
A Whole-Genome Sequencing Study Implicates GRAMD1B in Multiple Sclerosis Susceptibility
 , , , , , , , , and add
Show full author list
, , , , , , , , and add
Show full author list
Abstract
1. Introduction
2. Materials and Methods
2.1. Ascertainment and Samples Collection
2.2. Genotyping and Linkage Analysis
2.3. Whole-Genome Sequencing (WGS)
2.4. Sanger and Targeted NGS Resequencing
2.5. Replication in a Canadian Cohort
2.6. Conservation Analysis
2.7. Gene Expression Profiling of Familial Subjects
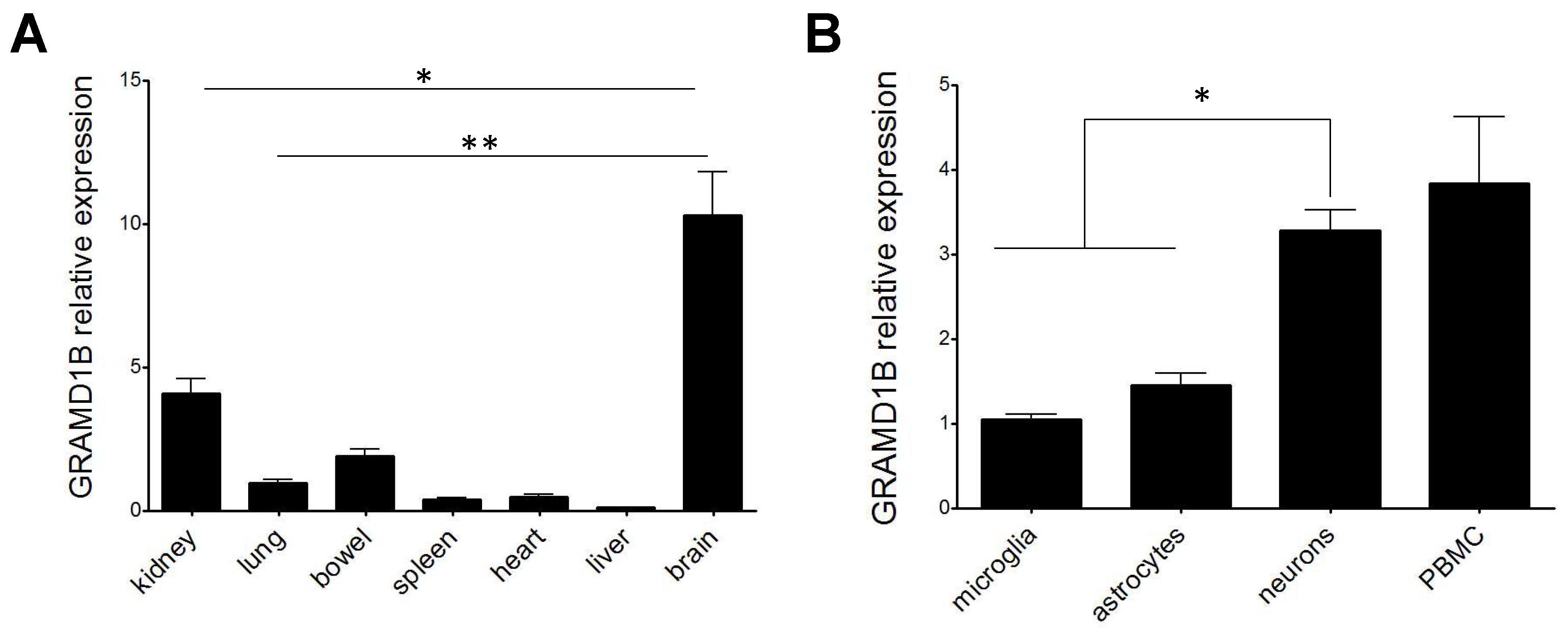
2.8. Assessment of GRAMD1B Expression in Rat Cells and Tissues
2.9. Construct Generation and Transfection
2.10. Reagents
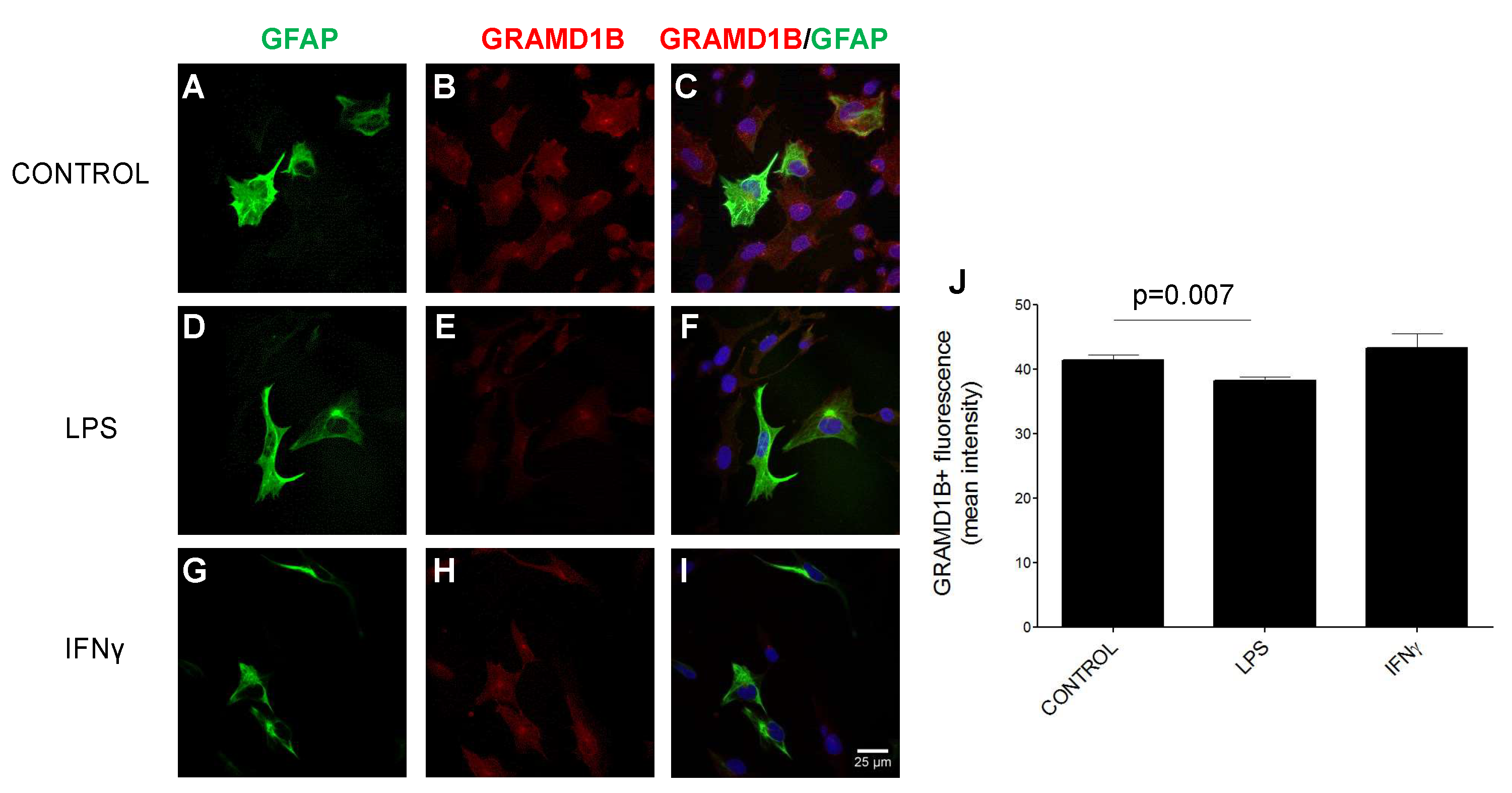
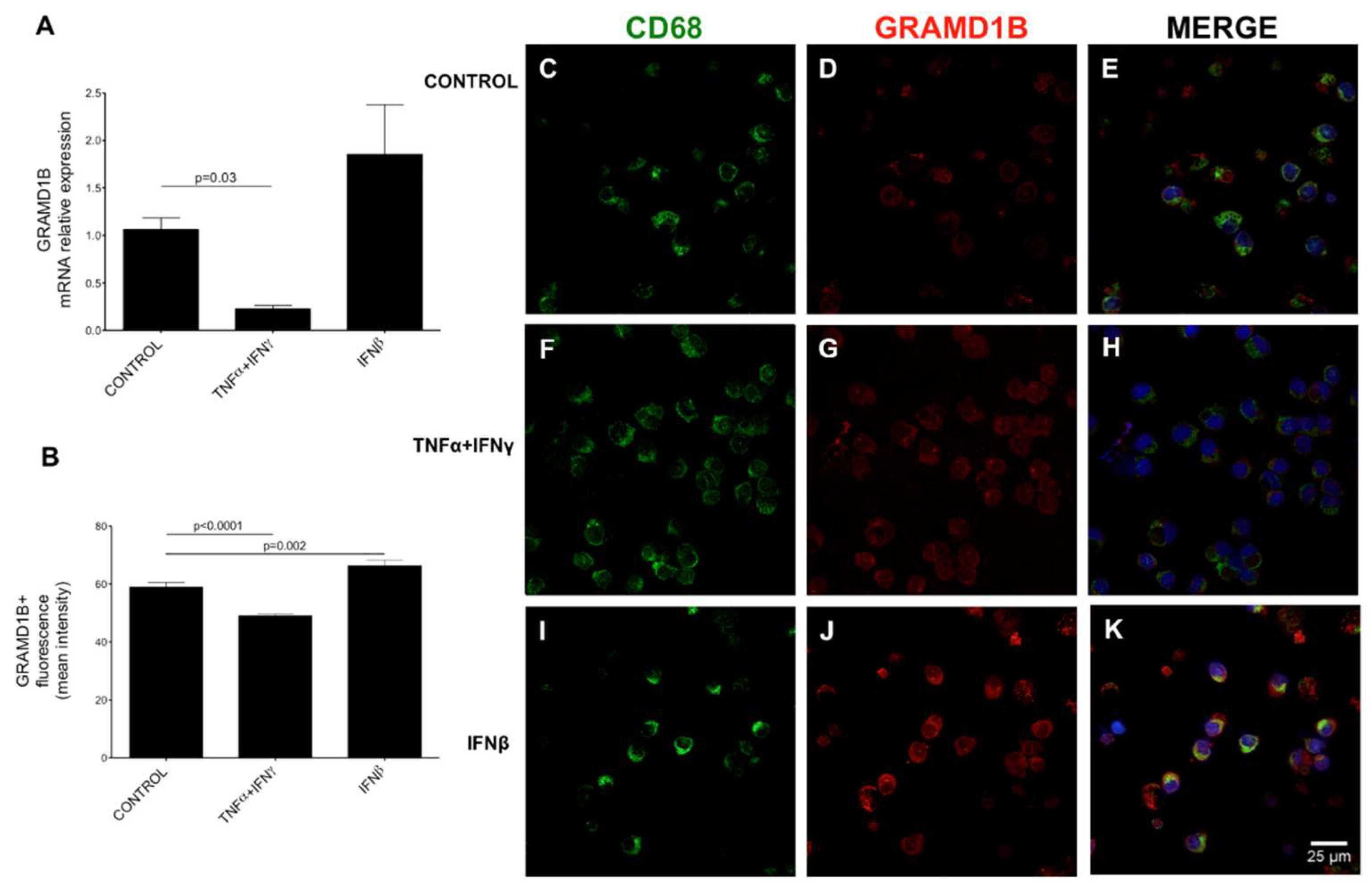
2.11. Cell Lines and Primary Cell Cultures, Stimulations, Immunocytochemistry Analyses and Assessment of GRAMD1B Expression
2.12. qRT-PCR Assays
2.13. Antibodies, Sources and Dilutions Used
2.14. Immunofluorescence Quantification and Statistical Analysis
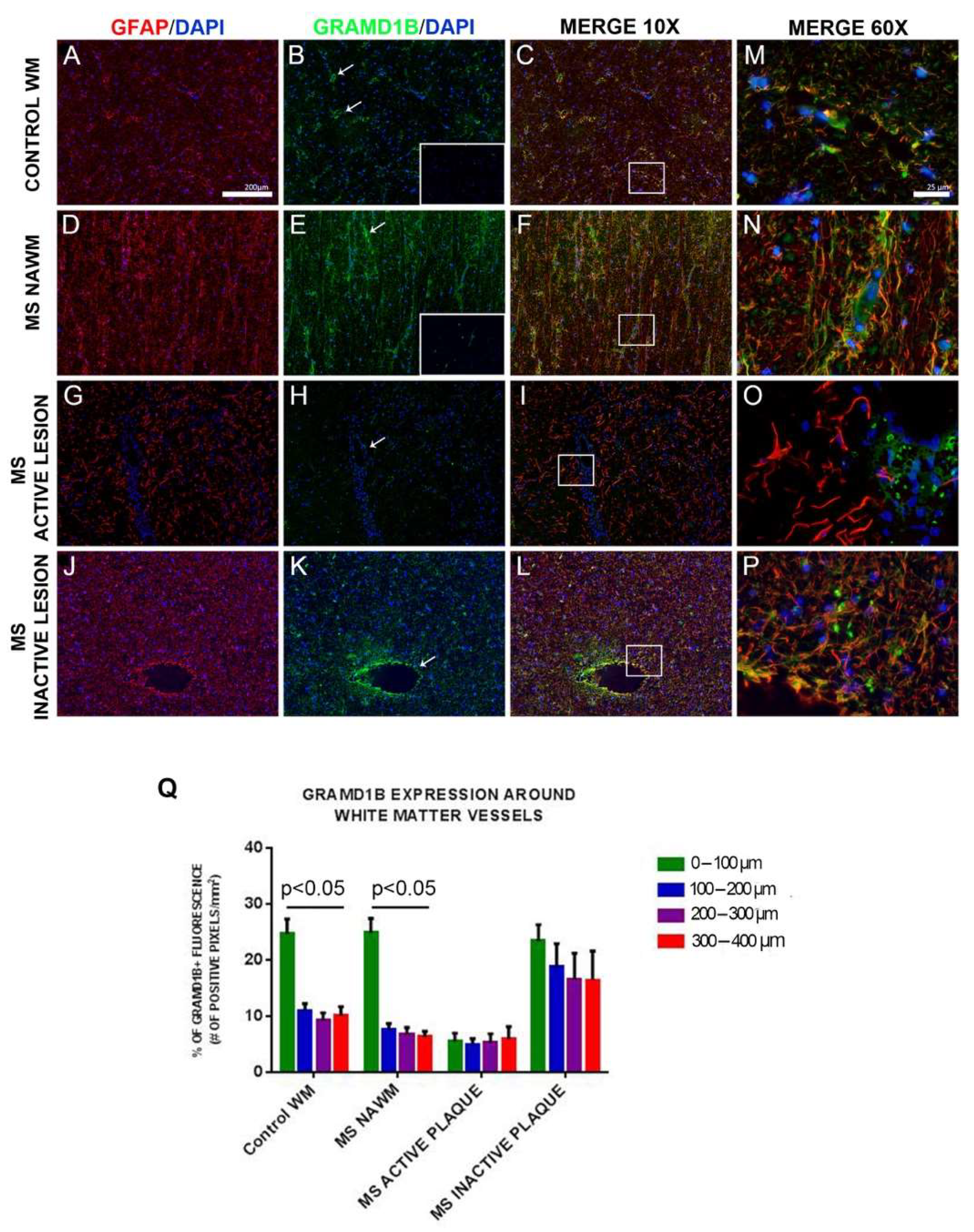
2.15. Human Brain Tissue Immunohistochemistry
2.16. Human Primary Cell Immunocytochemistry
3. Results
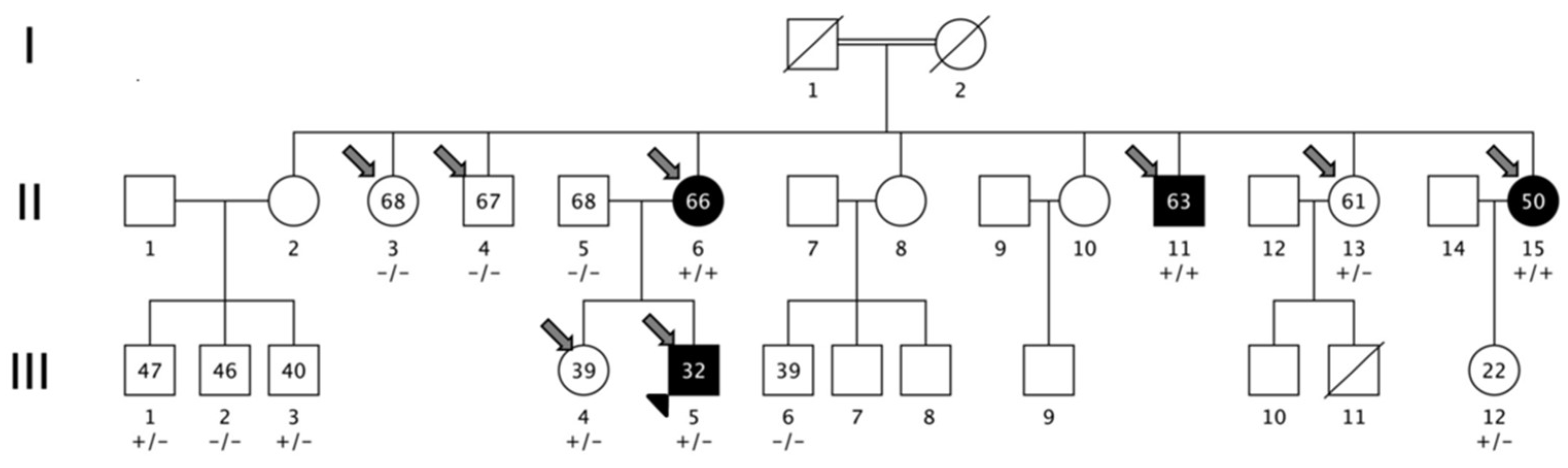
3.1. Family Description
3.2. Linkage Analysis
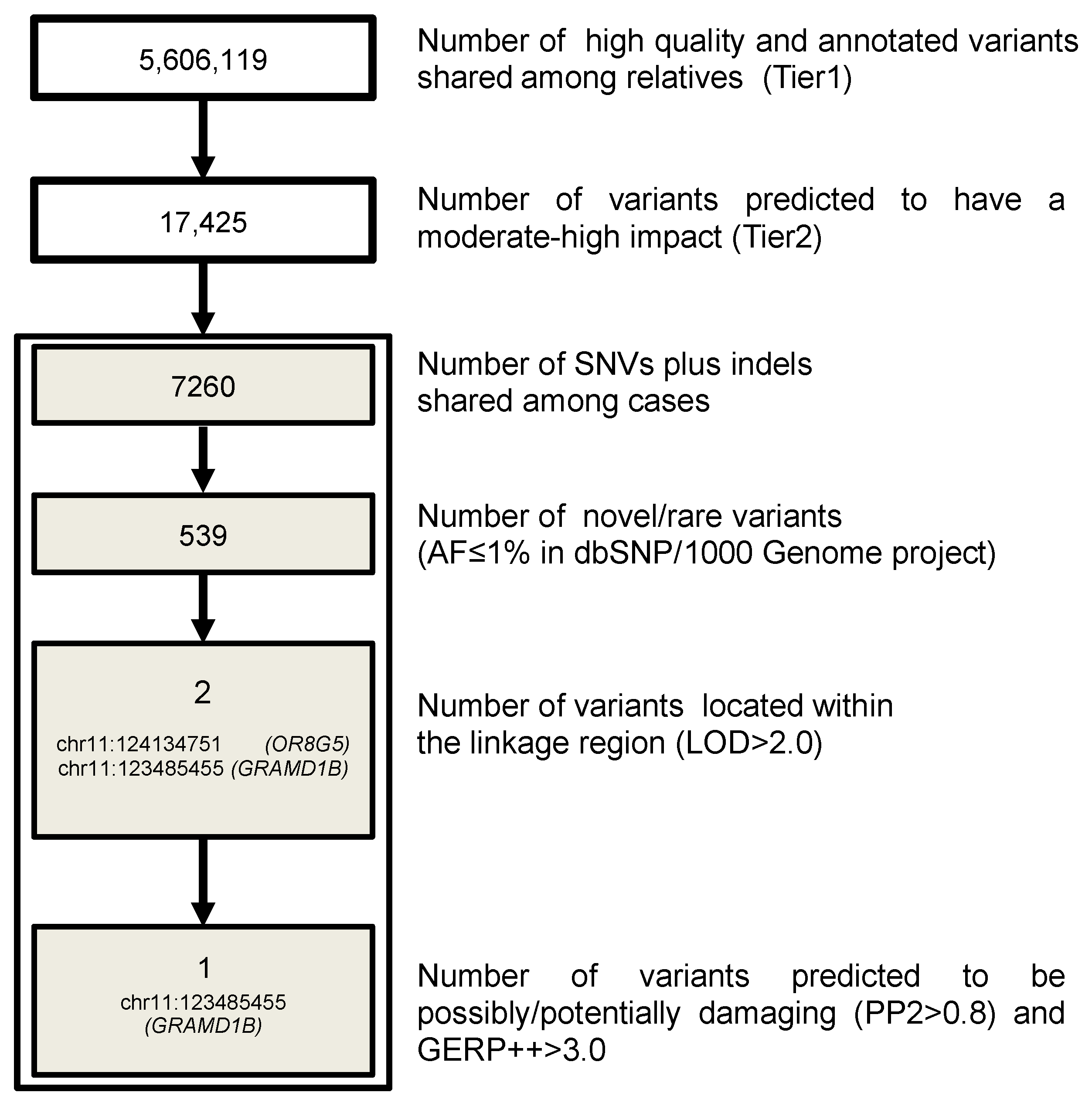
3.3. WGS Results
3.4. GRAMD1B Target Sequencing and Burden Test
3.5. Replication in Canadian Cohort
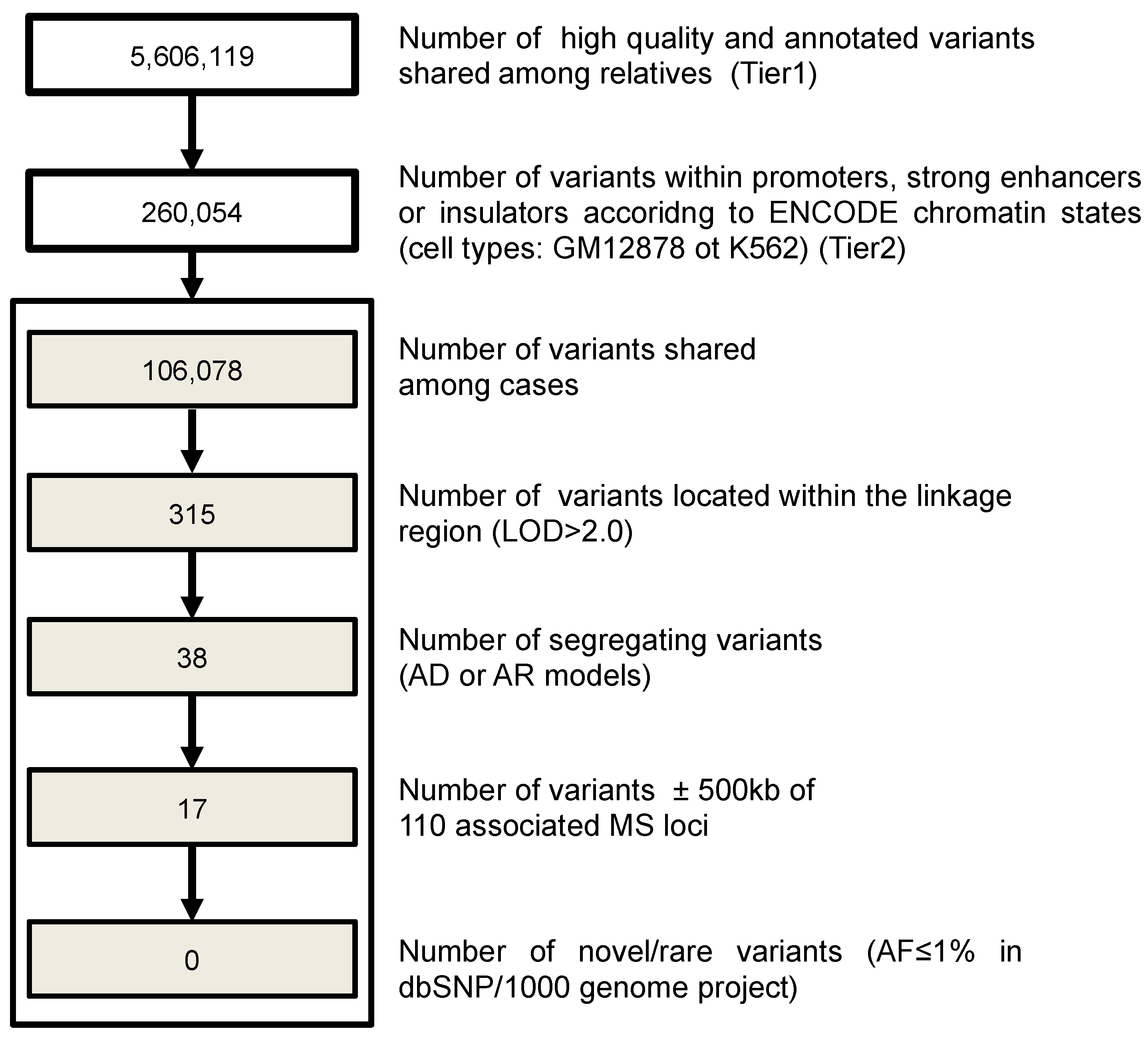
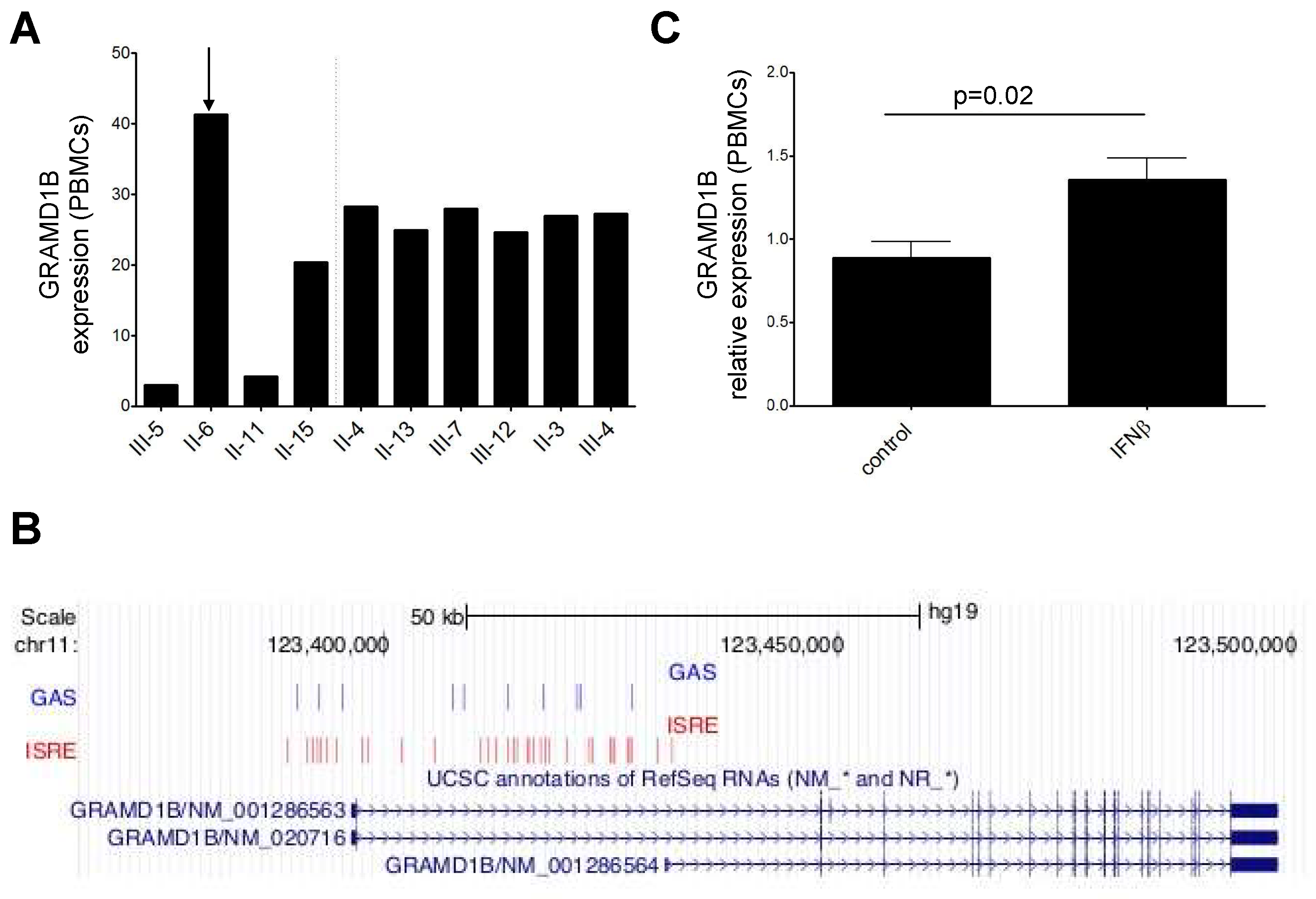
3.6. Investigation of Regulatory Variants
3.7. Transcriptomics Data and Interferon β (IFNβ) Stimulation
3.8. GRAMD1B RNA Expression in Rat Cells and Tissues
3.9. GRAMD1B Protein Expression in Human Brain Tissues
3.10. GRAMD1B Expression Modulation in Astrocytes in Inflammatory Conditions
3.11. Expression and Modulation of GRAMD1B in Human Peripheral Blood Immune Cells
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Compston, A. Genetic Epidemiology of Multiple Sclerosis. J. Neurol. Neurosurg. Psychiatry 1997, 62, 553–561. [Google Scholar] [CrossRef] [PubMed]
- Sadovnick, A.D.; Baird, P.A.; Ward, R.H. Multiple Sclerosis: Updated Risks for Relatives. Am. J. Med. Genet. 1988, 29, 533–541. [Google Scholar] [CrossRef] [PubMed]
- International Multiple Sclerosis Genetics Consortium. Multiple Sclerosis Genomic Map Implicates Peripheral Immune Cells and Microglia in Susceptibility. Science 2019, 365, eaav7188. [Google Scholar] [CrossRef] [PubMed]
- International Multiple Sclerosis Genetics Consortium. Low-Frequency and Rare-Coding Variation Contributes to Multiple Sclerosis Risk. Cell 2018, 175, 1679–1687.e7. [Google Scholar] [CrossRef] [PubMed]
- Jacobsen, M.; Schweer, D.; Ziegler, A.; Gaber, R.; Schock, S.; Schwinzer, R.; Wonigeit, K.; Lindert, R.B.; Kantarci, O.; Schaefer-Klein, J.; et al. A Point Mutation in PTPRC Is Associated with the Development of Multiple Sclerosis. Nat. Genet. 2000, 26, 495–499. [Google Scholar] [CrossRef]
- Saarela, J.; Kallio, S.P.; Chen, D.; Montpetit, A.; Jokiaho, A.; Choi, E.; Asselta, R.; Bronnikov, D.; Lincoln, M.R.; Sadovnick, A.D.; et al. PRKCA and Multiple Sclerosis: Association in Two Independent Populations. PLoS Genet. 2006, 2, e42. [Google Scholar] [CrossRef]
- Torre-Fuentes, L.; Matías-Guiu, J.A.; Pytel, V.; Montero-Escribano, P.; Maietta, P.; Álvarez, S.; Gómez-Pinedo, U.; Matías-Guiu, J. Variants of Genes Encoding TNF Receptors and Ligands and Proteins Regulating TNF Activation in Familial Multiple Sclerosis. CNS Neurosci. Ther. 2020, 26, 1178–1184. [Google Scholar] [CrossRef]
- Salehi, Z.; Keramatipour, M.; Talebi, S.; Arab, S.S.; Moghadasi, A.N.; Sahraian, M.A.; Izad, M. Exome Sequencing Reveals Novel Rare Variants in Iranian Familial Multiple Sclerosis: The Importance of POLD2 in the Disease Pathogenesis. Genomics 2021, 113, 2645–2655. [Google Scholar] [CrossRef]
- Barizzone, N.; Cagliani, R.; Basagni, C.; Clarelli, F.; Mendozzi, L.; Agliardi, C.; Forni, D.; Tosi, M.; Mascia, E.; Favero, F.; et al. An Investigation of the Role of Common and Rare Variants in a Large Italian Multiplex Family of Multiple Sclerosis Patients. Genes 2021, 12, 1607. [Google Scholar] [CrossRef]
- Fazia, T.; Marzanati, D.; Carotenuto, A.L.; Beecham, A.; Hadjixenofontos, A.; McCauley, J.L.; Saddi, V.; Piras, M.; Bernardinelli, L.; Gentilini, D. Homozygosity Haplotype and Whole-Exome Sequencing Analysis to Identify Potentially Functional Rare Variants Involved in Multiple Sclerosis among Sardinian Families. Curr. Issues Mol. Biol. 2021, 43, 1778–1793. [Google Scholar] [CrossRef]
- Mascia, E.; Clarelli, F.; Zauli, A.; Guaschino, C.; Sorosina, M.; Barizzone, N.; Basagni, C.; Santoro, S.; Ferrè, L.; Bonfiglio, S.; et al. Burden of Rare Coding Variants in an Italian Cohort of Familial Multiple Sclerosis. J. Neuroimmunol. 2022, 362, 577760. [Google Scholar] [CrossRef] [PubMed]
- Everest, E.; Ahangari, M.; Uygunoglu, U.; Tutuncu, M.; Bulbul, A.; Saip, S.; Duman, T.; Sezerman, U.; Reich, D.S.; Riley, B.P.; et al. Investigating the Role of Common and Rare Variants in Multiplex Multiple Sclerosis Families Reveals an Increased Burden of Common Risk Variation. Sci. Rep. 2022, 12, 16984. [Google Scholar] [CrossRef] [PubMed]
- Horjus, J.; van Mourik-Banda, T.; Heerings, M.A.P.; Hakobjan, M.; De Witte, W.; Heersema, D.J.; Jansen, A.J.; Strijbis, E.M.M.; de Jong, B.A.; Slettenaar, A.E.J.; et al. Whole Exome Sequencing in Multi-Incident Families Identifies Novel Candidate Genes for Multiple Sclerosis. Int. J. Mol. Sci. 2022, 23, 11461. [Google Scholar] [CrossRef] [PubMed]
- Ramagopalan, S.V.; Dyment, D.A.; Cader, M.Z.; Morrison, K.M.; Disanto, G.; Morahan, J.M.; Berlanga-Taylor, A.J.; Handel, A.; De Luca, G.C.; Sadovnick, A.D.; et al. Rare Variants in the CYP27B1 Gene Are Associated with Multiple Sclerosis. Ann. Neurol. 2011, 70, 881–886. [Google Scholar] [CrossRef]
- Dyment, D.A.; Cader, M.Z.; Chao, M.J.; Lincoln, M.R.; Morrison, K.M.; Disanto, G.; Morahan, J.M.; De Luca, G.C.; Sadovnick, A.D.; Lepage, P.; et al. Exome Sequencing Identifies a Novel Multiple Sclerosis Susceptibility Variant in the TYK2 Gene. Neurology 2012, 79, 406–411. [Google Scholar] [CrossRef]
- Sadovnick, A.D.; Traboulsee, A.L.; Bernales, C.Q.; Ross, J.P.; Forwell, A.L.; Yee, I.M.; Guillot-Noel, L.; Fontaine, B.; Cournu-Rebeix, I.; Alcina, A.; et al. Analysis of Plasminogen Genetic Variants in Multiple Sclerosis Patients. G3 2016, 6, 2073–2079. [Google Scholar] [CrossRef]
- Wang, Z.; Sadovnick, A.D.; Traboulsee, A.L.; Ross, J.P.; Bernales, C.Q.; Encarnacion, M.; Yee, I.M.; de Lemos, M.; Greenwood, T.; Lee, J.D.; et al. Nuclear Receptor NR1H3 in Familial Multiple Sclerosis. Neuron 2016, 90, 948–954. [Google Scholar] [CrossRef]
- Sadovnick, A.D.; Gu, B.J.; Traboulsee, A.L.; Bernales, C.Q.; Encarnacion, M.; Yee, I.M.; Criscuoli, M.G.; Huang, X.; Ou, A.; Milligan, C.J.; et al. Purinergic Receptors P2RX4 and P2RX7 in Familial Multiple Sclerosis. Hum. Mutat. 2017, 38, 736–744. [Google Scholar] [CrossRef]
- Maver, A.; Lavtar, P.; Ristić, S.; Stopinšek, S.; Simčič, S.; Hočevar, K.; Sepčić, J.; Drulović, J.; Pekmezović, T.; Novaković, I.; et al. Identification of Rare Genetic Variation of NLRP1 Gene in Familial Multiple Sclerosis. Sci. Rep. 2017, 7, 3715. [Google Scholar] [CrossRef]
- Mescheriakova, J.Y.; Verkerk, A.J.; Amin, N.; Uitterlinden, A.G.; van Duijn, C.M.; Hintzen, R.Q. Linkage Analysis and Whole Exome Sequencing Identify a Novel Candidate Gene in a Dutch Multiple Sclerosis Family. Mult. Scler. 2019, 25, 909–917. [Google Scholar] [CrossRef]
- Zrzavy, T.; Leutmezer, F.; Kristoferitsch, W.; Kornek, B.; Schneider, C.; Rommer, P.; Berger, T.; Zimprich, A. Exome-Sequence Analyses of Four Multi-Incident Multiple Sclerosis Families. Genes 2020, 11, 988. [Google Scholar] [CrossRef] [PubMed]
- Esposito, F.; Guaschino, C.; Sorosina, M.; Clarelli, F.; Ferre’, L.; Mascia, E.; Santoro, S.; Pagnesi, M.; Radaelli, M.; Colombo, B.; et al. Impact of MS Genetic Loci on Familial Aggregation, Clinical Phenotype, and Disease Prediction. Neurol. Neuroimmunol. Neuroinflammation 2015, 2, e129. [Google Scholar] [CrossRef] [PubMed]
- Sorosina, M.; Brambilla, P.; Clarelli, F.; Barizzone, N.; Lupoli, S.; Guaschino, C.; Osiceanu, A.M.; Moiola, L.; Ghezzi, A.; Coniglio, G.; et al. Genetic Burden of Common Variants in Progressive and Bout-Onset Multiple Sclerosis. Mult. Scler. J. 2014, 20, 802–811. [Google Scholar] [CrossRef]
- Kong, A.; Cox, N.J. Allele-Sharing Models: LOD Scores and Accurate Linkage Tests. Am. J. Hum. Genet. 1997, 61, 1179–1188. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Li, H.; Durbin, R. Fast and Accurate Long-Read Alignment with Burrows-Wheeler Transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef] [PubMed]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce Framework for Analyzing next-Generation DNA Sequencing Data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef]
- DePristo, M.A.; Banks, E.; Poplin, R.; Garimella, K.V.; Maguire, J.R.; Hartl, C.; Philippakis, A.A.; del Angel, G.; Rivas, M.A.; Hanna, M.; et al. A Framework for Variation Discovery and Genotyping Using Next-Generation DNA Sequencing Data. Nat. Genet. 2011, 43, 491–498. [Google Scholar] [CrossRef]
- Van der Auwera, G.A.; Carneiro, M.O.; Hartl, C.; Poplin, R.; Del Angel, G.; Levy-Moonshine, A.; Jordan, T.; Shakir, K.; Roazen, D.; Thibault, J.; et al. From FastQ Data to High Confidence Variant Calls: The Genome Analysis Toolkit Best Practices Pipeline. Curr. Protoc. Bioinform. 2013, 43, 11.10.1–11.10.33. [Google Scholar] [CrossRef]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A Program for Annotating and Predicting the Effects of Single Nucleotide Polymorphisms, SnpEff: SNPs in the Genome of Drosophila Melanogaster Strain W1118; Iso-2; Iso-3. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef]
- Cingolani, P.; Patel, V.M.; Coon, M.; Nguyen, T.; Land, S.J.; Ruden, D.M.; Lu, X. Using Drosophila Melanogaster as a Model for Genotoxic Chemical Mutational Studies with a New Program, SnpSift. Front. Genet. 2012, 3, 35. [Google Scholar] [CrossRef]
- Rentzsch, P.; Witten, D.; Cooper, G.M.; Shendure, J.; Kircher, M. CADD: Predicting the Deleteriousness of Variants throughout the Human Genome. Nucleic Acids Res. 2019, 47, D886–D894. [Google Scholar] [CrossRef] [PubMed]
- Davydov, E.V.; Goode, D.L.; Sirota, M.; Cooper, G.M.; Sidow, A.; Batzoglou, S. Identifying a High Fraction of the Human Genome to Be under Selective Constraint Using GERP++. PLoS Comput. Biol. 2010, 6, e1001025. [Google Scholar] [CrossRef] [PubMed]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A Method and Server for Predicting Damaging Missense Mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [PubMed]
- Raney, B.J.; Cline, M.S.; Rosenbloom, K.R.; Dreszer, T.R.; Learned, K.; Barber, G.P.; Meyer, L.R.; Sloan, C.A.; Malladi, V.S.; Roskin, K.M.; et al. ENCODE Whole-Genome Data in the UCSC Genome Browser (2011 Update). Nucleic Acids Res. 2011, 39, D871–D875. [Google Scholar] [CrossRef]
- International Multiple Sclerosis Genetics Consortium (IMSGC). Analysis of Immune-Related Loci Identifies 48 New Susceptibility Variants for Multiple Sclerosis. Nat. Genet. 2013, 45, 1353–1360. [Google Scholar] [CrossRef]
- Qu, W.; Shen, Z.; Zhao, D.; Yang, Y.; Zhang, C. MFEprimer: Multiple Factor Evaluation of the Specificity of PCR Primers. Bioinformatics 2009, 25, 276–278. [Google Scholar] [CrossRef]
- Wang, Z.; Sadovnick, A.D.; Traboulsee, A.L.; Ross, J.P.; Bernales, C.Q.; Encarnacion, M.; Yee, I.M.; de Lemos, M.; Greenwood, T.; Lee, J.D.; et al. Nuclear Receptor NR1H3 in Familial Multiple Sclerosis. Neuron 2016, 92, 555. [Google Scholar] [CrossRef]
- Lek, M.; Karczewski, K.J.; Minikel, E.V.; Samocha, K.E.; Banks, E.; Fennell, T.; O’Donnell-Luria, A.H.; Ware, J.S.; Hill, A.J.; Cummings, B.B.; et al. Analysis of Protein-Coding Genetic Variation in 60,706 Humans. Nature 2016, 536, 285–291. [Google Scholar] [CrossRef]
- Trinh, J.; Vilariño-Güell, C.; Donald, A.; Shah, B.; Yu, I.; Szu-Tu, C.; Aasly, J.O.; Wu, R.-M.; Hentati, F.; Rajput, A.H.; et al. STX6 Rs1411478 Is Not Associated with Increased Risk of Parkinson’s Disease. Park. Relat. Disord. 2013, 19, 563–565. [Google Scholar] [CrossRef]
- Sadovnick, A.D.; Risch, N.J.; Ebers, G.C. Canadian Collaborative Project on Genetic Susceptibility to MS, Phase 2: Rationale and Method. Canadian Collaborative Study Group. Can. J. Neurol. Sci. 1998, 25, 216–221. [Google Scholar] [CrossRef]
- Polman, C.H.; Reingold, S.C.; Banwell, B.; Clanet, M.; Cohen, J.A.; Filippi, M.; Fujihara, K.; Havrdova, E.; Hutchinson, M.; Kappos, L.; et al. Diagnostic Criteria for Multiple Sclerosis: 2010 Revisions to the McDonald Criteria. Ann. Neurol. 2011, 69, 292–302. [Google Scholar] [CrossRef] [PubMed]
- Chenna, R.; Sugawara, H.; Koike, T.; Lopez, R.; Gibson, T.J.; Higgins, D.G.; Thompson, J.D. Multiple Sequence Alignment with the Clustal Series of Programs. Nucleic Acids Res. 2003, 31, 3497–3500. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma Powers Differential Expression Analyses for RNA-Sequencing and Microarray Studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- Consonni, A.; Morara, S.; Codazzi, F.; Grohovaz, F.; Zacchetti, D. Inhibition of Lipopolysaccharide-Induced Microglia Activation by Calcitonin Gene Related Peptide and Adrenomedullin. Mol. Cell. Neurosci. 2011, 48, 151–160. [Google Scholar] [CrossRef] [PubMed]
- Esposito, F.; Sorosina, M.; Ottoboni, L.; Lim, E.T.; Replogle, J.M.; Raj, T.; Brambilla, P.; Liberatore, G.; Guaschino, C.; Romeo, M.; et al. A Pharmacogenetic Study Implicates SLC9a9 in Multiple Sclerosis Disease Activity. Ann. Neurol. 2015, 78, 115–127. [Google Scholar] [CrossRef] [PubMed]
- Morga, E.; Faber, C.; Heuschling, P. Regional Heterogeneity of the Astroglial Immunoreactive Phenotype: Effect of Lipopolysaccharide. J. Neurosci. Res. 1999, 57, 941–952. [Google Scholar] [CrossRef]
- Sénécal, V.; Deblois, G.; Beauseigle, D.; Schneider, R.; Brandenburg, J.; Newcombe, J.; Moore, C.S.; Prat, A.; Antel, J.; Arbour, N. Production of IL-27 in Multiple Sclerosis Lesions by Astrocytes and Myeloid Cells: Modulation of Local Immune Responses. Glia 2016, 64, 553–569. [Google Scholar] [CrossRef]
- Cannella, B.; Raine, C.S. The Adhesion Molecule and Cytokine Profile of Multiple Sclerosis Lesions. Ann. Neurol. 1995, 37, 424–435. [Google Scholar] [CrossRef]
- Cannella, B.; Sizing, I.D.; Benjamin, C.D.; Browning, J.L.; Raine, C.S. Antibodies to Lymphotoxin Alpha (LT Alpha) and LT Beta Recognize Different Glial Cell Types in the Central Nervous System. J. Neuroimmunol. 1997, 78, 172–179. [Google Scholar] [CrossRef]
- Abecasis, G.R.; Cherny, S.S.; Cookson, W.O.; Cardon, L.R. Merlin--Rapid Analysis of Dense Genetic Maps Using Sparse Gene Flow Trees. Nat. Genet. 2002, 30, 97–101. [Google Scholar] [CrossRef]
- Khanna, P.; Chua, P.J.; Wong, B.S.E.; Yin, C.; Thike, A.A.; Wan, W.K.; Tan, P.H.; Baeg, G.H. GRAM Domain-Containing Protein 1B (GRAMD1B), a Novel Component of the JAK/STAT Signaling Pathway, Functions in Gastric Carcinogenesis. Oncotarget 2017, 8, 115370–115383. [Google Scholar] [CrossRef] [PubMed]
- Khanna, P.; Lee, J.S.; Sereemaspun, A.; Lee, H.; Baeg, G.H. GRAMD1B Regulates Cell Migration in Breast Cancer Cells through JAK/STAT and Akt Signaling. Sci. Rep. 2018, 8, 9511. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.Y.; Yang, X.; Gharpure, K.M.; Hatakeyama, H.; Egli, M.; McGuire, M.H.; Nagaraja, A.S.; Miyake, T.M.; Rupaimoole, R.; Pecot, C.V.; et al. 2’-OMe-Phosphorodithioate-Modified SiRNAs Show Increased Loading into the RISC Complex and Enhanced Anti-Tumour Activity. Nat. Commun. 2014, 5, 3459. [Google Scholar] [CrossRef] [PubMed]
- Santos-Cortez, R.L.P.; Khan, V.; Khan, F.S.; Mughal, Z.-U.-N.; Chakchouk, I.; Lee, K.; Rasheed, M.; Hamza, R.; Acharya, A.; Ullah, E.; et al. Novel Candidate Genes and Variants Underlying Autosomal Recessive Neurodevelopmental Disorders with Intellectual Disability. Hum. Genet. 2018, 137, 735–752. [Google Scholar] [CrossRef] [PubMed]
- Han, F.; Pan, W. A Data-Adaptive Sum Test for Disease Association with Multiple Common or Rare Variants. Hum. Hered. 2010, 70, 42–54. [Google Scholar] [CrossRef] [PubMed]
- Neale, B.M.; Rivas, M.A.; Voight, B.F.; Altshuler, D.; Devlin, B.; Orho-Melander, M.; Kathiresan, S.; Purcell, S.M.; Roeder, K.; Daly, M.J. Testing for an Unusual Distribution of Rare Variants. PLoS Genet. 2011, 7, e1001322. [Google Scholar] [CrossRef]
- Tsukahara, T.; Kim, S.; Taylor, M.W. REFINEMENT: A Search Framework for the Identification of Interferon-Responsive Elements in DNA Sequences--a Case Study with ISRE and GAS. Comput. Biol. Chem. 2006, 30, 134–147. [Google Scholar] [CrossRef]
- Bailey, T.L.; Boden, M.; Buske, F.A.; Frith, M.; Grant, C.E.; Clementi, L.; Ren, J.; Li, W.W.; Noble, W.S. MEME SUITE: Tools for Motif Discovery and Searching. Nucleic Acids Res. 2009, 37, W202–W208. [Google Scholar] [CrossRef]
- Grant, C.E.; Bailey, T.L.; Noble, W.S. FIMO: Scanning for Occurrences of a given Motif. Bioinformatics 2011, 27, 1017–1018. [Google Scholar] [CrossRef]
- Correale, J.; Villa, A. The Blood-Brain-Barrier in Multiple Sclerosis: Functional Roles and Therapeutic Targeting. Autoimmunity 2007, 40, 148–160. [Google Scholar] [CrossRef]
- Yatagai, Y.; Sakamoto, T.; Masuko, H.; Kaneko, Y.; Yamada, H.; Iijima, H.; Naito, T.; Noguchi, E.; Hirota, T.; Tamari, M.; et al. Genome-Wide Association Study for Levels of Total Serum IgE Identifies HLA-C in a Japanese Population. PLoS ONE 2013, 8, e80941. [Google Scholar] [CrossRef] [PubMed]
- Kiu, H.; Nicholson, S.E. Biology and Significance of the JAK/STAT Signalling Pathways. Growth Factors 2012, 30, 88–106. [Google Scholar] [CrossRef] [PubMed]
- Fabbri, C.; Serretti, A. Role of 108 Schizophrenia-Associated Loci in Modulating Psychopathological Dimensions in Schizophrenia and Bipolar Disorder. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2017, 174, 757–764. [Google Scholar] [CrossRef] [PubMed]
- Reuter, M.S.; Tawamie, H.; Buchert, R.; Gebril, O.H.; Froukh, T.; Thiel, C.; Uebe, S.; Ekici, A.B.; Krumbiegel, M.; Zweier, C.; et al. Diagnostic Yield and Novel Candidate Genes by Exome Sequencing in 152 Consanguineous Families with Neurodevelopmental Disorders. JAMA psychiatry 2017, 74, 293–299. [Google Scholar] [CrossRef]
- Thyme, S.B.; Pieper, L.M.; Li, E.H.; Pandey, S.; Wang, Y.; Morris, N.S.; Sha, C.; Choi, J.W.; Herrera, K.J.; Soucy, E.R.; et al. Phenotypic Landscape of Schizophrenia-Associated Genes Defines Candidates and Their Shared Functions. Cell 2019, 177, 478–491.e20. [Google Scholar] [CrossRef]
- Ercan, B.; Naito, T.; Koh, D.H.Z.; Dharmawan, D.; Saheki, Y. Molecular Basis of Accessible Plasma Membrane Cholesterol Recognition by the GRAM Domain of GRAMD1b. EMBO J. 2021, 40, e106524. [Google Scholar] [CrossRef]
- Howie, D.; Bokum, A.T.; Necula, A.S.; Cobbold, S.P.; Waldmann, H. The Role of Lipid Metabolism in T Lymphocyte Differentiation and Survival. Front. Immunol. 2017, 8, 1949. [Google Scholar] [CrossRef]
- Caprariello, A.V.; Adams, D.J. The Landscape of Targets and Lead Molecules for Remyelination. Nat. Chem. Biol. 2022, 18, 925–933. [Google Scholar] [CrossRef]
- Pelizzoni, I.; Zacchetti, D.; Smith, C.P.; Grohovaz, F.; Codazzi, F. Expression of divalent metal transporter 1 in primary hippocampal neurons: Reconsidering its role in non-transferrin-bound iron influx. J. Neurochem. 2022, 120, 269–278. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics of MS Patients | Individual II-6 | Individual II-15 | Individual II-11 | Individual III-5 |
|---|---|---|---|---|
| Gender | Female | Female | Male | Male |
| Age (years) | 66 | 50 | 63 | 32 |
| MS course | SP | SP | SP | RR |
| Age at MS onset (years) | 41 | 33 | 33 | 25 |
| Age at MS diagnosis (years) | 49 | 35 | 39 | 31 |
| Disease duration (years) | 24 | 17 | 30 | 7 |
| EDSS | 6.5 | 7.0 | 6.5 | 2.0 |
| MSSS | 6.362 | 7.765 | 5.608 | 3.17 |
| MS treatment | IFN 1β | No treatment | Chronic steroid | GA |
| POS | 11:123464786 | 11:123476158 | 11:123484303 | 11:123484370 | 11:123485455 |
|---|---|---|---|---|---|
| SNP | rs118067934 | rs140366389 | rs755488531 | rs769527838 | . |
| REF | G | C | G | T | T |
| ALT | A | G | A | G | C |
| AF fMS (n = 91) | 0.0205 | 0.005 | 0.005 | 0.006 | 0.005 |
| AF sMS (n = 192) | 0.0104 | 0.005 | 0 | 0 | 0 |
| AF HC cohort 1 (n = 296) | 0.172 | 0.009 | 0 | 0 | 0 |
| AF HC cohort 2 (n = 1096) | 0.024 | 0.001 | 0 | 0 | 0 |
| IMPACT | LOW | MODERATE | MODERATE | LOW | MODERATE |
| CODON | c.256-4G > A | c.866C > G | c.1735G > A | c.1798 + 4T > G | c.1801T > C |
| AA change | . | p.Ser289Cys | p.Val579Met | . | p.Ser601Pro |
| GERP++ RS | . | 4.83 | 5.52 | . | 4.72 |
| Polyphen2 HDIV | . | 0.986 (D) | 0.978 (D) | . | 1.0 (D) |
| SIFT | . | 0.005(D) | 0.274(T) | . | 0.006 (D) |
| CADD PHRED Score | 8.347 | 24.5 | 24.8 | 10.66 | 24.0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Esposito, F.; Osiceanu, A.M.; Sorosina, M.; Ottoboni, L.; Bollman, B.; Santoro, S.; Bettegazzi, B.; Zauli, A.; Clarelli, F.; Mascia, E.; et al. A Whole-Genome Sequencing Study Implicates GRAMD1B in Multiple Sclerosis Susceptibility. Genes 2022, 13, 2392. https://doi.org/10.3390/genes13122392
Esposito F, Osiceanu AM, Sorosina M, Ottoboni L, Bollman B, Santoro S, Bettegazzi B, Zauli A, Clarelli F, Mascia E, et al. A Whole-Genome Sequencing Study Implicates GRAMD1B in Multiple Sclerosis Susceptibility. Genes. 2022; 13(12):2392. https://doi.org/10.3390/genes13122392
Chicago/Turabian StyleEsposito, Federica, Ana Maria Osiceanu, Melissa Sorosina, Linda Ottoboni, Bryan Bollman, Silvia Santoro, Barbara Bettegazzi, Andrea Zauli, Ferdinando Clarelli, Elisabetta Mascia, and et al. 2022. "A Whole-Genome Sequencing Study Implicates GRAMD1B in Multiple Sclerosis Susceptibility" Genes 13, no. 12: 2392. https://doi.org/10.3390/genes13122392
APA StyleEsposito, F., Osiceanu, A. M., Sorosina, M., Ottoboni, L., Bollman, B., Santoro, S., Bettegazzi, B., Zauli, A., Clarelli, F., Mascia, E., Calabria, A., Zacchetti, D., Capra, R., Ferrari, M., Provero, P., Lazarevic, D., Cittaro, D., Carrera, P., Patsopoulos, N., ... Martinelli Boneschi, F. (2022). A Whole-Genome Sequencing Study Implicates GRAMD1B in Multiple Sclerosis Susceptibility. Genes, 13(12), 2392. https://doi.org/10.3390/genes13122392