2.2. Spatial and Temporal Expression during Neurodevelopment
Comprehensive transcriptional analyses using RT-PCR by Noor et al. revealed that the
PTCHD1 full-length (isoform a) transcript exhibits varying levels of expression in the human brain, as well as in numerous peripheral tissues, with the highest levels of transcription being evident in the cerebellum. A subsequent northern blot analysis revealed that, in addition to the cerebellum, the
PTCHD1 transcript was also detected in all of the four major lobes of the brain. The authors then employed qRT-PCR to compare the expression of
PTCHD1 in multiple brain subregions, confirming the highest relative levels of transcription in both the cerebellum and the pituitary gland. Noor et al. also used RT-PCR to characterize the expression of
PTCHD1-AS1 and
PTCHD1-AS2 and reported detectable levels of both transcripts in the cerebellum, parietal and occipital lobes, spinal cord, and fibroblasts [
11].
Studies employing RNA in situ hybridization and fluorescent in situ hybridization (FISH) characterized the spatial expression profile of
Ptchd1 in mice during embryogenesis and postnatally. Noor et al. first reported extensive expression of the
Ptchd1 transcript throughout the developing brain during embryogenesis (E9 and E14) through RNA in situ hybridization [
11]. Furthermore, at birth (P0),
Ptchd1 mRNA appears to be primarily restricted to the thalamic reticular nucleus (TRN). Beginning in adolescence (P15), however,
Ptchd1 expression becomes detectable in the cortex, striatum, cerebellum, and dentate gyrus of the hippocampus [
31]. Consistent with these reports, Tora et al. confirmed high relative levels of
Ptchd1 expression in both the TRN and the cerebellum at P12, as well as in the dentate gyrus at P21. Interestingly,
Ptchd1 transcription in the dentate gyrus at P21 appears to be almost entirely restricted to dentate granule cells, with very low expression observed in the pyramidal cells in other hippocampal subfields (CA1–3). Furthermore, considerable
Ptchd1 expression was observed in the dentate gyrus of adult (P60) mice [
32]. In addition to the dentate gyrus and TRN,
Ptchd1 exhibits strong expression in the anterodorsal subdivision (AD) of the thalamus in adult mice [
13].
Tora et al. subsequently used qRT-PCR in order to quantitatively profile
Ptchd1 expression throughout early postnatal development and into adulthood (P5–P60) in these relevant brain subregions. These analyses revealed that
Ptchd1 expression declined in the thalamus after P5 and, conversely, increased in the cerebellum after P15; no postnatal changes in expression were observed in the cortex, hippocampus, or striatum [
32]. Ung et al. also report variable embryonic and postnatal expression of
Ptchd1 between numerous additional brain subregions, such as the midbrain, pons, medulla, olfactory bulb, and hypothalamus, as well as dynamic expression within these subregions between the embryonic (E13–E18) and postnatal (P7 and P35) ages that were assessed [
33].
2.5. Functional Characterization of PTCHD1
Studying the function of the PTCHD1 protein will enable a better understanding of the biological pathways leading to ASD and ID, as well as providing the possibility of more accurate clinical prediction for PTCHD1 sequence variants, and in particular for missense variants, and the first steps towards considering therapeutic strategies.
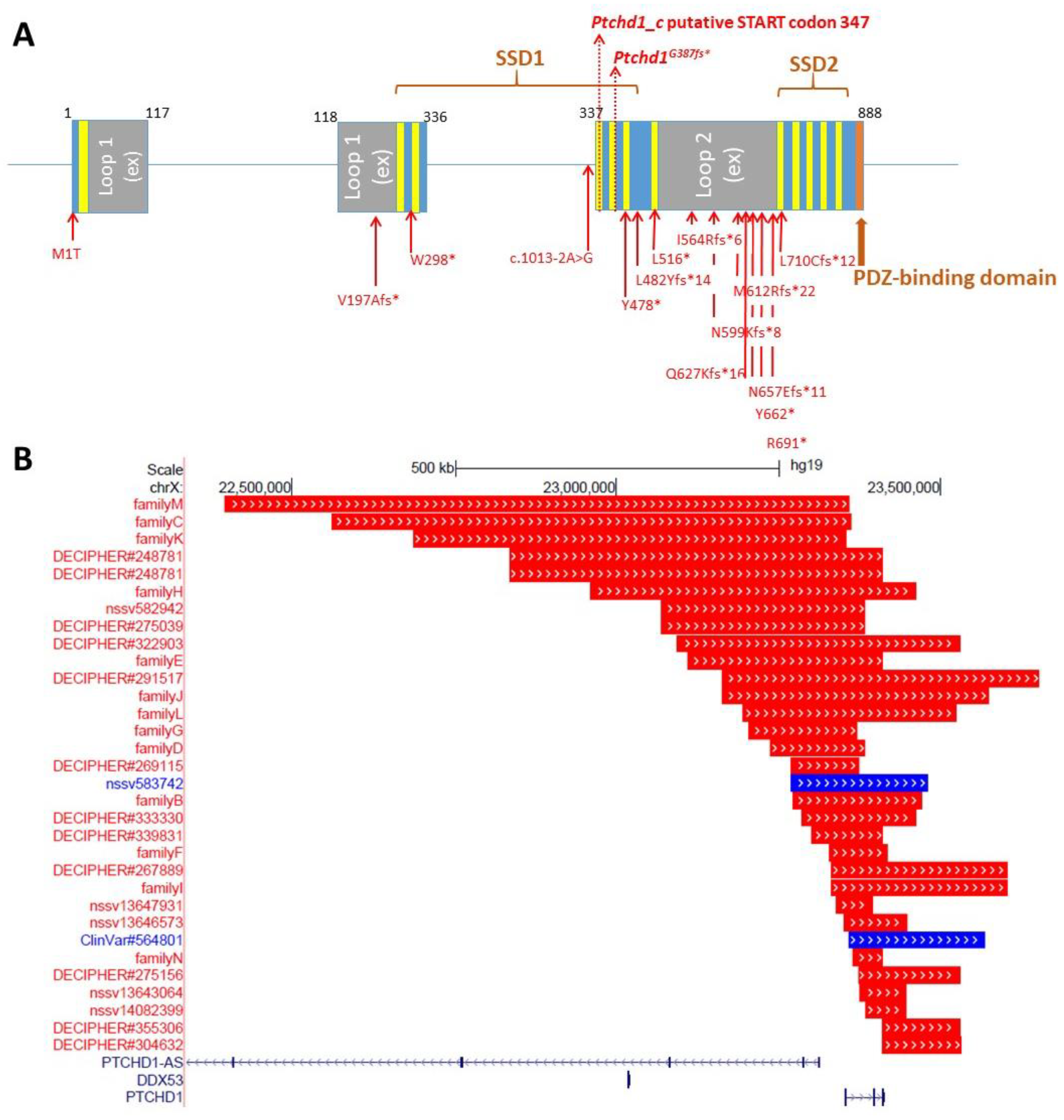
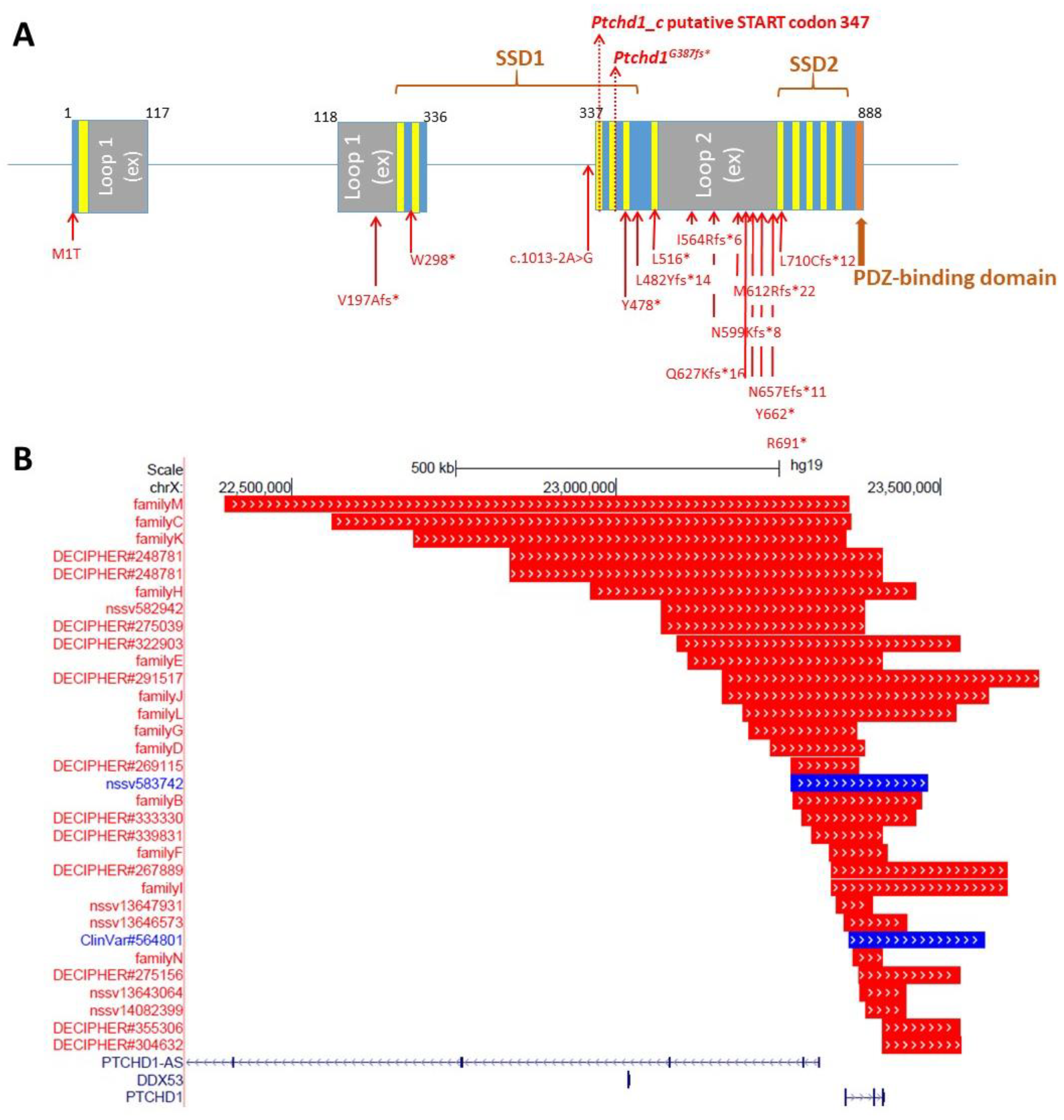
Preliminary in silico analyses indicate that PTCHD1 is predicted to be a transmembrane protein that encodes the 12 transmembrane helices that form two modules, similar to the sterol-sensing domains in NPC-1, as well as a C-terminal PDZ-binding motif encoded by the final four amino acids (Ile-Thr-Thr-Val, or ITTV; see
Figure 3). In order to acquire an initial understanding of the function of PTCHD1, Noor et al. first sought to study its subcellular localization. The authors reported that a C-terminal PTCHD1-GFP fusion protein primarily exhibited localization to the plasma membrane in both COS7 and SK-N-SH (human neuroblastoma) cells in vitro [
11]. Ung et al. characterized the subcellular localization of Ptchd1 in neuronal cells by fusing GFP to either its N-terminus (GFP-Ptchd1) or C-terminus (Ptchd1-GFP) and then transiently expressing these fusion proteins in primary hippocampal neuronal cultures ex vivo. These analyses indicated that this exogenously expressed Ptchd1-GFP exhibits a distinct pattern of membrane localization within dendritic spines, which was further corroborated by co-labelling experiments with the post-synaptic density protein Psd95. In addition, the authors report that a portion of the intracellular C-terminal tail encoded by amino acids 850–873 appears to be essential for dendritic and synaptic targeting. Interestingly, GFP-Ptchd1 displayed ubiquitous fluorescence throughout the neuronal cells, suggesting that post-translational processing of the N-terminus of Ptchd1 is necessary for appropriate subcellular localization [
33].
Although many
PTCHD1 missense variants have been reported (e.g., Noor et al.) or listed in ClinVar, and even though many of these are exceedingly rare variants, it is not currently possible to assign consequential pathogenicity to them. In order to enable more accurate clinical diagnoses for
PTCHD1 missense variants, it is important to establish empirical methods (rather than in silico predictions) that can identify a link between the variants and PTCHD1 function and thus etiopathological role. With this in mind, Halewa et al. sought to determine the likely pathogenicity of a number of reported
PTCHD1 missense variants by assessing both their protein stability and their plasma membrane localization in vitro. In order to evaluate protein stability, wildtype and missense PTCHD1-GFP fusion protein constructs were transiently overexpressed in HEK293T cells. Subsequent western blotting revealed substantial decreases in the protein expression of the missense variants Pro32Arg, Pro32Leu, Lys181Thr, Tyr213Cys, Gly300Arg, and Ala310Pro, likely indicating conformational instability and resultant degradation. Consistent with these findings, transient overexpression in both HEK293T and Neuro-2a cells, followed by immunostaining, demonstrated that the same six missense variants displayed weak localization at the plasma membrane. Interestingly, these mutations are all clustered within either the extracellular loop (Lys181Thr and Tyr213Cys) or one of the transmembrane domains (Pro32Arg, Pro32Leu, Gly300Arg, and Ala310Pro), implying that mutations in these locations are particularly deleterious and are likely to be retained by the endoplasmic reticulum and targeted for subsequent proteasomal degradation [
16]. In a similar vein, Xie et al. conducted cycloheximide chase assays following the transient overexpression of GFP-PTCHD1 in HEK293T cells and concluded that wild type PTCHD1 exhibits a half-life of greater than 12 h. This assay also inferred similar levels of protein stability for the Pro75Gln, Lys181Thr, Gly303Arg, and Tyr802Cys missense variants as well as the PDZ-binding domain deleting mutation I885*, in comparison with the marked instability of Pro32Arg and Phe549Cys. In addition, Xie et al. stably expressed GFP-PTCHD1 missense variants in HEK293T cells, and despite modest 5–10% attenuations in the mRNA expression of the Pro32Arg, Gly303Arg, and Phe549Cys transcripts, 40–80% decreases in their basal protein expression were observed, which shows further consistency with significant increased instability. However, in contrast with Halewa et al., Xie et al. were able to detect Lys181Thr, both basally and following transient overexpression [
37]. A summary of the functional studies through missense variants is provided in
Table 1.
Table 1.
Summary of functional analysis of missense PTCHD1 variants from Xie et al. [
37] and Halewa et al. [
16].
Table 1.
Summary of functional analysis of missense PTCHD1 variants from Xie et al. [
37] and Halewa et al. [
16].
| | | Xie et al. | Halewa et al. |
|---|
| PTCHD1 Variant | Minor Allele Frequency (gnomAD)/No. Hemizygotes | Source (ID) | Inheritance | Post-Translational Defect | Protein Stability Reduced | Protein Localization Affected | Source | Inheritance | Protein Stability Reduced | Impaired Plasma Membrane Localization |
|---|
| P32R | 0/0 | DECIPHER: 284363 | Mat | + | + | - | Lille | Mat | + | + |
| P32L | 0/0 | | | | | | Lyon | Mat | + | + |
| S51N | 0/0 | | | | | | Torrico | NR | - | - |
| L73F | 5.36 × 10−5/2 | | Mat | | | - | Noor | Mat | - | - |
| P75Q | 0/0 | MSSNG: AU3794302 | NR | + | - | - | | | | |
| I173V | 3.24 × 10−4/14 | | | | | | Noor | Mat | - | - |
| K181T | 0/0 | Clinvar | NR | + | + | - | Karaca, 2015 [17] | Mat | + | + |
| V195I | 0/0 | | | | | | Noor | Mat | - | - |
| Y213C | 0/0 | | | | | | Paris/
Strasbourg | Mat | + | + |
| G300R | 0/0 | | | | | | Lille | Mat | + | + |
| G303R | 0/0 | ClinVar ID 417957 | NR | + | - | - | | | | |
| A310P | 0/0 | | | | | | Paris | De novo | + | + |
| H359R | 0/0 | | | | | | Noor | Mat | - | - |
| A470D | 0/0 | | | | | | Noor | Mat | - | - |
| E479G | 0/0 | | | | | | Noor | Mat | - | - |
| F549C | 0/0 | Ptchd1-base.com | NR | + | + | - | | | | |
| Q884* | 0/0 | No subject | NA | | | - | | | | |
PTCHD1 shares homology (21.17% amino acid identity, as determined by multiple sequence alignment, using Clustal Omega,
www.ebi.ac.uk, accessed on 1 February 2022) with Niemann-Pick disease type C1 (NPC1), which is a multi-pass transmembrane protein that possesses 19 residues for N-linked glycosylation [
38]. Given this apparent structural homology, Xie et al. hypothesized that disease-associated missense variants of PTCHD1 may lead to aberrant protein processing by the endoplasmic reticulum and Golgi apparatus. Transient overexpression of GFP-PTCHD1 in HEK293T cells and subsequent treatment with either Endoglycosidase H or the amidase PNGase F, followed by SDS-PAGE, revealed that PTCHD1 consistently undergoes N-linked glycosylation [
37]. This finding is in agreement with in silico analyses, which predict 10 sites for N-linked glycosylation in PTCHD1 [
39]. Furthermore, in addition to N-linked glycosylation with mature complex oligosaccharides, PTCHD1 transiently exists in two putative intermediate states of N-linked glycosylation with immature mannose-rich oligosaccharides. Xie et al. next evaluated N-linked glycan processing in several PTCHD1 missense variants and reported that Pro75Gln, Lys181Thr, Tyr802Cys, and Ile884* all demonstrate the presence of N-linked glycosylation with complex oligosaccharides, at varying levels. In contrast, Pro32Arg, Gly303Arg, and Phe549Cys all fail to achieve N-linked glycosylation with mature glycans, although the two alleged intermediate N-linked glycosylation states were observed for Gly303Arg and Phe549Cys. Interestingly, only one of these supposed intermediate states was observed for Pro32Arg, indicating that this missense variant is particularly resistant to N-linked glycosylation. Regardless, defects in N-linked glycan processing of PTCHD1 missense variants did not appear to cause protein retention in either the endoplasmic reticulum or the Golgi apparatus, suggesting the possibility that PTCHD1 is trafficked via an unconventional secretory pathway [
40].
In addition to NPC1, PTCHD1 exhibits sequence homology with the Patched domain-containing proteins Patched-1 (PTCH1; 21.62% identity) and Patched-2 (PTCH2; 20.65% identity), both of which are transmembrane receptors that negatively regulate the Hedgehog (Hh) signalling pathway. Binding of the Hh ligand to PTCH1 inhibits its repression of the G protein-coupled receptor, Smoothened (SMO), and initiates a signalling cascade that ultimately leads to activation of the GLI family of transcription factors [
41]. Based on its sequence similarity, it was hypothesized that PTCHD1 may also behave similarly to Ptch1 in the Hh-signalling pathway. To explore this, Noor et al. transiently overexpressed a reporter vector with multimerized GLI transcription factor binding elements and either PTCH1, PTCH2, or PTCHD1 in the Hh-responsive cell line 10T1/2 and reported that PTCH1, PTCH2, and PTCHD1 all inhibit GLI-dependent transcription in vitro [
11]. However, Ung et al. showed that PTCHD1 was incapable of rescuing the canonical sonic hedgehog (SHH) pathway in cells depleted of PTCH1, which would suggest PTCHD1 to function in a separate pathway (Ung et al.). Hh-signalling has been implicated in early postnatal granule cell proliferation in both the cerebellum [
42] and the dentate gyrus [
43]. Moreover, the overproliferation of granule cell precursors leading to medulloblastoma has been reported in mice with homozygous or heterozygous deletions in
Ptch1, the mouse ortholog of
PTCH1 [
44]. In this regard, Tora et al. sought to assess the consequences of
Ptchd1 ablation on these populations of granule neuron precursors using the thymidine analogue BrdU. Unexpectedly, the authors indicate that the granule cell precursors in both the cerebellum and the dentate gyrus do not exhibit increased proliferation in
Ptchd1Δ2/Y mice, which contain a deletion of exon 2, during early postnatal development (P4), nor do the cells in the adult dentate gyrus. Immunocytochemistry subsequently revealed that Shh-binding to ectopically expressed Ptchd1 was undetectable in COS7 cells or mouse embryonic fibroblasts in vitro [
32]. Supporting these findings, Ung et al. report that exogenous Ptchd1 does not repress GLI-dependent transcription in mouse embryonic fibroblasts derived from
Ptch1−/− mice [
33]. Furthermore, in unpublished data from our lab, mouse embryonic hippocampal gene transcription was compared for
Ptchd1 and Hh-signalling pathway genes
Shh,
Smo, and
Ptch1. While the Hh pathway genes show decreasing transcription levels after E12 to birth,
Ptchd1 exhibits the opposite trajectory, with transcription increasing steadily from E12 to P2 (
Supplementary Figure S1). Collectively, these support the notion that PTCHD1 operates in pathways other than Hh signalling.
The predicted PDZ-binding motif at the C-terminus of PTCHD1 is also present in SEC8 (also known as EXOC4), a component of the exocyst complex, where it facilitates interaction with the PDZ domains of the postsynaptic proteins PSD95 and SAP102 [
45]. Correspondingly, Ung et al. sought to investigate if the PDZ-binding motif in Ptchd1 also mediates a similar interaction with both Psd95 and Sap102. Affinity purification experiments in synaptoneurosomal lysates from the adult mouse cortex confirmed that Ptchd1 interacts with both Psd95 and Sap102 in vitro and that this interaction is reliant on the PDZ-binding motif [
33]. In order to identify additional proteins agnostically that may be putatively interacting with Ptchd1 in vitro, Tora et al. performed affinity purification in adult mouse whole brain lysates, followed by liquid chromatography-mass spectrometry. As bait, the authors used the final 43 amino acids of Ptchd1 and also a C-terminal variant in which they deleted the PDZ-binding motif (ΔPDZ). This approach identified numerous additional proteins that preferentially interacted with the wildtype C-terminal bait fragment, including novel components of the postsynaptic density (Dlg1-3, Magi1, Magi3, and Lin7), as well as components of the retromer complex (Snx27 and Vps26b). Furthermore, the additional retromer complex protein Vps35 was found to interact equally with both the wildtype and the ΔPDZ C-terminal bait fragments. Subsequent western blot analyses confirmed binding between the Ptchd1 C-terminus and both Psd95 and Vps35, with the latter interaction being independent of the PDZ-binding motif. Lastly, endogenous Ptchd1 was reported to be enriched in the postsynaptic density, further supporting its putative interaction with these proteins under physiological conditions [
32].
Xie et al. employed a yeast two-hybrid screen to identify binding partners for the luminal loops of PTCHD1-binding proteins in vitro. Using bait fragments consisting of the first lumenal loop (F49-R270) and a chimera of both lumenal loops (F49-R270::Q521-S695), these authors found consistent interactions between PTCHD1 and the SNARE-associated protein (SNAPIN). This putative interaction was further corroborated by immunofluorescent studies in neuronally differentiated P19 cells, which demonstrated colocalization of exogenous PTCHD1 and SNAPIN in dendritic projections [
37]. SNAPIN is involved in synaptic vesicle docking and fusion and is also a component of the biogenesis of lysosome-related organelles complex 1 (BLOC-1) complex [
46], suggesting that PTCHD1 may play a role in the endosomal-lysosomal system.
2.6. PTCHD1-AS Function
As
PTCHD1-AS does not encode for a protein, its contribution to the etiology of ASD and ID is unclear. Emerging evidence suggests that lncRNAs govern the expression of coding genes by altering chromatin status, influencing the initiation of transcription, as well as by affecting mRNA stability post-transcriptionally [
47]. To investigate this, Ross et al. employed RNA fractionation and qRT-PCR to conclude that
PTCHD1-AS was almost entirely localized to the nucleus, where it was also found to primarily associate with chromatin. In addition, given the proximity of the 5′ exons, the common bi-directional promoter, and their modest sequence homology, it seems plausible that
PTCHD1-AS could be governing the expression of
PTCHD1 in
cis to some extent. However, data from Ross et al. report that basal
PTCHD1 transcription was unchanged in male iPSC-derived cortical neurons from a patient with a 125 Kb microdeletion that encompassed exon 3 of
PTCHD1-AS (PTCHD1-ASΔ3/Y). Next, in order to prematurely terminate transcription, these authors used CRISPR/Cas9 and homology-directed repair to replace exon 3 of
PTCHD1-AS with two tandem polyadenylation sequences in iPSCs derived from an unaffected male
(PTCHD1-ASΔ3-pA/Y) and subsequently observed a profound decrease in neuronal
PTCHD1 expression. These data suggest that, while
PTCHD1-AS exon 3 may not necessarily be implicated in regulating
PTCHD1 expression, downstream portions of the antisense transcript could be involved. Curiously, transcripts bearing exons 5 and 6 of
PTCHD1-AS were still detected in these CRISPR-edited cortical neurons, albeit at a reduced level, proposing the possibility of an additional TSS that is downstream of exon 3. To assess the global alterations in gene expression that are mediated by
PTCHD1-AS in
trans, Ross et al. performed microarray-based analyses on cortical neurons from iPSCs derived from the aforementioned male proband, as well as from an additional male ASD case with a 167 Kb microdeletion that eliminates the first two exons of
PTCHD1-AS2 and
PTCHD1-AS3, as well as the first exon of
PTCHD1. These investigations identified a paucity of abnormally expressed genes in cortical neurons derived from either proband, implying that
PTCHD1-AS does not significantly affect global neuronal gene expression in
trans. In addition, of the few dysregulated genes that were identified, none had any annotated neuronal function [
35].
2.7. Ptchd1 Mutant Mouse Models for Cognitive and Metabolic Phenotypes
There is a considerable accumulation of evidence to suggest that
PTCHD1 is required for normal neurodevelopment; however, its mechanistic association with the etiology of ASD and ID remains poorly understood. To investigate this, numerous studies have used
Ptchd1 mutant (
Ptchd1-/Y) mice in order to evaluate the effects of
Ptchd1 on behaviour, cognition, metabolism, gene expression, and both neuronal and synaptic structure and function. In order to disrupt
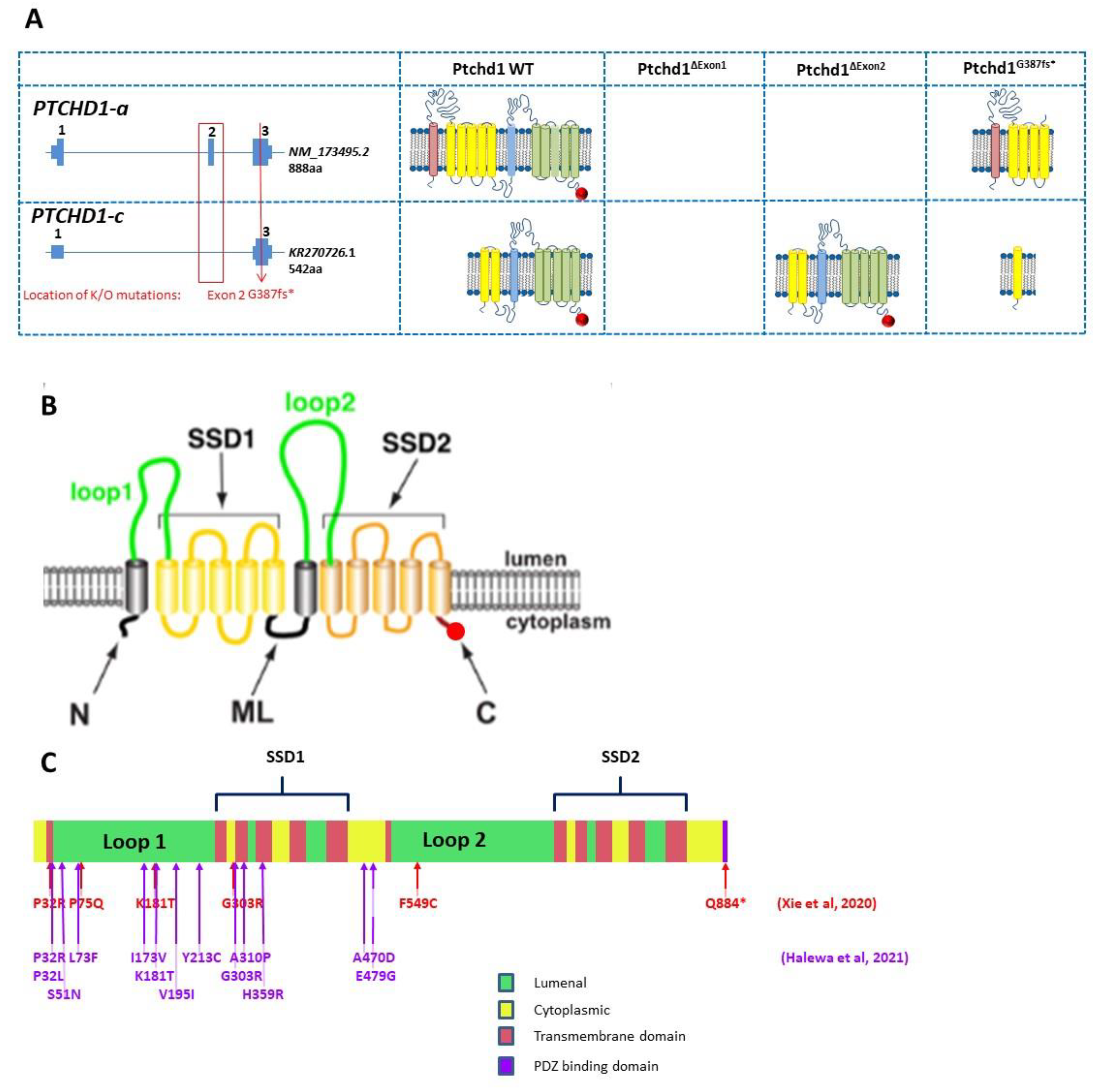
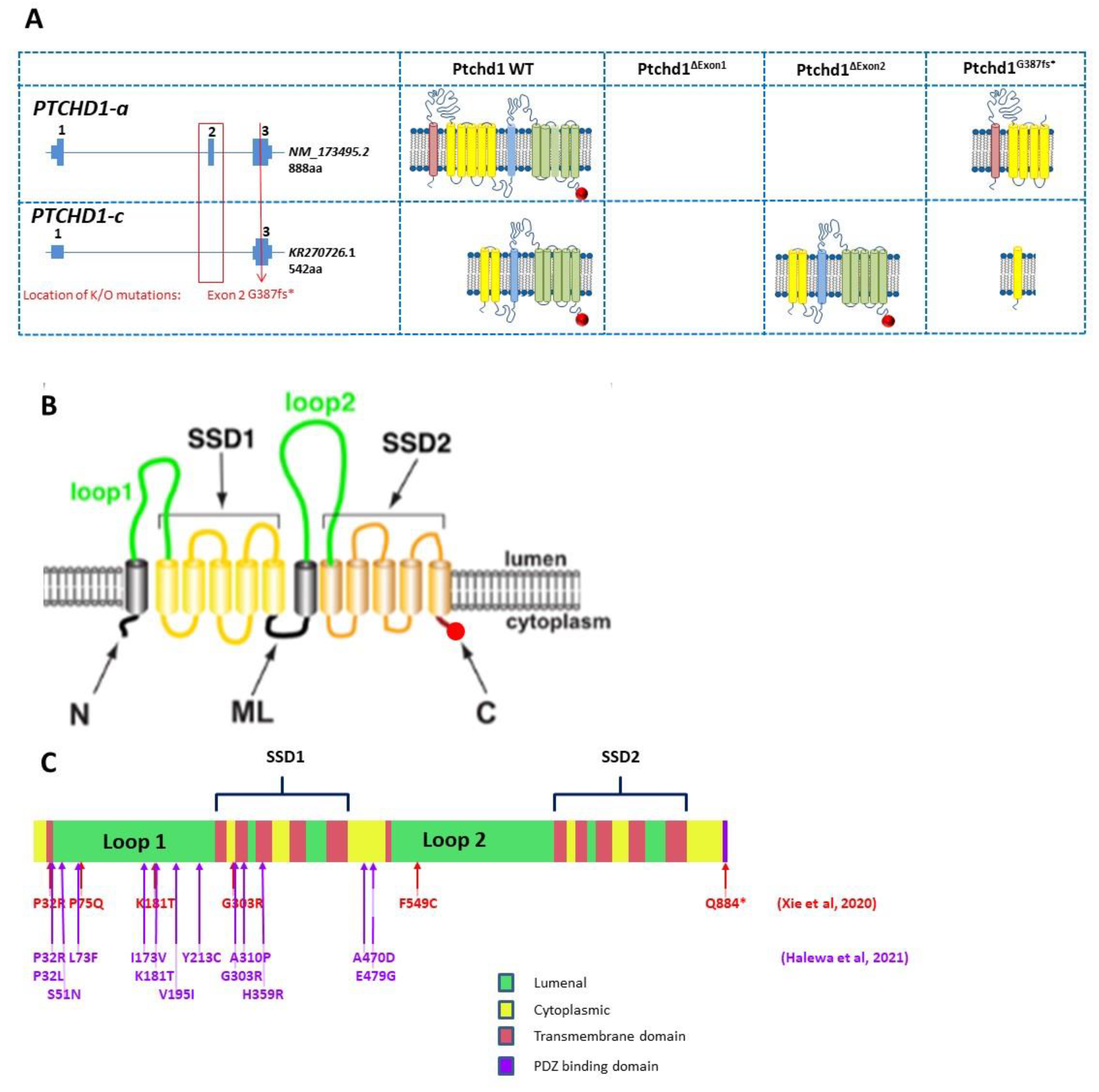
Ptchd1 in mice, multiple groups have independently generated a conditional allele by targeting exon 2 (
Ptchd1Δ2/Y), which encodes three of the 12 transmembrane domains and a portion of one of two predicted sterol-sensing domains. Exon 2 consists of 661 nucleotides, and therefore, the resulting
Ptchd1Δ2 transcript will have a premature truncation ahead of the final nine transmembrane domains, the sterol-sensing domain, and the PDZ-binding motif [
31,
32,
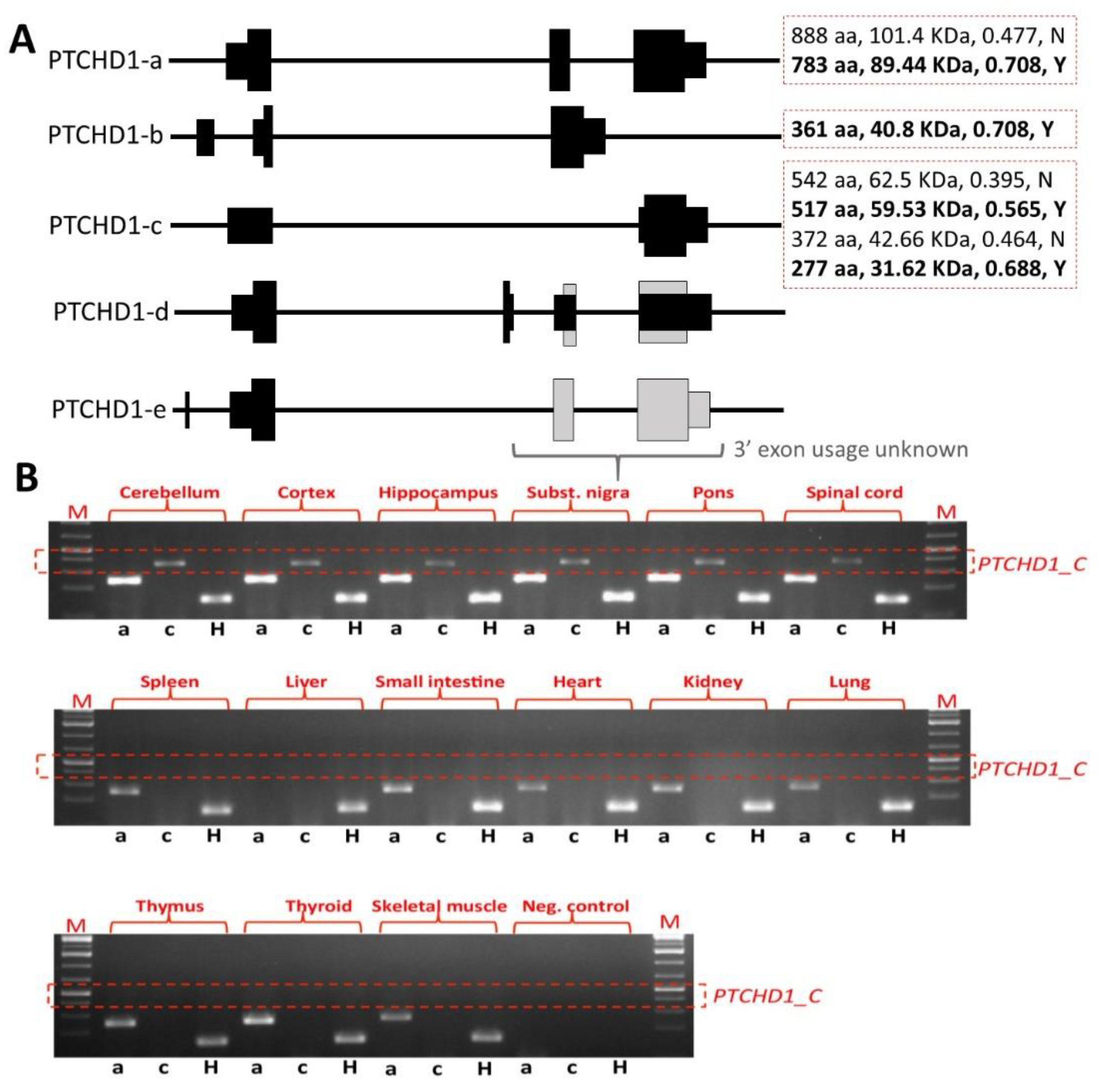
33]. However, it should be noted that the removal of exon 2 still permits the generation of the shorter transcript encoding the 542 amino acid ORF and other more C-terminal ORFs (
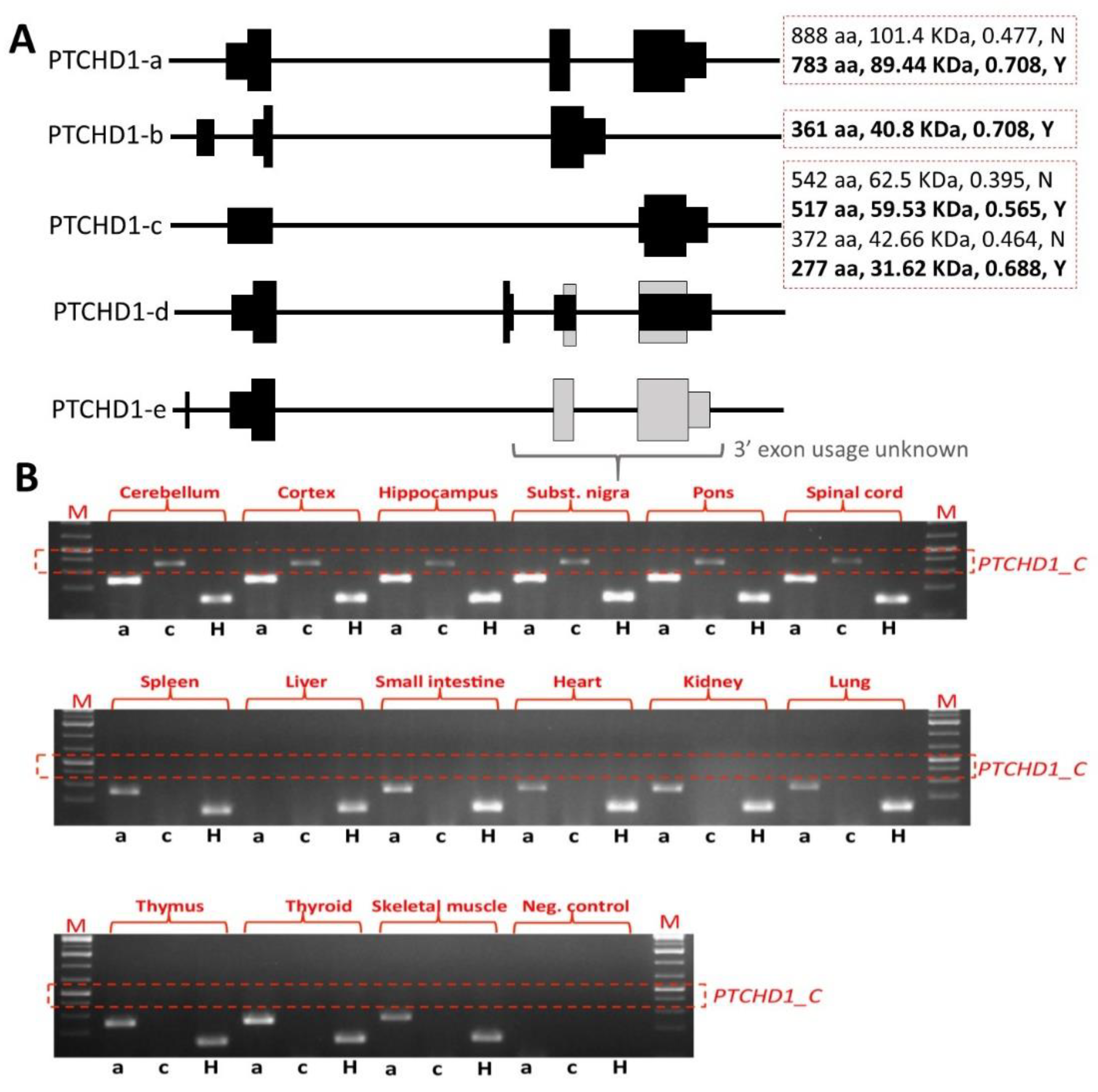
Figure 3A). Our own studies of brain tissue from the
Ptchd1-/Y mice, courtesy of Guoping Feng, indicate that there is no loss of
Ptchd1 transcripts; however, while the
Ptchd1-a transcript is lost, there is activation (~80-fold) of the shorter
Ptchd1-c transcript (Vincent lab,
unpublished data). Whether or not this transcript (or indeed the full-length transcript) is translated into protein has yet to be established.
In order to recapitulate clinical models where CNVs have encompassed just exon 1 or exon 3 of
PTCHD1 [
5,
6,
9,
12], exon-specific deletion mice have been generated. Murakami et al. generated mice with an exon 1 deletion (
Ptchd1Δ1/Y) [
48], while Ko et al. generated mice with an exon 3 deletion (
Ptchd1Δ3/Y) [
49]. Lastly, Roy et al. employed Cre-dependent SpCas9 adeno-associated viruses (AAVs) to knockdown
Ptchd1 (
Ptchd1KD) in specific brain subregions in vivo [
13]. The differences in the predicted effects of these knockout strategies on Ptchd1 protein manufacture are outlined in
Figure 4.
A multitude of studies investigating the behavioural and neuromotor phenotype of male
Ptchd1-/Y (i.e., Ptchd1
Δ1/Y, Ptchd1
Δ2/Y, and Ptchd1
Δ3/Y) mice have identified numerous perturbations, many of which recapitulate the clinical symptoms of ASD and/or ADHD.
Ptchd1-/Y mice demonstrate spontaneous hyperactivity, as inferred from an increase in the total distance travelled in a novel environment during an open field test (OFT) [
48,
49], and elevated locomotor activity [
31,
33].
The
Ptchd1Δ1/Y mice displayed ADHD-like behavior, exhibiting difficulty in habituating to new environments. Whereas control mice exhibit reduced exploration upon repeated exposure to an OFT, this reduction is attenuated in
Ptchd1Δ1/Y mice. Furthermore, the
Ptchd1Δ1/Y mice displayed heightened impulsivity, as determined by decreased jump latency in the cliff avoidance test [
48]. Interestingly, treatment of the
Ptchd1Δ1/Y mice with the norepinephrine reuptake inhibitor Atomoxetine, which has been used to treat ADHD, was found to reduce, but not abolish, spontaneous hyperlocomotor activity in both the OFT and the cliff avoidance tests. Furthermore, Atomoxetine was also observed to normalize habituation of exploration in the OFT and reduce impulsivity in the
Ptchd1Δ1/Y mice [
48].
The
Ptchd1Δ2/Y mice exhibited reduced anxiety, as evident from more time spent in the centre of the field during an OFT [
33], although this phenotype was not replicated in
Ptchd1Δ1/Y mice [
48]. Motor function defects were also reported in
Ptchd1Δ2/Y mice—Wells et al. report that
Ptchd1Δ2/Y mice exhibit altered gait parameters as well as signs of hypotonia, which was inferred from decreased grip strength, as assessed by either the hanging wire test [
31] or an isometric dynamometer [
33]. Ung et al. reported that
Ptchd1Δ2/Y mice presented with impaired motor coordination according to a rotarod test [
33], although this was not detected by Wells et al. [
31] Lastly, Wells et al. reported that
Ptchd1Δ2/Y mice exhibit a fragmented sleep pattern and hyper-aggressiveness, as determined by a longer attack duration and a shorter latency to attack during the resident intruder test, but the sensorimotor gating function remains unaffected (prepulse inhibition of the acoustic startle response [
31]).
Figure 4.
(
A) Genomic organization of Ptchd1 isoforms a and c and predicted translation products resulting from the Murakami et al. [
48] exon 1 knockout mice, Wells et al. [
31] and Ung et al. [
33] exon 2 knockout mice, and the CRISPR/cas9 knockout mouse generated by Frankland and Vincent [
49]. The PDZ-binding motif is indicated by a red circle. N.B. Ptchd1-c transcription is activated (>80-fold) in the brains of the Wells et al. mice [
31]. There are no predicted translation products for the exon 1 knockout [
48], as the predicted promoter is also disrupted, and no additional promoters are predicted (
genomatix.com, accessed on 2 August 2011). (
B) Cartoon of PTCHD1 illustrating the predicted topological orientation of specific regions in the membrane; putative sterol-sensing domains (SSD) 1 and 2, luminal loops 1 and 2, medial loop (ML), N and C-termini, are shown. (
C) Localization of missense variants studied in Halewa et al. [
16] and Xie [
37]. A more complete list of
PTCHD1 SNVs, including those identified in the MSSNG study, or reported by ClinVar, can be found on the
www.PTCHD1-base.com (accessed on 1 February 2022) website.
Figure 4.
(
A) Genomic organization of Ptchd1 isoforms a and c and predicted translation products resulting from the Murakami et al. [
48] exon 1 knockout mice, Wells et al. [
31] and Ung et al. [
33] exon 2 knockout mice, and the CRISPR/cas9 knockout mouse generated by Frankland and Vincent [
49]. The PDZ-binding motif is indicated by a red circle. N.B. Ptchd1-c transcription is activated (>80-fold) in the brains of the Wells et al. mice [
31]. There are no predicted translation products for the exon 1 knockout [
48], as the predicted promoter is also disrupted, and no additional promoters are predicted (
genomatix.com, accessed on 2 August 2011). (
B) Cartoon of PTCHD1 illustrating the predicted topological orientation of specific regions in the membrane; putative sterol-sensing domains (SSD) 1 and 2, luminal loops 1 and 2, medial loop (ML), N and C-termini, are shown. (
C) Localization of missense variants studied in Halewa et al. [
16] and Xie [
37]. A more complete list of
PTCHD1 SNVs, including those identified in the MSSNG study, or reported by ClinVar, can be found on the
www.PTCHD1-base.com (accessed on 1 February 2022) website.
![Genes 13 00527 g004]()
Cognitive assessments of male
Ptchd1Δ1/Y and
Ptchd1Δ2/Y mice reveal impairments related to learning and memory. Both
Ptchd1Δ1/Y and
Ptchd1Δ2/Y mice display diminished short-term working memory, which was inferred from fewer spontaneous alternations during the Y-maze test [
33,
48]. The Morris water maze test did not reveal deficits in spatial learning and memory in
Ptchd1Δ2/Y mice [
31], although a targeted knockdown of
Ptchd1 in the anterodorsal (AD) thalamus suggested diminished spatial working memory, as inferred from fewer correct alternations during the T-maze test. Interestingly, this deficit was only present when the sample and choice trials were separated by a longer (60 s) interval, but a similar performance was observed when the inter-trial duration was short (10 s). Furthermore, these AD thalamus
Ptchd1KD mice also demonstrated impaired long-term memory recall, which was evident from a significantly reduced freezing when tested 24 h after training in a contextual fear conditioning paradigm [
13]. Notably, a reduced discrimination index on the novel object recognition test (NORT) provides evidence of weakened recognition memory in both
Ptchd1Δ1/Y and
Ptchd1Δ2/Y mice [
32,
33,
48]. Interestingly, Atomoxetine treatment rescued this impairment in recognition memory, as evident by less interaction time with the familiar object on the NORT in
Ptchd1Δ1/Y mice [
48]. Moreover,
Ptchd1Δ2/Y mice demonstrated additional learning impairments, as indicated by a diminished latency to cross during the inhibitory avoidance task [
31] and decreased freezing in contextual and cued conditioning paradigms [
31,
33] (similar to the AD thalamus
Ptchd1KD mice).
Interestingly,
Ptchd1 disruption appears to be associated with deficits in attentional engagement and sensory filtering. These deficits presumably contribute to learning and memory impairments, as
Ptchd1Δ2/Y mice are impaired in a cognitive task in the presence of a visual distractor, suggesting sensory-related distractibility in these mice [
31]. Similarly,
Ptchd1Δ2/Y mice demonstrated a markedly poorer ability to discriminate between different auditory stimuli when provided with high levels of background noise, both in the presence and absence of preceding visual cues [
50].
The studies by Wells et al. have implicated the TRN as a key brain region in several of the behavioural and cognitive abnormalities observed in
Ptchd1Δ2/Y mice. When the deletion of
Ptchd1 exon 2 was principally confined to the TRN using
Somatostatin-driven
Cre mice (
Som-Cre+:Ptchd1fl/Y), hyperactivity, problems with attentional engagement, and fragmented sleep all persisted, indicating that
Ptchd1 expression in the TRN is critical for these functions. Notably, another type of learning and memory (i.e., inhibitory avoidance task) was found to improve, and both hyper-aggression and hypotonia were no longer detected in these
Som-Cre+:Ptchd1fl/Y mice, suggesting that
Ptchd1 expression elsewhere in the brain influences these traits [
31].
Murakami et al. investigated the possible relationship between
Ptchd1-mediated metabolic dysregulation and ADHD-like behavioral phenotypes. These authors specifically focused on the effects of
Ptchd1 on the kynurenine pathway (KP), which metabolizes tryptophan to generate nicotinamide adenine dinucleotide (NAD
+), a coenzyme that is critical for redox reactions in both metabolism and energy production. In 11-week-old
Ptchd1Δ1/Y mice, these authors detected increased serum concentrations of kynurenine, as well as an increased presumptive activity of the enzyme indoleamine-2,3-oxygenase1 (Ido1), the first and rate-limiting KP enzyme. Notably, atomoxetine, a selective norepinephrine reuptake inhibitor, typically used as a treatment for ADHD, ameliorated both the heightened activity of Ido1 and the elevated concentration of kynurenine in the serum of the
Ptchd1Δ1/Y mice. In addition to the serum, elevated levels of the KP metabolites 3-hydroxykynurenine (3-HK), anthranilic acid (AA), and 3-hydroxyanthranilic acid (3-HAA) were all detected in the
Ptchd1Δ1/Y frontal cortex. Atomoxetine administraton also reduced the levels of AA in the
Ptchd1Δ1/Y frontal cortex, although the concentrations of both 3-HK and 3-HAA remained abnormally high. Interestingly, atomoxetine was observed to significantly raise the concentration of an additional KP metabolite, kynurenic acid (KYNA), in the
Ptchd1Δ1/Y frontal cortex. Collectively, these data suggest an association between the dysregulation of tryptophan metabolism and
PTCHD1-related ASD and/or ID; however, this association has yet to be confirmed either in
PTCHD1-deficient patients or in other
Ptchd1 mouse models [
20].
Unexpectedly, given the
PTCHD1 involvement in ASD, no social abnormalities were apparent in either
Ptchd1Δ1/Y or
Ptchd1Δ2/Y mice as inferred from the three-chambered social interaction assay [
31,
48] and the social recognition test [
33]. Despite this, evidence of possible stereotypic behaviours was reported in
Ptchd1Δ2/Y mice, including more frequent rearing and increased locomotor activity in the back portion of the cage during the active part of the circadian cycle [
33], although repetitive grooming was not observed [
31].
However, a preliminary study by Ko et al. suggests potential PTCHD1/Ptchd1 involvement in both ASD and ID, with respect to both social abnormalities and learning deficits in mice [
49]. Based on loss CNV and nonsense mutation cases reported in PTCHD1-related ASD patients [
6], they generated
Ptchd1 exon 3 truncating mutation mice using CRISPR-Cas9 technology (
Ptchd1Δ3/Y).
Ptchd1Δ3/Y mice exhibited significant reductions in both
Ptchd1 full-length (i.e., the transcript derived from exons 1 to 3) and shorter transcripts (i.e., the transcript derived from exons 1 and 3), and social interaction and communication behaviors were abnormal in these mice, as inferred from the reduced male-female interaction time in the three-chambered social interaction assay, the reduced emission of ultrasonic vocalization during the social interaction, and the reduced sniffing behavior in the social odor cue reactivity task, respectively. Furthermore,
Ptchd1Δ3/Y mice also exhibit impaired learning and memory in contextual fear conditioning [
49]. Given that
Ptchd1Δ2/Y mice display significant enrichment of the shorter alternatively spliced form of the
Ptchd1 transcript (
Ptchd1-c) and that
Ptchd1Δ3/Y mice lack expression of both full-length (
Ptchd1-a) and shorter form (
Ptchd1-c) transcripts, it seems plausible that the more complete phenotypic picture in the
Ptchd1Δ3/Y mice (i.e., including social deficits) relates to the loss of both major alternative transcripts (
Ptchd1-a and
-c). In other words, it is plausible that the increased expression of
Ptchd1-c in the
Ptchd1Δ2/Y mice rescues the social deficit phenotypes.
2.8. Neurodevelopmental Implications of PTCHD1 and PTCHD1-AS
Extensive efforts have sought to determine the neurophysiological mechanisms of
Ptchd1-mediated phenotypic abnormalities. Pioneering work by Wells et al. in 2016 implicated the TRN as being a critical region in the brain that underlies many of the behavioural and cognitive abnormalities displayed in
Ptchd1Δ2/Y mice. In TRN neurons of
Ptchd1Δ2/Y mice, whole-cell patch clamp recordings reveal a decrease in repetitive bursting. In addition,
Ptchd1Δ2/Y TRN neurons exhibited a lower rate of burst firing during sleep, which translated into an overall reduction in sleep spindles and a fragmented sleep pattern. Subsequent analyses indicate that this lowered burst rate may be a consequence of reduced ion currents traveling through small conductance Ca
2+-activated K
+ (SK) channels. Furthermore, a two-fold lower basal concentration of intracellular Ca
2+ was observed, which may also contribute to the deficits observed in SK channels [
31]. Moreover, the insufficient hyperpolarization that results from these reduced K
+ currents likely impairs the voltage-gated recruitment of T-type Ca
2+ channels [
51], which may affect the excitability of the TRN. The TRN is a GABAergic group of neurons that provides the principal source of inhibition to the thalamic relay nuclei, and therefore, diminished TRN function could contribute to attenuated thalamic inhibition [
52]. One possible implication of reduced thalamic inhibition is an inability to suppress unwanted sensory stimuli [
53], which may help to explain the heightened distractibility in
Ptchd1Δ2/Y mice [
31]. Interestingly, pharmacological augmentation of SK currents in
Ptchd1Δ2/Y mice using the agonist 1-ethyl-2-benzimidazolinone (1-EBIO) significantly increased sensory-evoked thalamic inhibition, which mitigated their heightened distractibility and improved cognitive performance in the presence of visual distractors [
31].
To elaborate on the apparent thalamic dysregulation and distractibility in
Ptchd1Δ2/Y mice, Nakajima et al. further probed the affiliation between
Ptchd1 disruption and thalamic-mediated noise hypersensitivity by examining the auditory subnetwork of the TRN (audTRN). These analyses reveal that the sound-evoked firing rates of audTRN neurons were diminished in
Ptchd1Δ2/Y mice. Interestingly, the impairment in the ability of
Ptchd1Δ2/Y mice to discriminate between auditory stimuli in the presence of high levels of background noise was fully rescued by 1-EBIO supplementation, but only when anticipatory visual cues were not provided before the auditory stimulus. Unexpectedly, when prior visual cues were given, 1-EBIO only elicited a marginal improvement in the ability of
Ptchd1Δ2/Y mice to filter out unwanted background noise. This finding suggests a deficit of executive control over sensory filtering in
Ptchd1Δ2/Y mice, which may be mediated by the prefrontal cortex (PFC). Remarkably, a synergistic approach that used the cognitive enhancer modafinil in combination with 1-EBIO was observed to fully restore both the PFC and the audTRN function, as well as to rescue the attenuated discrimination performance in the
Ptchd1Δ2/Y mice when visual cues were provided [
50].
Roy et al. subsequently used ex vivo electrophysiology to examine neuronal properties in the
Ptchd1KD AD thalamus and identified a reduction in action potential half-width and a corresponding increase in firing frequency [
13]. These authors also report that a contextual fear-conditioning paradigm did not elicit synaptic strengthening in the
Ptchd1KD AD thalamus, as evidenced by a stagnant AMPA-to-NMDA (A/N) ratio in the AD thalamus-retrosplenial cortex (RSC) circuit. This diminished synaptic strengthening appears to be a consequence of the hyperexcitability of neurons in the
Ptchd1KD AD thalamus. Synaptic strengthening during learning is dependent upon a stimulus-induced increase in the excitability of AD neurons, and the abnormally high basal excitability of
Ptchd1KD AD thalamic neurons may occlude learning-induced changes. They showed that
Ptchd1KD impairs the learning-dependent A/N ration and the
c-fos expression, but pharmacological inhibition of AD thalamic- > RSC circuits in
Ptchd1KD mice rescued the A/N ratio and
c-fos levels in the RSC but not in the AD thalamus. Next, to determine the molecular mechanism for this apparent hyperexcitability in
Ptchd1KD AD thalamic neurons, Roy et al. used FISH and elucidated that the expression of two transcripts,
Cacna1a and
Cacna1b, that encode for voltage-gated P-type calcium channel subunits alpha-1A (which contain the Ca
2+ pore of Ca
v2.1 channels) and alpha-1B (Ca
v2.2 channels), respectively, were both markedly lower in these neurons. Subsequent ex vivo electrophysiology revealed reduced Ca
v2.1 and Ca
v2.2 current amplitudes in
Ptchd1KD AD thalamic neurons [
13].
In addition to the thalamus, considerable research has been devoted to another brain subregion, the hippocampus, in an effort to comprehend the memory impairments that have been reported in
Ptchd1-/Y (exon 2 deletion) mice. In the hippocampus, Tora et al. were unable to detect differences in the density or morphology of glutamatergic dendritic spines in dentate granule cells in P21 and P60
Ptchd1Δ2/Y mice [
32]. In contrast, Ung et al. used transmission electron microscopy to show a decrease in the density of excitatory synapses, as well as ultrastructural attenuations in both postsynaptic density thickness and synaptic cleft width in the hippocampal synapses of
Ptchd1Δ2/Y mice that were 13–15 weeks old. Interestingly, these authors also report a significant accumulation of neurotransmitter vesicles in
Ptchd1Δ2/Y synaptic boutons. Lastly, Ung et al. evaluated the morphology of GFP-labelled
Ptchd1Δ2/Y primary hippocampal neuronal cells that were differentiated for 18 days ex vivo and identified decreased dendritic length and branching arborisation [
33]. Functionally, whole-cell voltage clamping of P21–P24 hippocampal slices revealed a reduction in the excitation-to-inhibition ratio of
Ptchd1Δ2/Y dentate granule cells, as well as a basal increase in the frequency of both spontaneous excitatory (sEPSC) and inhibitory (sIPSC) potentials [
32]. Despite this apparent increase in basal currents in dentate granule cells, a substantial decrease in the frequency of evoked miniature excitatory postsynaptic currents (mEPSCs) was observed in the surrounding
Ptchd1Δ2/Y CA1 pyramidal neurons. Subsequently, the paired-pulse ratio indicates that
Ptchd1Δ2/Y Schaffer collateral axons, the primary excitatory input onto CA1 pyramidal cells, have a higher probability of vesicular release, which may mitigate the reduced frequency of mEPSCs [
33].
To evaluate the effects of
Ptchd1 exon 2 deletion on the hippocampal transcriptome, Ung et al. performed RNA sequencing on hippocampal samples from P30
Ptchd1Δ2/Y mice and identified a large number of aberrantly expressed genes [
33]. These authors further used published single-cell transcriptomics data from the P21–P30 mouse hippocampus [
54] in order to perform gene set enrichment analyses to identify specific hippocampus cell subtypes that may be particularly affected in the
Ptchd1Δ2/Y mice. This bioinformatic approach revealed that upregulated genes in the
Ptchd1Δ2/Y hippocampus were significantly enriched in the markers of neuronal genes, in particular pyramidal neurons and interneurons. Interestingly, downregulated genes in the
Ptchd1Δ2/Y hippocampus were significantly enriched in the markers of glial cells, specifically astrocytes and oligodendrocytes, as well as endothelial cells. A subsequent gene ontology enrichment analysis demonstrated that the array of upregulated neuronal genes in the
Ptchd1Δ2/Y hippocampus was significantly enriched in genes encoding synaptic proteins. Specifically, abnormal upregulation was observed for approximately 20% of genes that encode presynaptic proteins, such as
Syt1,
Bsn,
Vamp3, and
Syn1-3. Similarly, the expression of almost 25% of the genes encoding the postsynaptic proteins was likewise found to be augmented, including
Dlg4 (which encodes for Psd95), as well as
Camk2a,
Syngap1, and
Shank1-3. In addition to synaptic protein-coding genes, the authors also report an upregulation of genes encoding proteins involved in different aspects of nervous system development, such as axonogenesis and dendritogenesis. Lastly, increases in the expression of genes encoding the neuronal activity-dependent transcription factors
Npas4 and
Egr1 were also discovered in the
Ptchd1Δ2/Y hippocampus. Taken in aggregate, these data suggest that the absence of
Ptchd1 in hippocampal neurons may lead to the dysregulation of neuronal and synaptic structure and function [
33].
In order to examine the neurophysiological function of
PTCHD1 and
PTCHD1-AS in a human context, Ross et al. sought to characterize the neuronal properties of iPSCs reprogrammed from male ASD probands with previously characterized microdeletions in Xp22.11 [
11]. No apparent alteration in the dendritic morphology was observed in cortical neurons from iPSCs derived from either of the two aforementioned male ASD probands (with 125 Kb (
PTCHD1-AS exon 3) and 167 Kb (
PTCHD1-AS exons plus exon 1 of
PTCHD1) loss CNVs). Interestingly,
PTCHD1-ASΔ3/Y cortical neurons appeared to display an increased density of excitatory synapses within dendrites. Furthermore, in vitro electrophysiological analyses indicated that CRISPR/cas9-edited
PTCHD1-ASΔ3-pA/Y cortical neurons (in which
PTCHD1-AS exon 3 has been replaced with tandem polyadenylation sites), as well as those derived from both the 125 Kb and 167 Kb loss CNV probands, all exhibited marked attenuations in AMPAR-mediated mEPSC frequency, with an additional reduction in mEPSC amplitude observed in the CRISPR-edited cortical neurons. Lastly, cortical neurons from both probands also demonstrated a reduction in NMDA-evoked current amplitude [
35].


 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}