozygous POLR2A Variant. Genes, 13(3), 470. https://doi.org/10.3390/genes13030470