
Genotype-Phenotype Correlation in Familial BAG3 Mutation Dilated Cardiomyopathy

Abstract
:1. Introduction
2. Case Presentation
2.1. Clinical Presentation
2.2. Genetic Test
3. Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dominguez, F.; Cuenca, S.; Bilinska, Z.; Toro, R.; Villard, E.; Vila-Barriales, R.; Ochoa, J.P.; Asselbergs, F.; Sammani, A.; Franaszczyj, M.; et al. Dilated Cardiomyopathy Due to BLC2-Associated Athanogene 3 (BAG3) Mutations. JACC 2018, 72, 20. [Google Scholar] [CrossRef]
- Al-Khatib, S.M.; Stevenson, W.G.; Ackerman, M.J.; Bryant, W.J.; Callans, D.J.; Curtis, A.B.; Deal, B.J.; Dickfeld, T.; Field, M.E.; Fonarow, G.C.; et al. 2017 AHA/ACC/HRS guideline for management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: A report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines and the Heart Rhythm Society. Circulation 2018, 138, 13. [Google Scholar]
- Orphanou, N.; Papatheodorou, E.; Anastasakis, A.A. Dilated cardiomyopathy in the era of precision medicine: Latest concepts and developments. Heart Fail Rev. 2021, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Monda, E.; Palmiero, G.; Rubino, M.; Verrillo, F.; Amodio, F.; Di Fraia, F.; Pacileo, R.; Fimiani, F.; Esposito, A.; Cirillo, A.; et al. Molecular Basis of Inflammation in the Pathogenesis of Cardiomyopathies. Int. J. Mol. Sci. 2020, 21, 6462. [Google Scholar] [CrossRef] [PubMed]
- Japp, A.G.; Gulati, A.; Cook, S.A.; Cowie, M.R.; Prasad, S.K. The Diagnosis and Evaluation of Dilated Cardiomyopathy. J. Am. Coll Cardiol. 2016, 67, 2996–3010. [Google Scholar] [CrossRef]
- Reichart, D.; Magnussen, C.; Zeller, T.; Blankenberg, S. Dilated cardiomyopathy: From epidemiologic to genetic phenotypes. Intern. Med. 2019, 286, 362–372. [Google Scholar] [CrossRef] [Green Version]
- Seferović, P.M.; Polovina, M.; Bauersachs, J.; Arad, M.; Gal, T.B.; Lund, L.H.; Felix, S.B.; Arbustini, E.; Caforio, A.L.P.; Farmakis, D.; et al. Heart failure in cardiomyopathies: A position paper from the Heart Failure Association of the European Society of Cardiology. Eur. J. Heart Fail. 2019, 21, 553–576. [Google Scholar] [CrossRef] [Green Version]
- Peña-Peña, M.L.; Monserrat, L. Risk Stratification in Patients with Nonisquemic Dilated Cardiomyopathy. The Role of Genetic Testing. Rev. Esp. Cardiol. 2019, 72, 4. [Google Scholar] [CrossRef] [PubMed]
- Mason, S.; Quinn, E.; Halliday, B.P. The promise and challenges of precision medicine in dilated cardiomyopathy. Eur. Heart J. -Case Rep. 2021, 5, ytab391. [Google Scholar] [CrossRef] [PubMed]
- Parvari, R.; Levitas, A. The Mutations Associated with Dilated Cardiomyopathy. Int. J. Biochem Res. Rev. 2012, 12, 2012. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Sun, K.; Zhang, X.; Tang, Y.; Xu, D. Advances in the role and mechanism of BAG3 in dilated cardiomyopathy. Heart Fail Rev. 2021, 26, 183–194. [Google Scholar] [CrossRef] [PubMed]
- Martin, T.G.; Myers, V.D.; Dubey, P.; Dubey, S.; Perez, E.; Moravec, C.S.; Willis, M.S.; Feldman, A.M.; Kirk, J.A. Cardiomyocyte contractile impairment in heart failure results from reduced BAG3-mediated sarcomere protein turnover. Nat. Commun. 2021, 12, 2942. [Google Scholar] [CrossRef] [PubMed]
- Shaw, R.M.; Nikolova, A.P. The Clinical Course of a Genetic Dilated Cardiomyopathy: Letting the Cat Out of the BAG3. J. Am. Coll Cardiol. 2018, 72, 2482–2484. [Google Scholar] [CrossRef]
- Martin, T.G.; Tawik, S.; Moravec, C.S.; Pak, T.R.; Kirk, J.A. BAG3 expression and sarcomere localization in the human heart are linked to HSF-1 and are differentially affected by sex and disease. Am. J. Physiol. Heart Circ. Physiol. 2021, 320, H2339–H2350. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Research | Description |
|---|---|
| Laboratory blood test | Within the normal range (creatinine 108 mcmol/L, K 5.5 mmol/L, Mg 0.85 mmol/L, RBC 5.16 × 10′12/L, WBC 9.74 × 10′9/L, HGB 160 g/L, PLT 274 × 10′9/L, BNP 4 ng/L). |
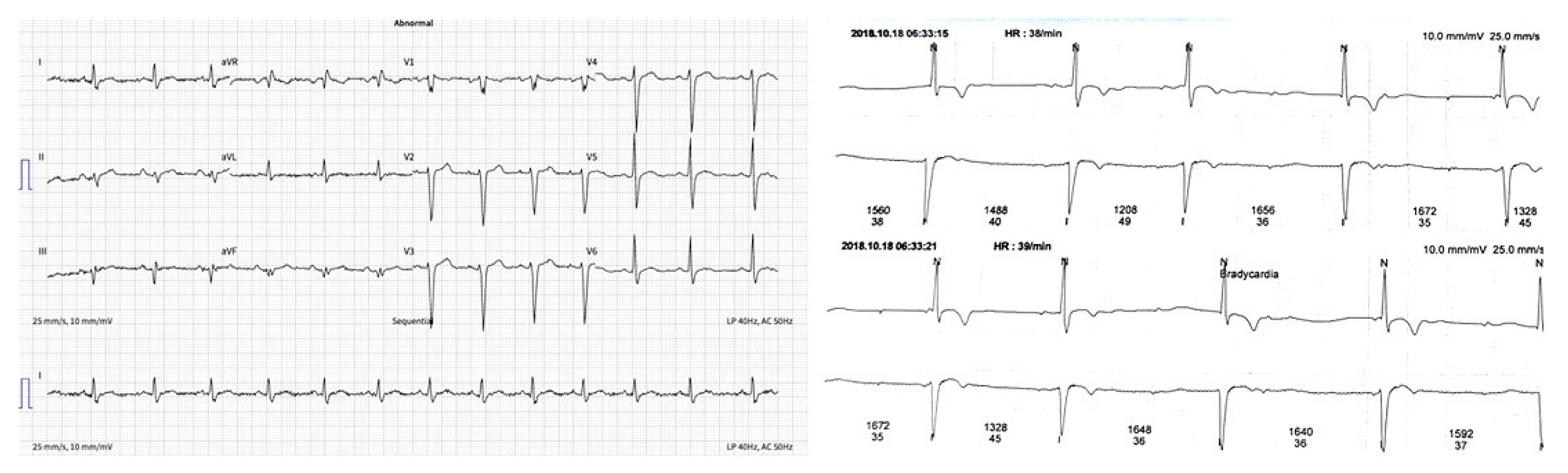
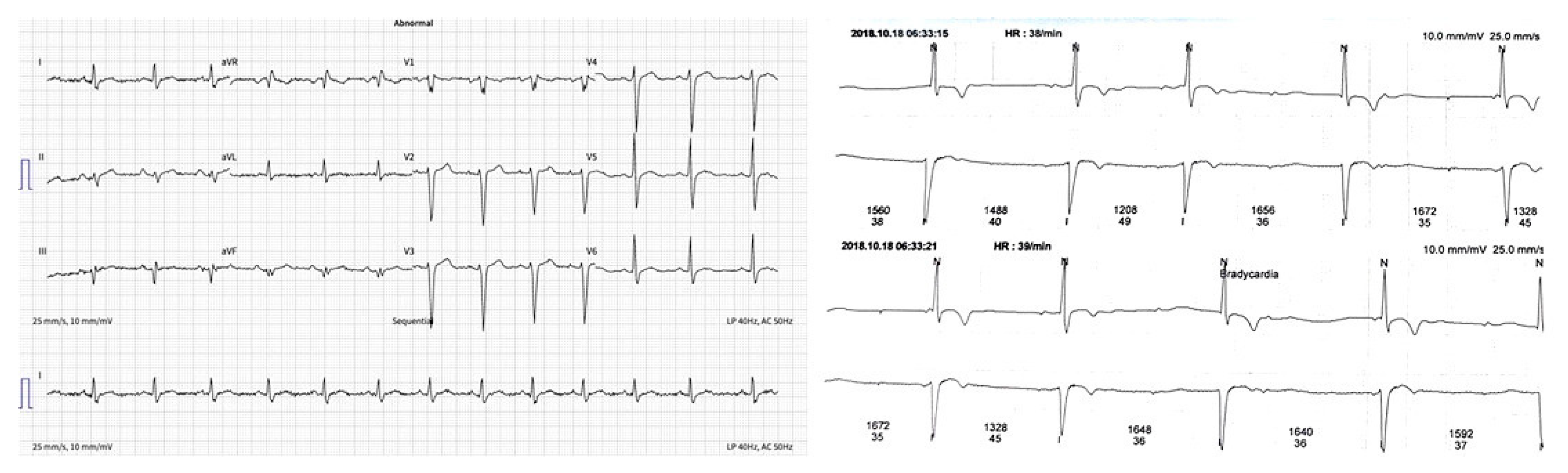
| ECG | Sinus rhythm, heart rate 80 bpm (Figure 1a). |
| Holter monitoring | Sinus rhythm, rare supraventricular extrasystoles, an intermittent second degree AV block, maximal R–R interval 1752 ms (Figure 1b). |
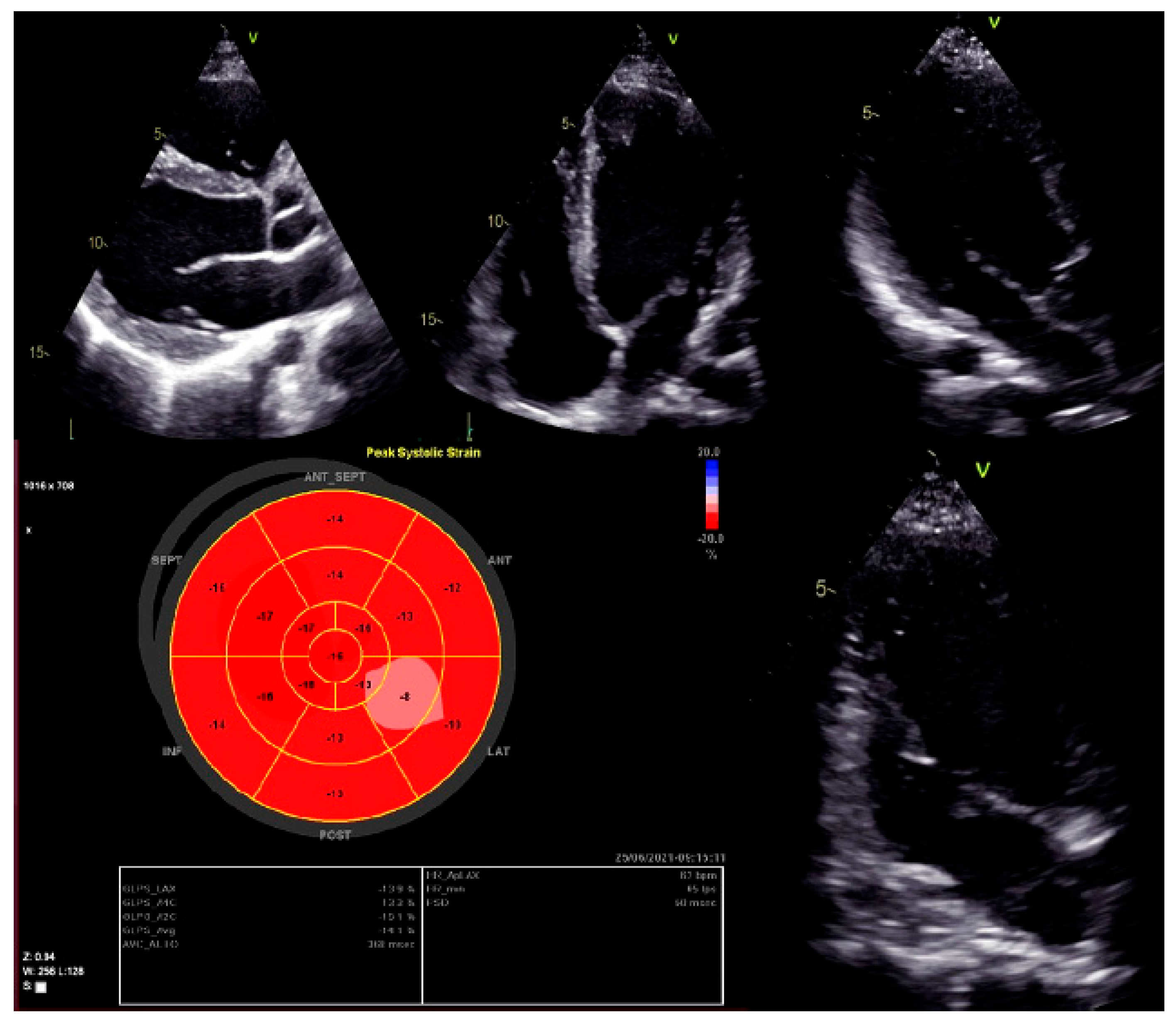
| 2D TTE echocardiography | LVEDD 58 mm (LVEDDi 30.69 mm/m2), LVEDV 133 mL (LVEDVi 70.37 mL/m2), LV EF 45%, LV longitudinal strain −12%. RV 38 mm, RA 34 mm, RV longitudinal strain −19.4 proc. Conclusions: normal Ao valve morphology and function. LA non-dilated, normal MV function. Dilated LV, decreased contractility function of the posterolateral LV wall and global longitudinal LV myocardial strain. Decreased systolic LV function. RV size and function within normal limits. |
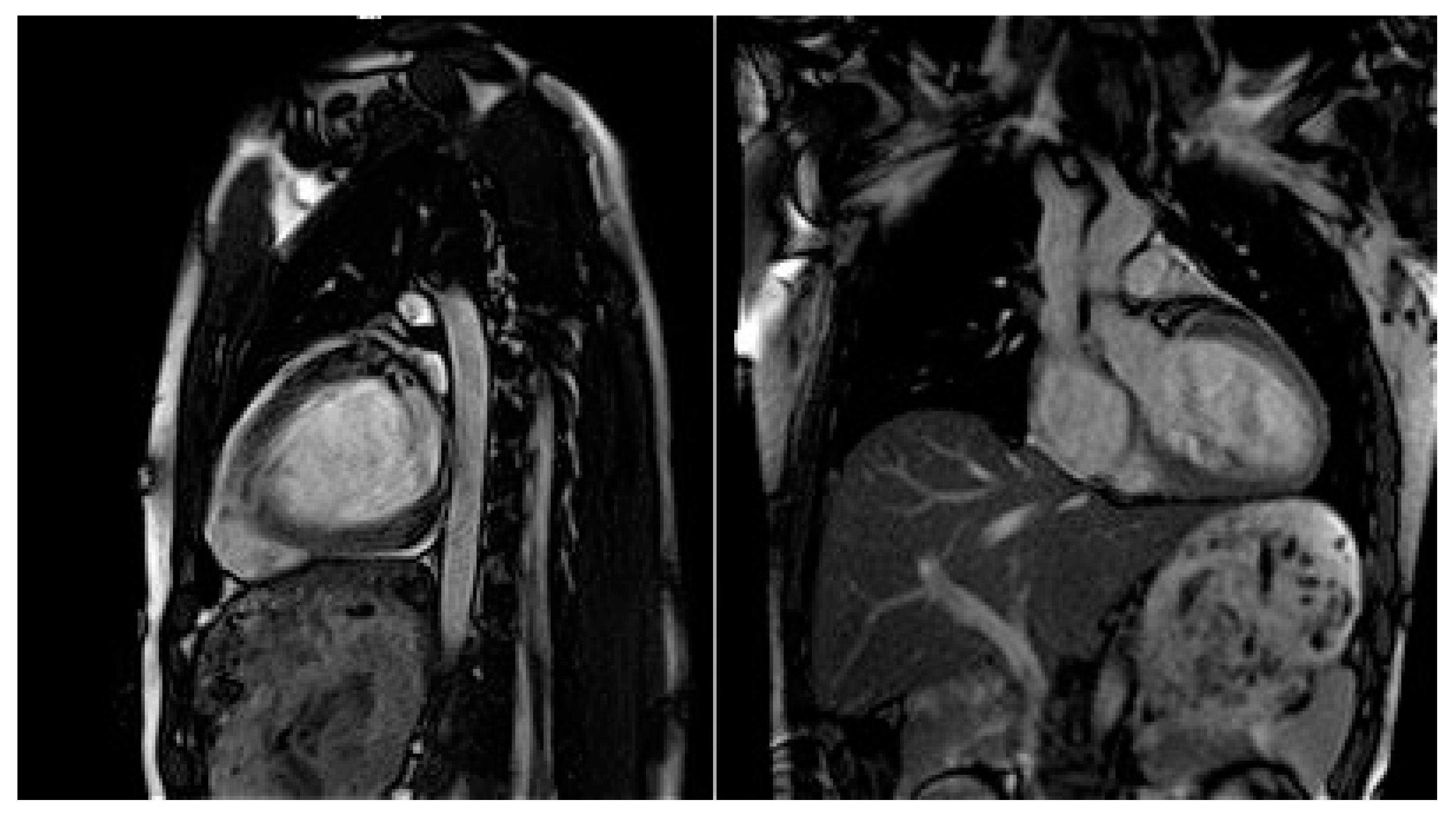
| Cardiac MRI | LVEDD 60 mm (LVEDDi 32.61 mm/m2), LVEDV 213 mL (LVEDVi 115.76 mL/m2), LV EF 43.2%. RV EF 50.8%. Conclusions: moderately dilated LV, decreased global systolic LV function. Preserved RV systolic function. |
| Parameters | Baseline | After 1 Year |
|---|---|---|
| BNP ng/L | 4 | 4 |
| 2D echocardiography | ||
| LVEDD (LVEDDi) | 58 mm (30.69 mm/m2) | 57 mm (30.89 mm/m2) |
| LVEDV (LVEDVi) | 133 mL (70.37 mL/m2) | 190 mL (103.26 mL/m2) |
| LVESV (LVESVi) | 73 mL (38.62 mL/m2) | 109 mL (59.23 mL/m2) |
| LV EF | 45% | 43% |
| LV longitudinal strain | −12% | −10% |
| RV | 38 mm | 38 mm |
| RA | 34 mm | 42 mm |
| RV longitudinal strain | −19.4% | −19% |
| Cardiac magnetic resonance | ||
| LVEDD (LVEDDi) | 61 mm (33.8 mm/m2) | 60 mm(32.61 mm/m2) |
| LVEDV (LVEDVi) | 213 mL (113 mL/m2) | 213 mL (115.76 mL/m2) |
| LVESV (LVESVi) | 124 mL (66 mL/m2) | 121 mL (65.76 mL/m2) |
| LVSV (LVSVi) | 89 mL (49.4 mL/m2) | 92 mL (50 mL/m2) |
| LAA LAAi | 20 cm3 (10.87 cm3) | 20 cm3 (10.87 cm3) |
| LV EF | 42% | 43.2% |
| RV EF | 50.8% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mėlinytė-Ankudavičė, K.; Šukys, M.; Plisienė, J.; Jurkevičius, R.; Ereminienė, E. Genotype-Phenotype Correlation in Familial BAG3 Mutation Dilated Cardiomyopathy. Genes 2022, 13, 363. https://doi.org/10.3390/genes13020363
Mėlinytė-Ankudavičė K, Šukys M, Plisienė J, Jurkevičius R, Ereminienė E. Genotype-Phenotype Correlation in Familial BAG3 Mutation Dilated Cardiomyopathy. Genes. 2022; 13(2):363. https://doi.org/10.3390/genes13020363
Chicago/Turabian StyleMėlinytė-Ankudavičė, Karolina, Marius Šukys, Jurgita Plisienė, Renaldas Jurkevičius, and Eglė Ereminienė. 2022. "Genotype-Phenotype Correlation in Familial BAG3 Mutation Dilated Cardiomyopathy" Genes 13, no. 2: 363. https://doi.org/10.3390/genes13020363
APA StyleMėlinytė-Ankudavičė, K., Šukys, M., Plisienė, J., Jurkevičius, R., & Ereminienė, E. (2022). Genotype-Phenotype Correlation in Familial BAG3 Mutation Dilated Cardiomyopathy. Genes, 13(2), 363. https://doi.org/10.3390/genes13020363