
Small RNA and Degradome Sequencing Reveal Important MicroRNA Function in Nicotiana tabacum Response to Bemisia tabaci



Abstract
:1. Introduction
2. Materials and Methods
2.1. Plants, Insects and Experiment Design
2.2. Small RNA Library Construction and Sequencing
2.3. Small RNA Identification and Prediction
2.4. Degradome Sequencing Analysis
2.5. Function Classification Based on GO and KEGG Analysis
2.6. Data Analysis
3. Results
3.1. Deep Sequencing of Small RNAs in N. tabacum
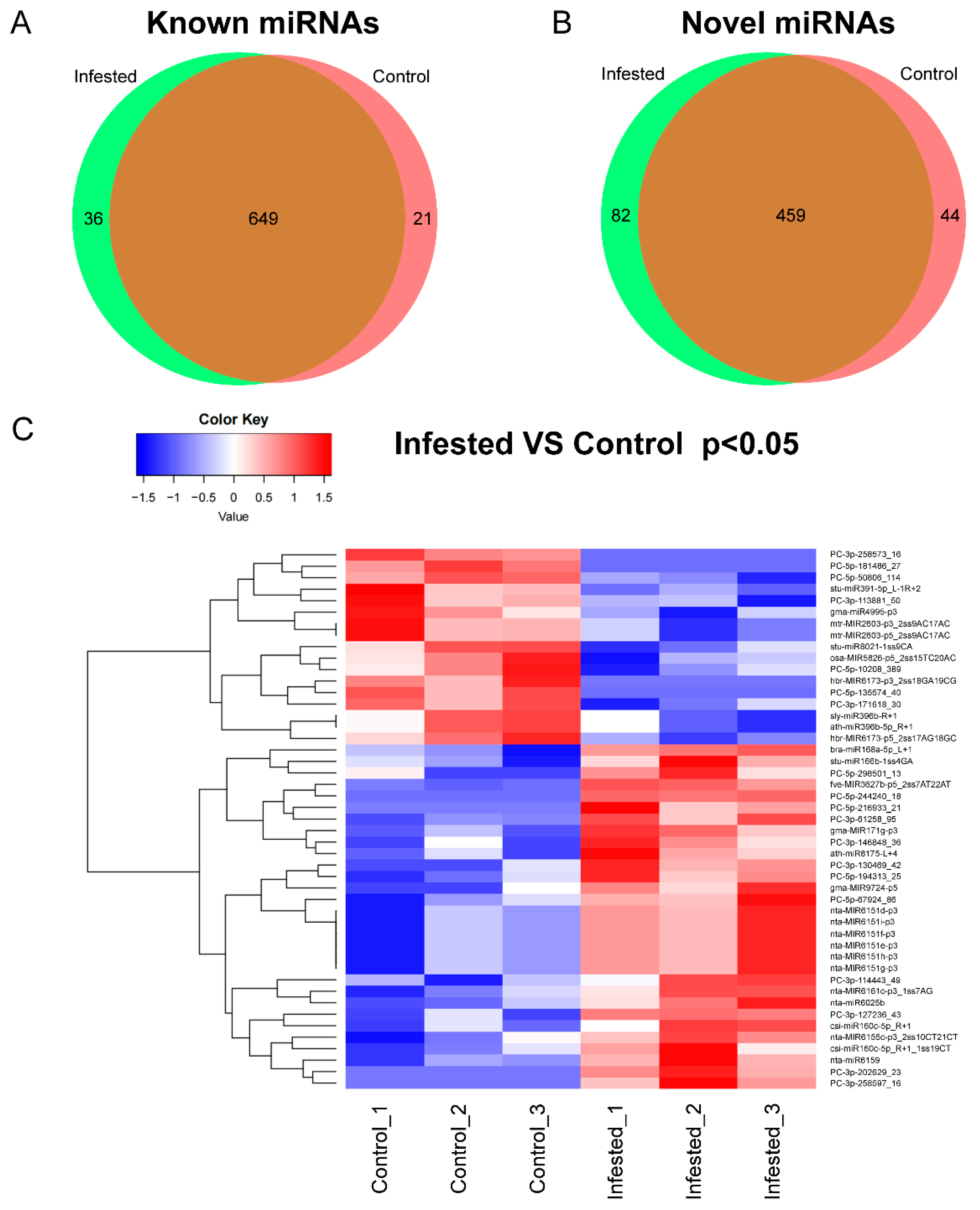
3.2. Identification and Their Expression Patterns of Known miRNAs in N. tabacum
3.3. Identification and Expression Patterns of Novel miRNAs in N. tabacum
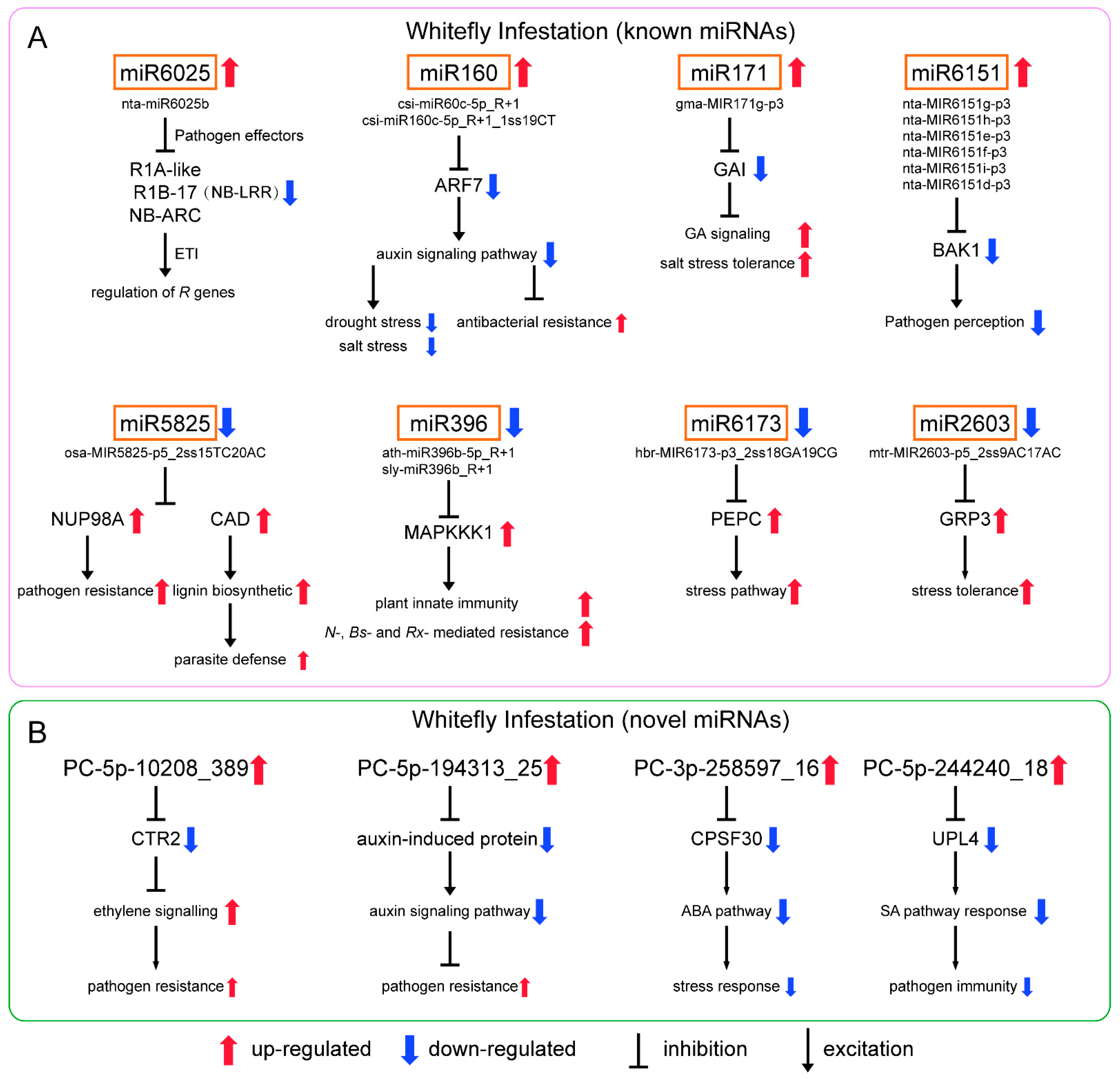
3.4. Prediction of Differential miRNA Target Genes
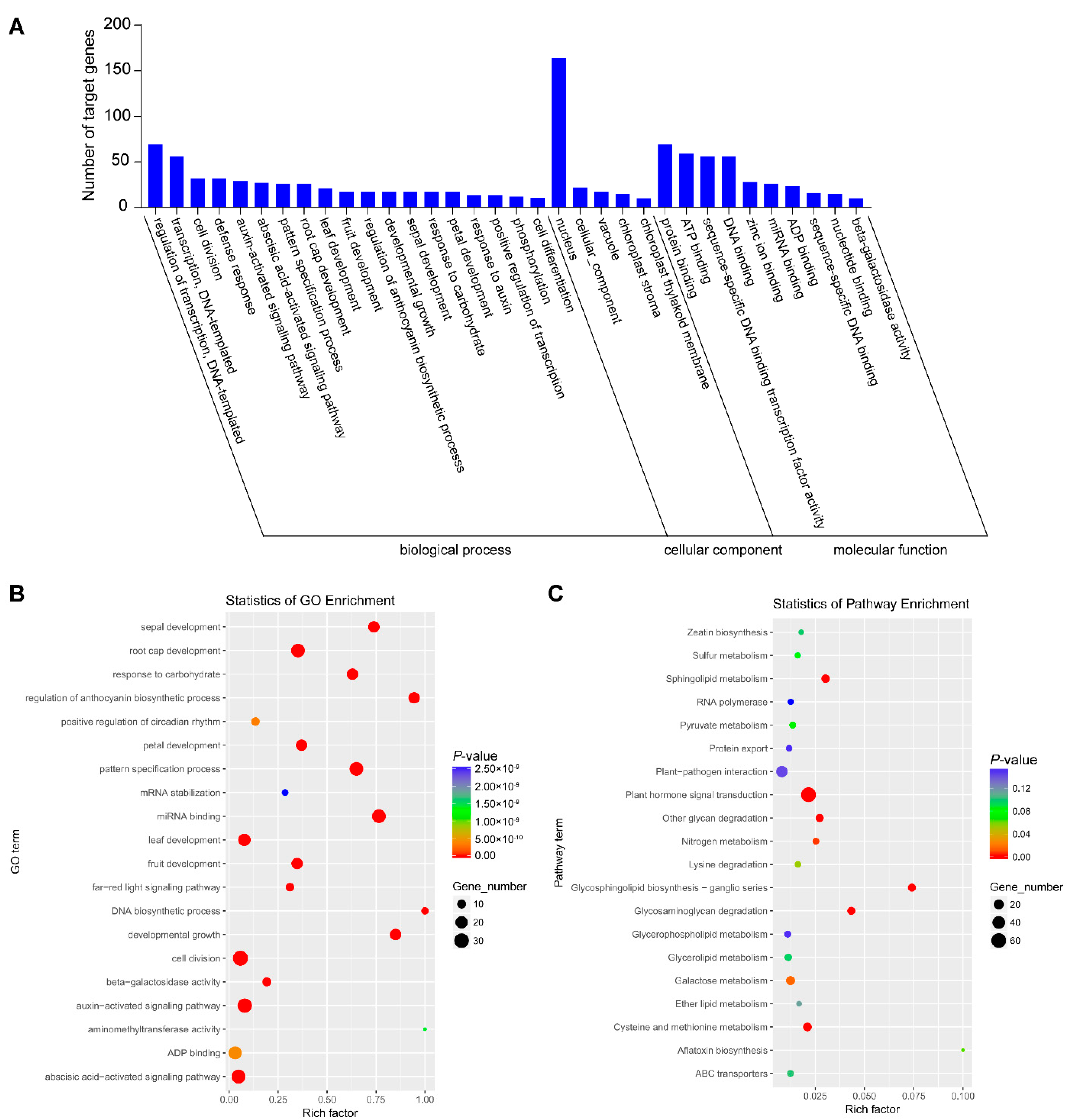
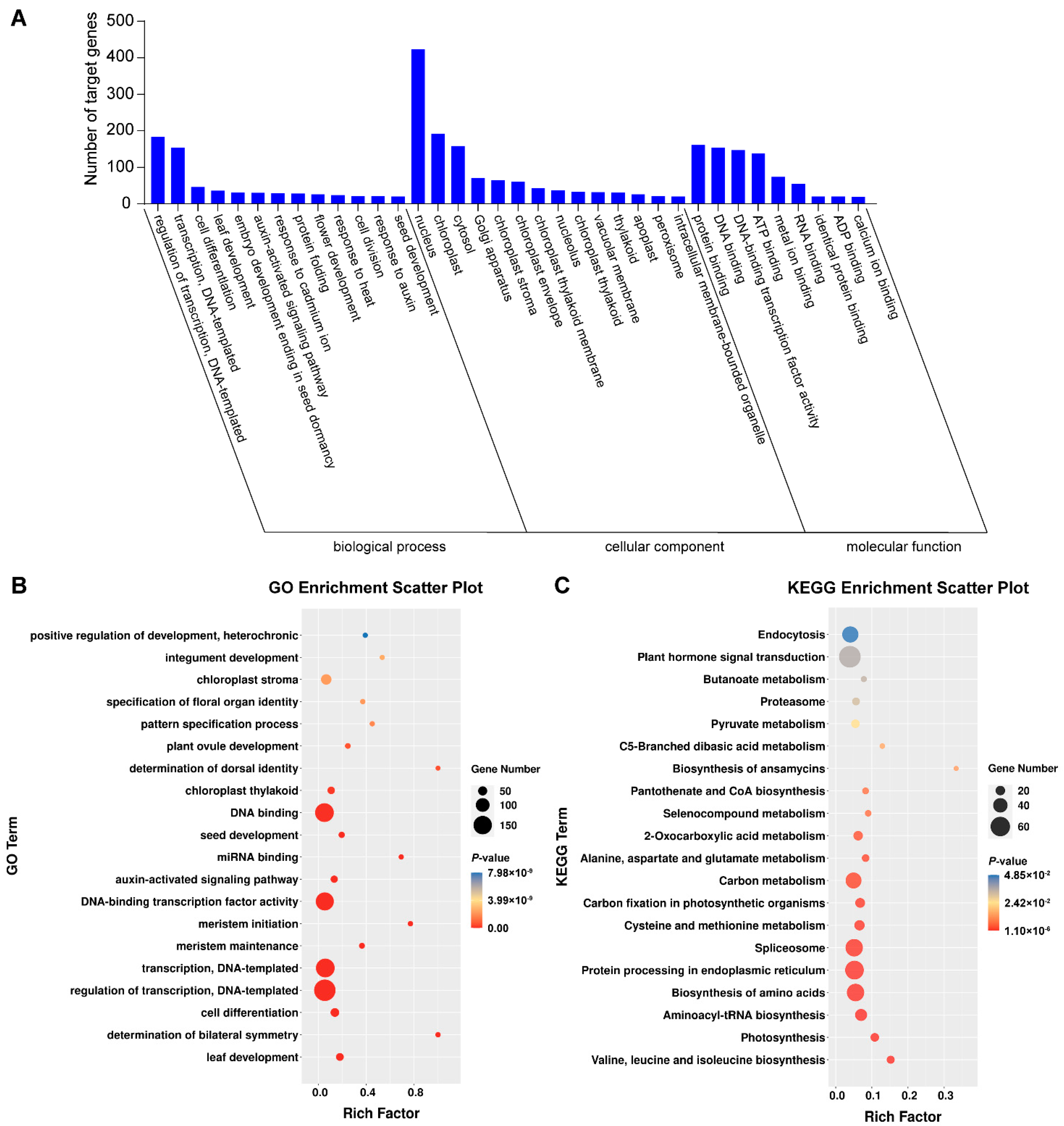
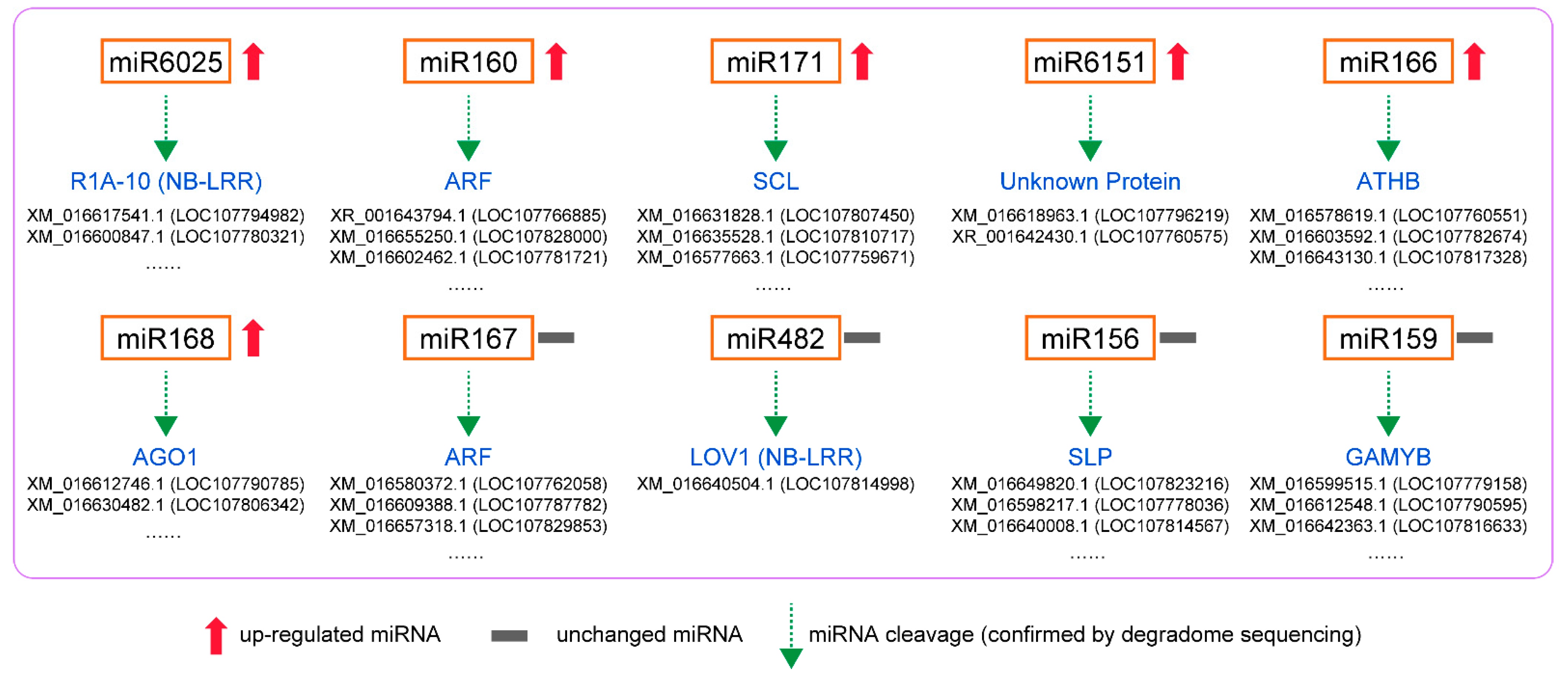
3.5. Degradome Sequencing of Small RNAs of N. tabacum Infested by B. tabaci
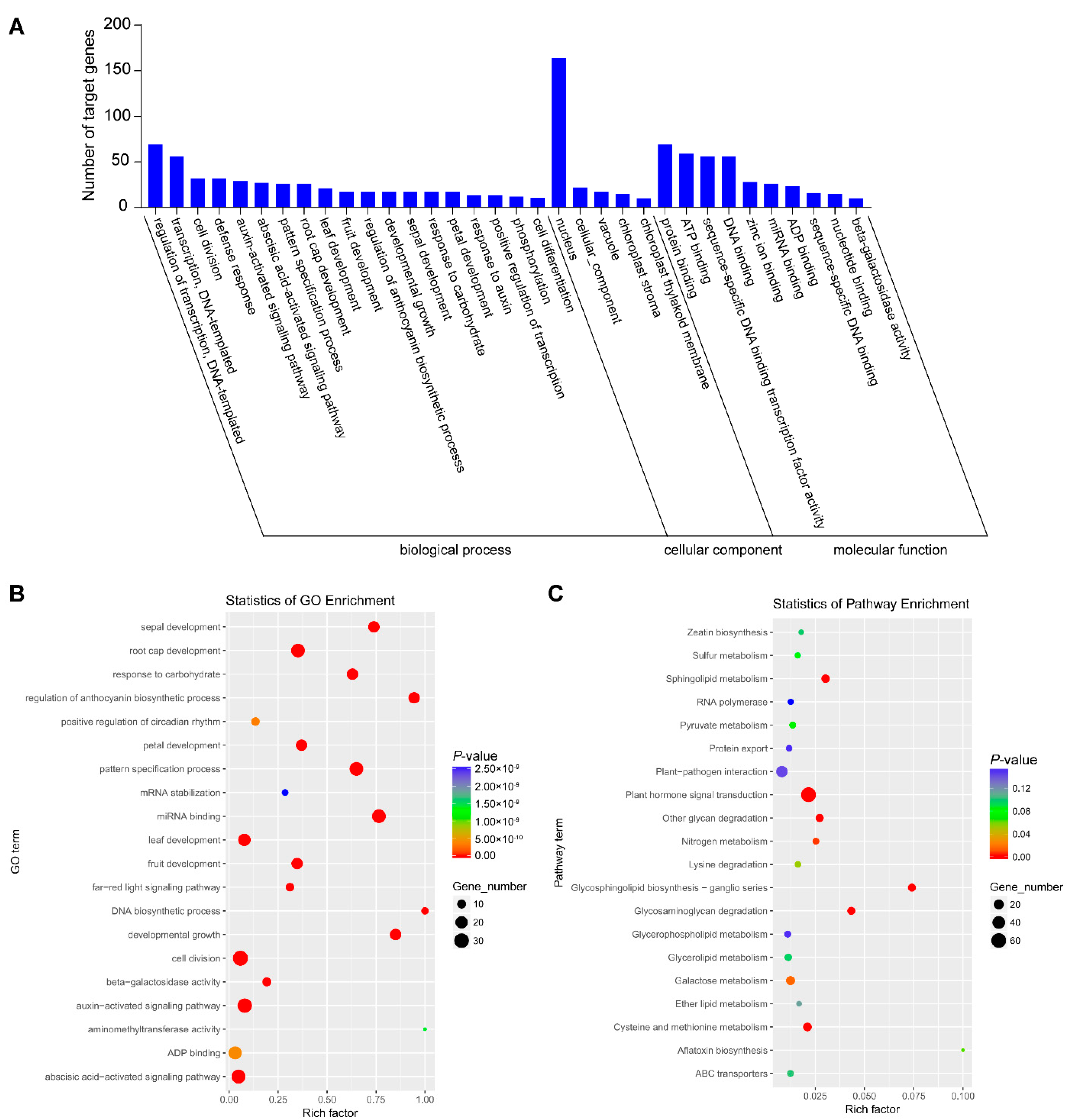
3.6. GO and KEGG Pathway Analysis of Targeted Genes in Degradome Sequencing
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bartel, D.P. MicroRNAs: Genomics, Biogenesis, Mechanism, and Function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef] [Green Version]
- Song, X.; Li, Y.; Cao, X.; Qi, Y. MicroRNAs and Their Regulatory Roles in Plant-Environment Interactions. Annu. Rev. Plant Biol. 2019, 70, 489–525. [Google Scholar] [CrossRef] [PubMed]
- Curaba, J.; Singh, M.B.; Bhalla, P.L. miRNAs in the Crosstalk between Phytohormone Signalling Pathways. J. Exp. Bot. 2014, 65, 1425–1438. [Google Scholar] [CrossRef]
- Pradhan, M.; Pandey, P.; Gase, K.; Sharaff, M.; Singh, R.K.; Sethi, A.; Baldwin, I.T.; Pandey, S.P. Argonaute 8 (AGO8) Mediates the Elicitation of Direct Defenses against Herbivory. Plant Physiol. 2017, 175, 927–946. [Google Scholar] [CrossRef]
- Hogenhout, S.A.; Bos, J.I. Effector Proteins That Modulate Plant-Insect Interactions. Curr. Opin. Plant Biol. 2011, 14, 422–428. [Google Scholar] [CrossRef] [PubMed]
- Ge, Y.; Han, J.; Zhou, G.; Xu, Y.; Ding, Y.; Shi, M.; Guo, C.; Wu, G. Silencing of MiR156 Confers Enhanced Resistance to Brown Planthopper in Rice. Planta 2018, 248, 813–826. [Google Scholar] [CrossRef] [PubMed]
- Kettles, G.J.; Drurey, C.; Schoonbeek, H.; Maule, A.J.; Hogenhout, S.A. Resistance of Arabidopsis thaliana to the Green Peach Aphid, Myzus persicae, Involves Camalexin and Is Regulated by MicroRNAs. New Phytol. 2013, 198, 1178–1190. [Google Scholar] [CrossRef] [Green Version]
- Sattar, S.; Song, Y.; Anstead, J.A.; Sunkar, R.; Thompson, G.A. Cucumis melo MicroRNA Expression Profile During Aphid Herbivory in a Resistant and Susceptible Interaction. Mol. Plant Microbe. Interact. 2012, 25, 839–848. [Google Scholar] [CrossRef] [Green Version]
- De Barro, P.J.; Liu, S.S.; Boykin, L.M.; Dinsdale, A.B. Bemisia tabaci: A Statement of Species Status. Annu. Rev. Entomol. 2011, 56, 1–19. [Google Scholar] [CrossRef]
- Liu, S.S.; Colvin, J.; De Barro, P.J. Species Concepts as Applied to the Whitefly Bemisia tabaci Systematics: How Many Species Are There? J. Integr. Agric. 2012, 11, 176–186. [Google Scholar] [CrossRef]
- Barbosa, L.d.F.; Marubayashi, J.M.; De Marchi, B.R.; Yuki, V.A.; Pavan, M.A.; Moriones, E.; Navas-Castillo, J.; Krause-Sakate, R. Indigenous American Species of the Bemisia tabaci Complex Are Still Widespread in the Americas: Indigenous B. tabaci Species Are Still Widespread in the Americas. Pest Manag. Sci. 2014, 70, 1440–1445. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.K.; Frohlich, D.R.; Rosell, R.C. The Sweetpotato or Silverleaf Whiteflies: Biotypes of Bemisia tabaci or a Species Complex? Annu. Rev. Entomol. 1995, 40, 511–534. [Google Scholar] [CrossRef]
- Navas-Castillo, J.; Fiallo-Olivé, E.; Sánchez-Campos, S. Emerging Virus Diseases Transmitted by Whiteflies. Annu. Rev. Phytopathol. 2011, 49, 219–248. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.S.; De Barro, P.J.; Xu, J.; Luan, J.B.; Zang, L.S.; Ruan, Y.M.; Wan, F.H. Asymmetric Mating Interactions Drive Widespread Invasion and Displacement in a Whitefly. Science 2007, 318, 1769–1772. [Google Scholar] [CrossRef]
- Kempema, L.A.; Cui, X.; Holzer, F.M.; Walling, L.L. Arabidopsis Transcriptome Changes in Response to Phloem-Feeding Silverleaf Whitefly Nymphs. Similarities and Distinctions in Responses to Aphids. Plant Physiol. 2007, 143, 849–865. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Zhu, L.; Hull, J.J.; Liang, S.; Daniell, H.; Jin, S.; Zhang, X. Transcriptome Analysis Reveals a Comprehensive Insect Resistance Response Mechanism in Cotton to Infestation by the Phloem Feeding Insect Bemisia tabaci (Whitefly). Plant Biotechnol. J. 2016, 14, 1956–1975. [Google Scholar] [CrossRef]
- Qin, L.; Wang, J.; Bing, X.L.; Liu, S.S. Identification of nine cryptic species of Bemisia tabaci (Hemiptera: Aleyrodidae) from China by using the mtCOI PCR-RFLP technique. Kun Chong Xue Bao 2013, 56, 186–194. [Google Scholar] [CrossRef]
- Meyers, B.C.; Axtell, M.J.; Bartel, B.; Bartel, D.P.; Baulcombe, D.; Bowman, J.L.; Cao, X.; Carrington, J.C.; Chen, X.; Green, P.J.; et al. Criteria for Annotation of Plant MicroRNAs. Plant Cell 2008, 20, 3186–3190. [Google Scholar] [CrossRef]
- Peng, T.; Sun, H.Z.; Du, Y.X.; Zhang, J.; Li, J.Z.; Liu, Y.X.; Zhao, Y.F.; Zhao, Q.Z. Characterization and Expression Patterns of MicroRNAs Involved in Rice Grain Filling. PLoS ONE 2013, 8, e54148. [Google Scholar] [CrossRef] [Green Version]
- Dai, X.; Zhao, P.X. PsRNATarget: A Plant Small RNA Target Analysis Server. Nucleic Acids Res. 2011, 39, W155–W159. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y. MiRU: An Automated Plant miRNA Target Prediction Server. Nucleic Acids Res. 2005, 33, W701–W704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- German, M.A.; Pillay, M.; Jeong, D.H.; Hetawal, A.; Luo, S.; Janardhanan, P.; Kannan, V.; Rymarquis, L.A.; Nobuta, K.; German, R.; et al. Global Identification of MicroRNA-Target RNA Pairs by Parallel Analysis of RNA Ends. Nat. Biotechnol. 2008, 26, 941–946. [Google Scholar] [CrossRef] [PubMed]
- Addo-Quaye, C.; Miller, W.; Axtell, M.J. CleaveLand: A Pipeline for Using Degradome Data to Find Cleaved Small RNA Targets. Bioinformatics 2009, 25, 130–131. [Google Scholar] [CrossRef] [PubMed]
- Addo-Quaye, C.; Eshoo, T.W.; Bartel, D.P.; Axtell, M.J. Endogenous siRNA and miRNA Targets Identified by Sequencing of the Arabidopsis Degradome. Curr. Biol. 2008, 18, 758–762. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Shahid, M.; Wu, J.; Wang, L.; Liu, X.; Lu, Y. Comparative Small RNA Analysis of Pollen Development in Autotetraploid and Diploid Rice. Int. J. Mol. Sci 2016, 17, 499. [Google Scholar] [CrossRef]
- Mestdagh, P.; Van Vlierberghe, P.; De Weer, A.; Muth, D.; Westermann, F.; Speleman, F.; Vandesompele, J. A Novel and Universal Method for MicroRNA RT-qPCR Data Normalization. Genome Biol. 2009, 10, R64. [Google Scholar] [CrossRef] [Green Version]
- Iwakawa, H.; Tomari, Y. The Functions of MicroRNAs: mRNA Decay and Translational Repression. Trends Cell Biol. 2015, 25, 651–665. [Google Scholar] [CrossRef]
- Changkwian, A.; Venkatesh, J.; Lee, J.H.; Han, J.W.; Kwon, J.K.; Siddique, M.I.; Solomon, A.M.; Choi, G.J.; Kim, E.; Seo, Y.; et al. Physical Localization of the Root-Knot Nematode (Meloidogyne incognita) Resistance Locus Me7 in Pepper (Capsicum annuum). Front. Plant Sci. 2019, 10, 886. [Google Scholar] [CrossRef]
- Pineda, A.; Zheng, S.J.; van Loon, J.J.A.; Dicke, M. Rhizobacteria Modify Plant-Aphid Interactions: A Case of Induced Systemic Susceptibility: Interactions between Rhizobacteria and Aphids. Plant Biol. 2012, 14, 83–90. [Google Scholar] [CrossRef]
- Toyota, M.; Spencer, D.; Sawai-Toyota, S.; Wang, J.; Zhang, T.; Koo, A.J.; Howe, G.A.; Gilroy, S. Glutamate Triggers Long-Distance, Calcium-Based Plant Defense Signaling. Science 2018, 361, 1112–1115. [Google Scholar] [CrossRef]
- Engelhardt, S.; Boevink, P.C.; Armstrong, M.R.; Ramos, M.B.; Hein, I.; Birch, P.R.J. Relocalization of Late Blight Resistance Protein R3a to Endosomal Compartments Is Associated with Effector Recognition and Required for the Immune Response. Plant Cell 2013, 24, 5142–5158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Ooijen, G.; Mayr, G.; Kasiem, M.M.A.; Albrecht, M.; Cornelissen, B.J.C.; Takken, F.L.W. Structure-Function Analysis of the NB-ARC Domain of Plant Disease Resistance Proteins. J. Exp. Bot. 2008, 59, 1383–1397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, J.M.; Zhang, Y. Plant Immunity: Danger Perception and Signaling. Cell 2020, 181, 978–989. [Google Scholar] [CrossRef] [PubMed]
- Jones, J.D.G.; Dangl, J.L. The Plant Immune System. Nature 2006, 444, 323–329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Vries, S.; Kloesges, T.; Rose, L.E. Evolutionarily Dynamic, but Robust, Targeting of Resistance Genes by the MiR482/2118 Gene Family in the Solanaceae. Genome Biol. Evol. 2015, 7, 3307–3321. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Xia, R.; Kuang, H.; Meyers, B.C. The Diversification of Plant NBS-LRR Defense Genes Directs the Evolution of MicroRNAs That Target Them. Mol. Biol. Evol. 2016, 33, 2692–2705. [Google Scholar] [CrossRef] [Green Version]
- Li, F.; Pignatta, D.; Bendix, C.; Brunkard, J.O.; Cohn, M.M.; Tung, J.; Sun, H.; Kumar, P.; Baker, B. MicroRNA Regulation of Plant Innate Immune Receptors. Proc. Natl. Acad. Sci. USA 2012, 109, 1790–1795. [Google Scholar] [CrossRef] [Green Version]
- Ouyang, S.; Park, G.; Atamian, H.S.; Han, C.S.; Stajich, J.E.; Kaloshian, I.; Borkovich, K.A. MicroRNAs Suppress NB Domain Genes in Tomato That Confer Resistance to Fusarium oxysporum. PLoS Pathog. 2014, 10, e1004464. [Google Scholar] [CrossRef]
- Shivaprasad, P.V.; Chen, H.-M.; Patel, K.; Bond, D.M.; Santos, B.A.C.M.; Baulcombe, D.C. A MicroRNA Superfamily Regulates Nucleotide Binding Site-Leucine-Rich Repeats and Other MRNAs. Plant Cell 2012, 24, 859–874. [Google Scholar] [CrossRef] [Green Version]
- Züst, T.; Agrawal, A.A. Mechanisms and Evolution of Plant Resistance to Aphids. Nat. Plants 2016, 2, 15206. [Google Scholar] [CrossRef]
- Kaloshian, I. Gene-for-gene Disease Resistance: Bridging Insect Pest and Pathogen Defense. J. Chem. Ecol. 2004, 30, 2419–2438. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Guo, G.; Wang, Z.; Ji, H.; Mu, F.; Li, X. Auxin in Plant Growth and Stress Responses. In Phytohormones: A Window to Metabolism, Signaling and Biotechnological Applications; Tran, L.S.P., Pal, S., Eds.; Springer: New York, NY, USA, 2014; pp. 1–35. ISBN 978-1-4939-0490-7. [Google Scholar]
- Shi, H.T.; Chen, L.; Ye, T.T.; Liu, X.D.; Ding, K.J.; Chan, Z.L. Modulation of auxin content in Arabidopsis confers improved drought stress resistance. Plant Physiol. Biochem. 2014, 82, 209–217. [Google Scholar] [CrossRef]
- Ribba, T.; Garrido-Vargas, F.; O’Brien, J.A. Auxin-Mediated Responses under Salt Stress: From Developmental Regulation to Biotechnological Applications. J. Exp. Bot. 2020, 71, 3843–3853. [Google Scholar] [CrossRef] [PubMed]
- Navarro, L.; Dunoyer, P.; Jay, F.; Arnold, B.; Dharmasiri, N.; Estelle, M.; Voinnet, O.; Jones, J.D.G. A Plant miRNA Contributes to Antibacterial Resistance by Repressing Auxin Signaling. Science 2006, 312, 436–439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sunkar, R.; Li, Y.F.; Jagadeeswaran, G. Functions of MicroRNAs in Plant Stress Responses. Trends Plant Sci. 2012, 17, 196–203. [Google Scholar] [CrossRef] [PubMed]
- Fahlgren, N.; Howell, M.D.; Kasschau, K.D.; Chapman, E.J.; Sullivan, C.M.; Cumbie, J.S.; Givan, S.A.; Law, T.F.; Grant, S.R.; Dangl, J.L.; et al. High-Throughput Sequencing of Arabidopsis MicroRNAs: Evidence for Frequent Birth and Death of MIRNA Genes. PLoS ONE 2007, 2, e219. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.J.; Liu, L.; Cheng, C.H.; Ren, Z.Y.; Xu, S.M.; Li, X. GAI Functions in the Plant Response to Dehydration Stress in Arabidopsis thaliana. Int. J. Mol. Sci. 2020, 21, 819. [Google Scholar] [CrossRef] [Green Version]
- Ma, Z.; Hu, X.; Cai, W.; Huang, W.; Zhou, X.; Luo, Q.; Yang, H.; Wang, J.; Huang, J. Arabidopsis MiR171-Targeted Scarecrow-Like Proteins Bind to GT Cis-Elements and Mediate Gibberellin-Regulated Chlorophyll Biosynthesis under Light Conditions. PLoS Genet. 2014, 10, e1004519. [Google Scholar] [CrossRef] [Green Version]
- Chinchilla, D.; Shan, L.; He, P.; de Vries, S.; Kemmerling, B. One for All: The Receptor-Associated Kinase BAK1. Trends Plant Sci. 2009, 14, 535–541. [Google Scholar] [CrossRef] [Green Version]
- Wong, J.; Gao, L.; Yang, Y.; Zhai, J.; Arikit, S.; Yu, Y.; Duan, S.; Chan, V.; Xiong, Q.; Yan, J.; et al. Roles of Small RNAs in Soybean Defense against Phytophthora sojae Infection. Plant J. 2014, 79, 928–940. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.; Zhao, Y.L.; Zhao, J.-H.; Wang, S.; Jin, Y.; Chen, Z.Q.; Fang, Y.Y.; Hua, C.L.; Ding, S.W.; Guo, H.S. Cotton Plants Export MicroRNAs to Inhibit Virulence Gene Expression in a Fungal Pathogen. Nat. Plants 2016, 2, 16153. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Hou, Y.; Chen, W.; Wang, S.; Wang, P.; Qu, S. Malus hupehensis MiR168 Targets to ARGONAUTE1 and Contributes to the Resistance against Botryosphaeria dothidea Infection by Altering Defense Responses. Plant Cell Physiol. 2017, 58, 1541–1557. [Google Scholar] [CrossRef] [PubMed]
- Xiao, L.; Jiang, S.; Huang, P.; Chen, F.; Wang, X.; Cheng, Z.; Miao, Y.; Liu, L.; Searle, I.; Liu, C.; et al. Two Nucleoporin98 Homologous Genes Jointly Participate in the Regulation of Starch Degradation to Repress Senescence in Arabidopsis. BMC Plant Biol. 2020, 20, 292. [Google Scholar] [CrossRef] [PubMed]
- Bouvier d’Yvoire, M.; Bouchabke-Coussa, O.; Voorend, W.; Antelme, S.; Cézard, L.; Legée, F.; Lebris, P.; Legay, S.; Whitehead, C.; McQueen-Mason, S.J.; et al. Disrupting the Cinnamyl Alcohol Dehydrogenase 1 Gene (BdCAD1) Leads to Altered Lignification and Improved Saccharification in Brachypodium distachyon. Plant J. 2013, 73, 496–508. [Google Scholar] [CrossRef]
- Ning, J.; Li, X.; Hicks, L.M.; Xiong, L. A Raf-Like MAPKKK Gene DSM1 Mediates Drought Resistance through Reactive Oxygen Species Scavenging in Rice. Plant Physiol. 2010, 152, 876–890. [Google Scholar] [CrossRef] [Green Version]
- Jin, H.; Axtell, M.J.; Dahlbeck, D.; Ekwenna, O.; Zhang, S.; Staskawicz, B.; Baker, B. NPK1, an MEKK1-like Mitogen-Activated Protein Kinase Kinase Kinase, Regulates Innate Immunity and Development in Plants. Dev. Cell 2002, 3, 291–297. [Google Scholar] [CrossRef] [Green Version]
- Cao, J.; Cheng, G.; Wang, L.; Maimaitijiang, T.; Lan, H. Genome-Wide Identification and Analysis of the Phosphoenolpyruvate Carboxylase Gene Family in Suaeda aralocaspica, an Annual Halophyte with Single-Cellular C4 Anatomy. Front. Plant Sci. 2021, 12, 665279. [Google Scholar] [CrossRef]
- Lin, Z.; Alexander, L.; Hackett, R.; Grierson, D. LeCTR2, a CTR1-like Protein Kinase from Tomato, Plays a Role in Ethylene Signalling, Development and Defence. Plant J. 2008, 54, 1083–1093. [Google Scholar] [CrossRef] [Green Version]
- Luziatelli, F.; Gatti, L.; Ficca, A.G.; Medori, G.; Silvestri, C.; Melini, F.; Muleo, R.; Ruzzi, M. Metabolites Secreted by a Plant-Growth-Promoting Pantoea Agglomerans Strain Improved Rooting of Pyrus communis L. Cv Dar Gazi Cuttings. Front. Microbiol. 2020, 11, 539359. [Google Scholar] [CrossRef]
- Chakrabarti, M.; Hunt, A. CPSF30 at the Interface of Alternative Polyadenylation and Cellular Signaling in Plants. Biomolecules 2015, 5, 1151–1168. [Google Scholar] [CrossRef]
- Furniss, J.J.; Grey, H.; Wang, Z.; Nomoto, M.; Jackson, L.; Tada, Y.; Spoel, S.H. Proteasome-Associated HECT-Type Ubiquitin Ligase Activity Is Required for Plant Immunity. PLoS Pathog. 2018, 14, e1007447. [Google Scholar] [CrossRef] [PubMed]
- Pysh, L.D.; Wysocka-Diller, J.W.; Camilleri, C.; Bouchez, D.; Benfey, P.N. The GRAS Gene Family in Arabidopsis: Sequence Characterization and Basic Expression Analysis of the SCARECROW-LIKE Genes. Plant J. 1999, 18, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Lorang, J.M.; Sweat, T.A.; Wolpert, T.J. Plant Disease Susceptibility Conferred by a “Resistance” Gene. Proc. Natl. Acad. Sci. USA 2007, 104, 14861–14866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mao, Y.B.; Liu, Y.Q.; Chen, D.Y.; Chen, F.Y.; Fang, X.; Hong, G.J.; Wang, L.J.; Wang, J.W.; Chen, X.Y. Jasmonate Response Decay and Defense Metabolite Accumulation Contributes to Age-Regulated Dynamics of Plant Insect Resistance. Nat. Commun. 2017, 8, 13925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fleet, C.M.; Sun, T. A DELLAcate Balance: The Role of Gibberellin in Plant Morphogenesis. Curr. Opin. Plant Biol. 2005, 8, 77–85. [Google Scholar] [CrossRef]
- Omidbakhshfard, M.A.; Proost, S.; Fujikura, U.; Mueller-Roeber, B. Growth-Regulating Factors (GRFs): A Small Transcription Factor Family with Important Functions in Plant Biology. Mol. Plant 2015, 8, 998–1010. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Read Data | Control_1 | Control_2 | Control_3 | Infested_1 | Infested_2 | Infested_3 |
|---|---|---|---|---|---|---|
| Raw reads | 16,690,408 | 18,356,133 | 16,318,533 | 17,874,836 | 16,381,518 | 17,070,653 |
| Total reads (18–25 nt) | 2,767,459 | 3,214,769 | 1,780,433 | 2,762,136 | 1,712,723 | 2,467,299 |
| Unique reads (18–25 nt) | 907,674 | 1,004,236 | 892,304 | 1,242,553 | 977,307 | 639,789 |
| Valid total reads | 11,715,643 | 12,977,086 | 12,792,819 | 13,198,805 | 12,990,661 | 12,890,987 |
| Valid unique reads | 3,953,590 | 4,662,695 | 4,912,871 | 4,831,961 | 5,153,909 | 4,923,357 |
| miRNA Name | miRNA Sequence | Up/Down | Expression Level | Fold Change | p-Value | Number of Target Genes | |
|---|---|---|---|---|---|---|---|
| Control | Infested | ||||||
| fve-MIR3627b-p5_2ss7AT22AT | TGTCGCTGGAGAGATGGCACTT | up | 41 ± 3 | 117 ± 10 | 2.81 | 0.0036 | 0 |
| gma-MIR171g-p3 | TTGAGCCGTGCCAATATCATA | up | 18 ± 2 | 29 ± 3 | 1.59 | 0.0090 | 15 |
| hbr-MIR6173-p5_2ss17AG18GC | TACCCCAGTAGTCCTAGC | down | 15 ± 3 | 6 ± 2 | 0.43 | 0.0171 | 45 |
| nta-MIR6151g-p3 | AATCCGAGCCCCACATTCATC | up | 176 ± 16 | 224 ± 15 | 1.27 | 0.0193 | 5 |
| nta-MIR6151h-p3 | AATCCGAGCCCCACATTCATC | up | 176 ± 16 | 224 ± 15 | 1.27 | 0.0193 | 5 |
| nta-MIR6151e-p3 | AATCCGAGCCCCACATTCATC | up | 176 ± 16 | 224 ± 15 | 1.27 | 0.0193 | 5 |
| nta-MIR6151f-p3 | AATCCGAGCCCCACATTCATC | up | 176 ± 16 | 224 ± 15 | 1.27 | 0.0193 | 5 |
| nta-MIR6151i-p3 | AATCCGAGCCCCACATTCATC | up | 176 ± 16 | 224 ± 15 | 1.27 | 0.0193 | 5 |
| nta-MIR6151d-p3 | AATCCGAGCCCCACATTCATC | up | 176 ± 16 | 224 ± 15 | 1.27 | 0.0193 | 5 |
| nta-miR6159 | TAGCATAGAATTCTCGCACCTA | up | 1382 ± 112 | 1892 ± 180 | 1.37 | 0.0203 | 1 |
| stu-miR8021_1ss9CA | ATTCAAGGATCAAACTCGAGACCT | down | 9 ± 1 | 5 ± 1 | 0.57 | 0.0211 | 0 |
| nta-miR6025b | TGCCAACTATTGAGATGACATC | up | 2468 ± 258 | 3507 ± 375 | 1.42 | 0.0211 | 27 |
| nta-MIR6161c-p3_1ss7AG | GCACCTGTGTATGAACTTCCAGCA | up | 681 ± 124 | 1047 ± 122 | 1.54 | 0.0220 | 4 |
| bra-miR168a-5p_L+1 | CTCGCTTGGTGCAGGTCGGGAA | up | 2 ± 2 | 9 ± 1 | 3.79 | 0.0232 | 0 |
| mtr-MIR2603-p5_2ss9AC17AC | GTCCCTGCCCTTTGTACA | down | 10 ± 2 | 4 ± 2 | 0.42 | 0.0240 | 86 |
| mtr-MIR2603-p3_2ss9AC17AC | GTCCCTGCCCTTTGTACA | down | 10 ± 2 | 4 ± 2 | 0.42 | 0.0240 | 86 |
| hbr-MIR6173-p3_2ss18GA19CG | CGTAAACGATGGATACTAG | down | 7 ± 2 | 0 ± 0 | -inf | 0.0241 | 19 |
| ath-miR8175_L+4 | GTTCGATCCCCGGCAACGGCGCCA | up | 5 ± 1 | 9 ± 2 | 1.92 | 0.0311 | 5 |
| osa-MIR5825-p5_2ss15TC20AC | TTATTATTGTTTTCCACAACC | down | 9 ± 1 | 6 ± 1 | 0.67 | 0.0353 | 25 |
| csi-miR160c-5p_R+1 | TGCCTGGCTCCCTGTATGCTTT | up | 9 ± 2 | 16 ± 3 | 1.75 | 0.0375 | 33 |
| gma-MIR4995-p3 | CATAGGCAGTGGCTTGGTT | down | 11 ± 2 | 5 ± 2 | 0.49 | 0.0383 | 48 |
| gma-MIR9724-p5 | ACAATCCTCACCTCAAAAGCTAGC | up | 1 ± 1 | 4 ± 1 | 4.93 | 0.0386 | 5 |
| stu-miR166b_1ss4GA | TCGAACCAGGCTTCATTCCTC | up | 6 ± 1 | 9 ± 1 | 1.52 | 0.0450 | 1 |
| stu-miR391-5p_L-1R+2 | ACGCAGGAGAGATGATGCTGGA | down | 371 ± 53 | 249 ± 19 | 0.67 | 0.0456 | 0 |
| csi-miR160c-5p_R+1_1ss19CT | TGCCTGGCTCCCTGTATGTTTT | up | 11 ± 5 | 23 ± 6 | 2.17 | 0.0466 | 31 |
| ath-miR396b-5p_R+1 | TTCCACAGCTTTCTTGAACTTT | down | 157 ± 9 | 135 ± 10 | 0.86 | 0.0477 | 40 |
| sly-miR396b_R+1 | TTCCACAGCTTTCTTGAACTTT | down | 157 ± 9 | 135 ± 10 | 0.86 | 0.0477 | 40 |
| nta-MIR6155-p3_2ss10CT21CT | ATTCGAGAGTAAGGCTACCTTATG | up | 103 ± 20 | 145 ± 11 | 1.40 | 0.0495 | 0 |
| miRNA Name | miRNA Sequence | Up/Down | Expression Level | Fold Change | p-Value | Number of Target Genes | |
|---|---|---|---|---|---|---|---|
| Control | Infested | ||||||
| PC-5p-244240_18 | AAACCCGCTCCCGTCACTTTAGTT | up | 0 ± 0 | 6 ± 0 | inf | 0.0001 | 2 |
| PC-5p-50806_114 | TTTTCGATATCGCTGGCCTCC | down | 29 ± 2 | 14 ± 4 | 0.48 | 0.0062 | 4 |
| PC-5p-181486_27 | ACCCATTGTGGAGTTGTTGGGCTA | down | 7 ± 1 | 0 ± 0 | -inf | 0.0073 | 0 |
| PC-3p-258573_16 | AATGTCGTGTCCTAAAGTTTGAGC | down | 5 ± 1 | 0 ± 0 | -inf | 0.0082 | 0 |
| PC-3p-61258_95 | TTTACTTCCCACCGCTTAGCA | up | 11 ± 1 | 19 ± 2 | 1.67 | 0.0109 | 0 |
| PC-5p-135574_40 | TCGCCTGATAATGCTCTTAAA | down | 13 ± 3 | 0 ± 0 | -inf | 0.0156 | 1 |
| PC-3p-130469_42 | AGCACCTGTGTATGAACTTCTAGT | up | 2 ± 3 | 12 ± 3 | 6.15 | 0.0176 | 5 |
| PC-3p-171618_30 | TCTTCCATGATACACATATTA | down | 17 ± 2 | 7 ± 3 | 0.42 | 0.0195 | 49 |
| PC-3p-202629_23 | TTTTCTTGAGGCTGTTAGGGATGT | up | 0 ± 0 | 8 ± 2 | inf | 0.0206 | 3 |
| PC-5p-194313_25 | AAAAGATTTTGAACCTCCTTGACC | up | 6 ± 7 | 26 ± 6 | 4.21 | 0.0211 | 7 |
| PC-3p-113881_50 | AATTAATGTCAGTTGGGTGAGGCA | down | 16 ± 4 | 5 ± 4 | 0.30 | 0.0323 | 1 |
| PC-5p-67924_86 | AGGGCTGCTATTTAGAGATTAGTC | up | 6 ± 1 | 8 ± 1 | 1.43 | 0.0334 | 4 |
| PC-3p-127236_43 | ATGCCTCATACAACTAGTGTAAGT | up | 2 ± 3 | 12 ± 0 | 6.03 | 0.0361 | 3 |
| PC-5p-10208_389 | TGTATTCTTTCCGCTCAATTC | down | 75 ± 6 | 62 ± 5 | 0.83 | 0.0380 | 3 |
| PC-3p-258597_16 | GTATCCTGCATCTTCTCTTTC | up | 0 ± 0 | 3 ± 1 | inf | 0.0404 | 43 |
| PC-3p-114443_49 | ATTTCTGGAGAATCCGACACGAGT | up | 4 ± 3 | 12 ± 4 | 3.37 | 0.0421 | 5 |
| PC-3p-146848_36 | AGCGTATTATGTTAGAACTCCAGC | up | 2 ± 3 | 10 ± 3 | 4.89 | 0.0428 | 0 |
| PC-5p-216933_21 | TAGTTTCGCCCCTAGAGCATA | up | 0 ± 0 | 7 ± 3 | inf | 0.0464 | 3 |
| PC-5p-298501_13 | TGGCCCGTCAACATCATGTTC | up | 2 ± 3 | 8 ± 2 | 4.61 | 0.0490 | 2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Han, W.-H.; Wang, J.-X.; Zhang, F.-B.; Liu, Y.-X.; Wu, H.; Wang, X.-W. Small RNA and Degradome Sequencing Reveal Important MicroRNA Function in Nicotiana tabacum Response to Bemisia tabaci. Genes 2022, 13, 361. https://doi.org/10.3390/genes13020361
Han W-H, Wang J-X, Zhang F-B, Liu Y-X, Wu H, Wang X-W. Small RNA and Degradome Sequencing Reveal Important MicroRNA Function in Nicotiana tabacum Response to Bemisia tabaci. Genes. 2022; 13(2):361. https://doi.org/10.3390/genes13020361
Chicago/Turabian StyleHan, Wen-Hao, Jun-Xia Wang, Feng-Bin Zhang, Yu-Xiao Liu, He Wu, and Xiao-Wei Wang. 2022. "Small RNA and Degradome Sequencing Reveal Important MicroRNA Function in Nicotiana tabacum Response to Bemisia tabaci" Genes 13, no. 2: 361. https://doi.org/10.3390/genes13020361
APA StyleHan, W.-H., Wang, J.-X., Zhang, F.-B., Liu, Y.-X., Wu, H., & Wang, X.-W. (2022). Small RNA and Degradome Sequencing Reveal Important MicroRNA Function in Nicotiana tabacum Response to Bemisia tabaci. Genes, 13(2), 361. https://doi.org/10.3390/genes13020361