

Cuproptosis-Related lncRNA Gene Signature Establishes a Prognostic Model of Gastric Adenocarcinoma and Evaluate the Effect of Antineoplastic Drugs


Abstract
1. Introduction
2. Method
2.1. Data Collection
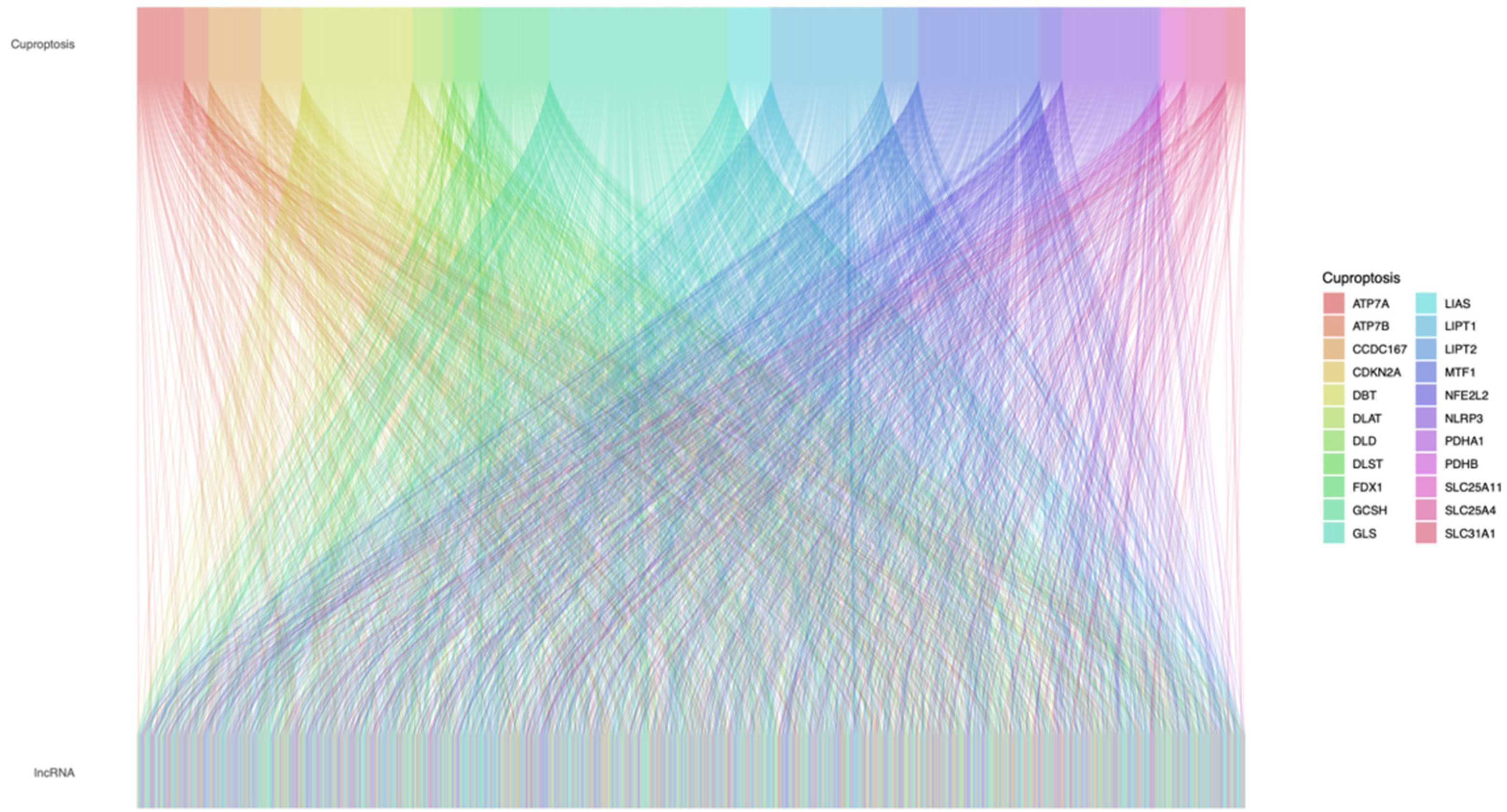
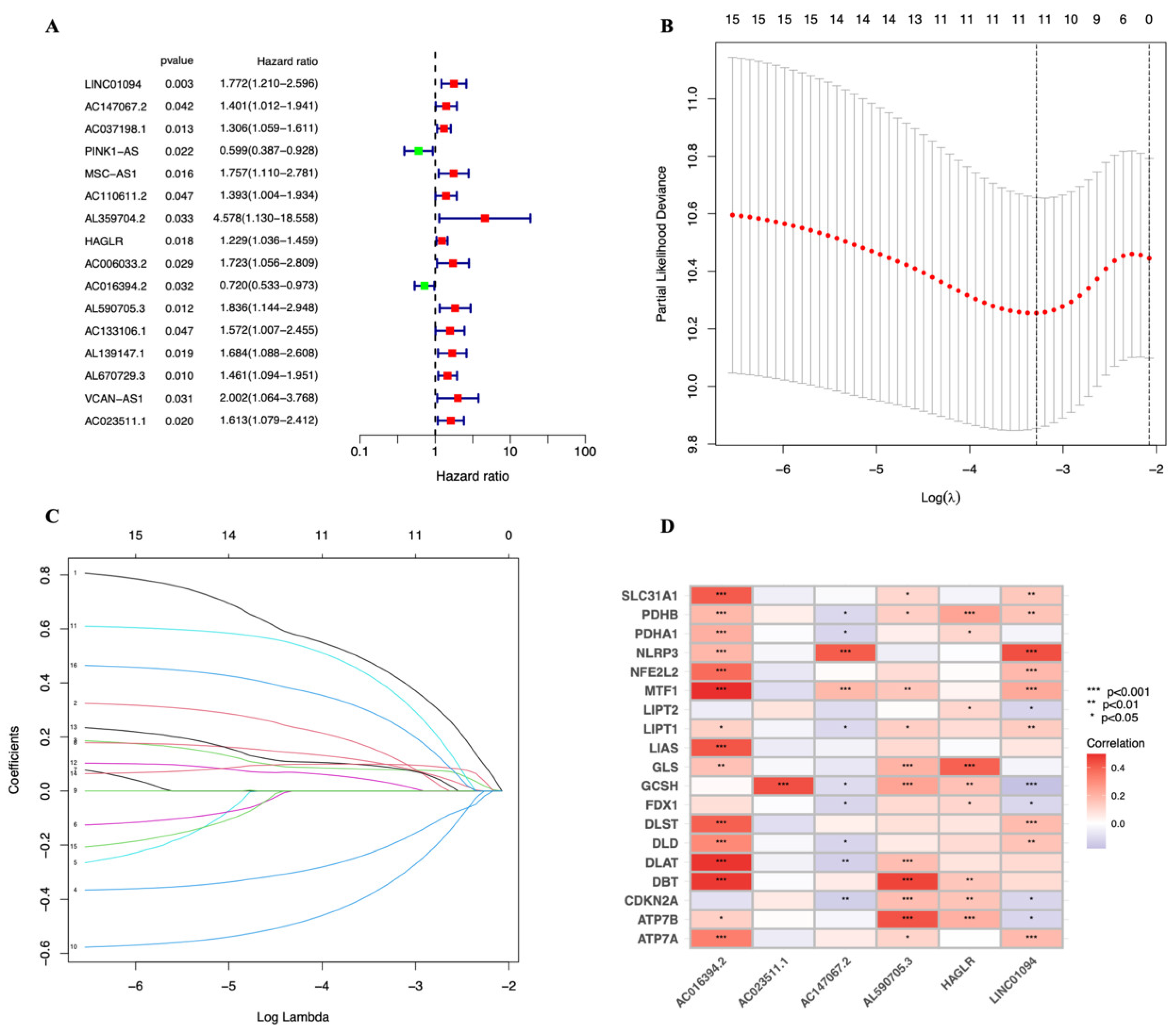
2.2. Construction of Cuproptosis-Related Prognostic Signature for STAD
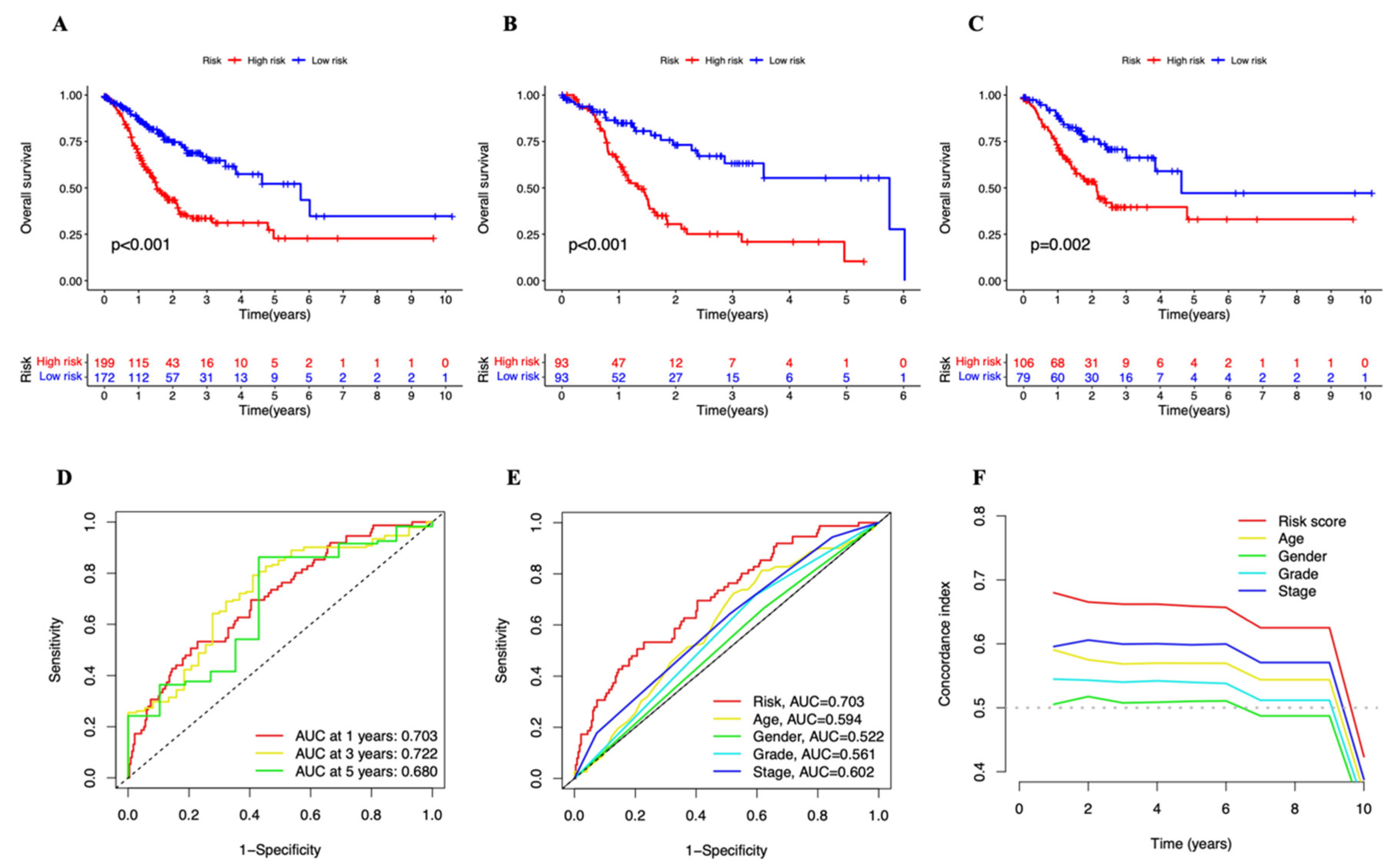
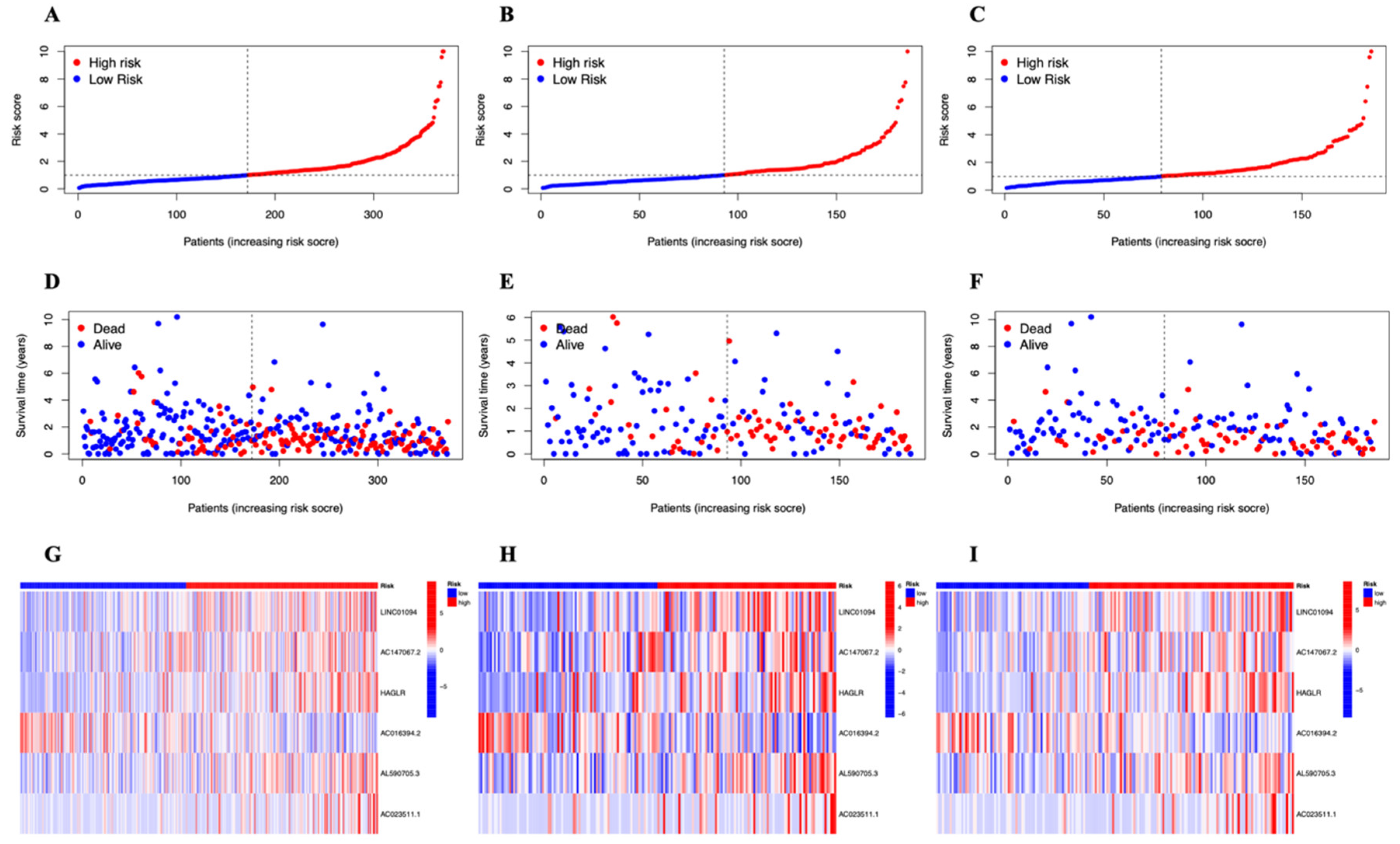
2.3. Establishment and Evaluation of the 6-lncRNA Signature
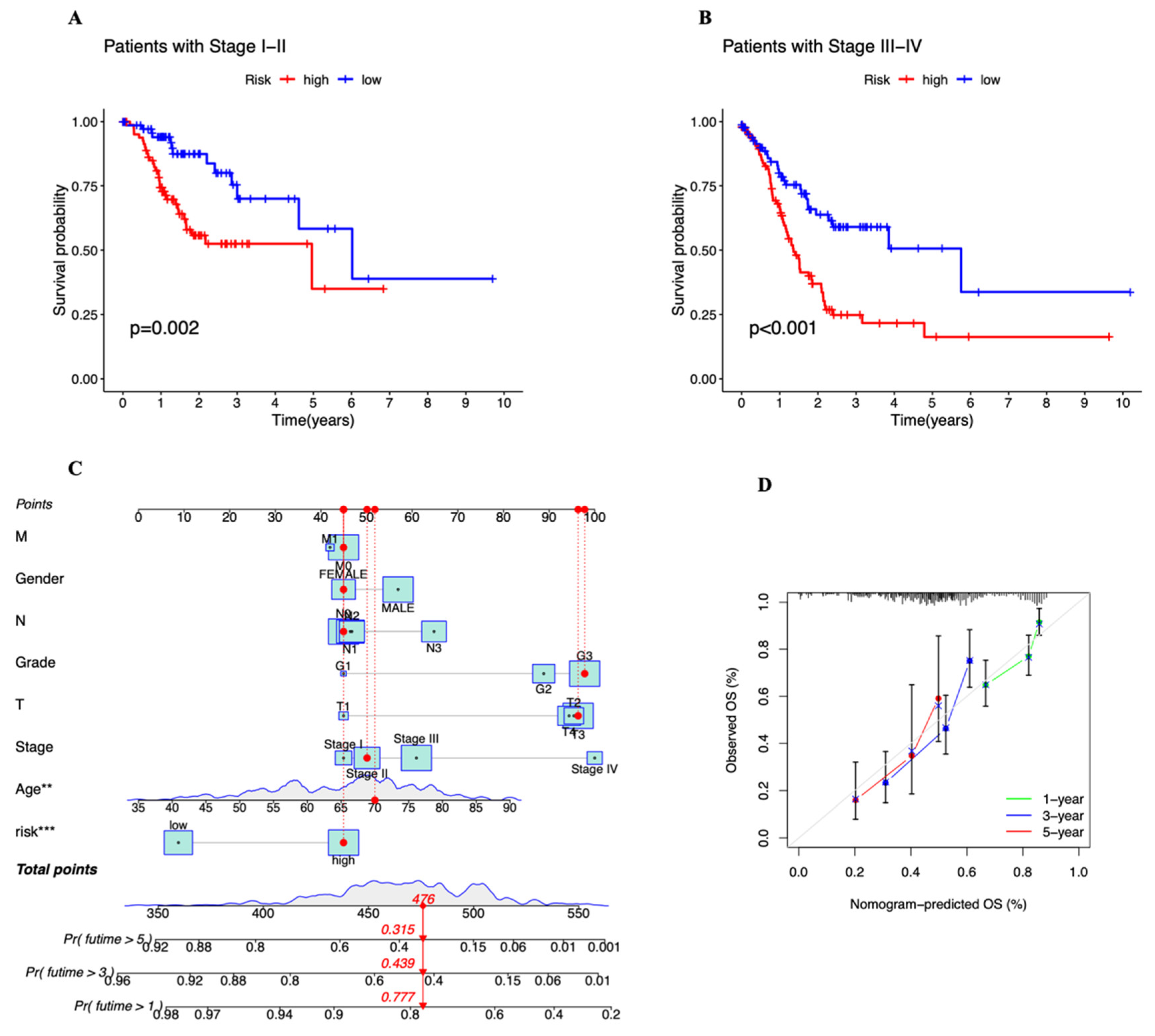
2.4. Exploration of the Relationship between the Prognostic Risk Score and Clinical Stage
2.5. Construction of Nomogram
2.6. Enrichment Functional Analysis
2.7. Estimation of Intratumoral Immune Cell Infiltration
2.8. Estimation of Tumor Mutation Burden
2.9. Evaluation of Drug Sensitivity
2.10. Statistical Analysis
3. Results
3.1. Data Processing
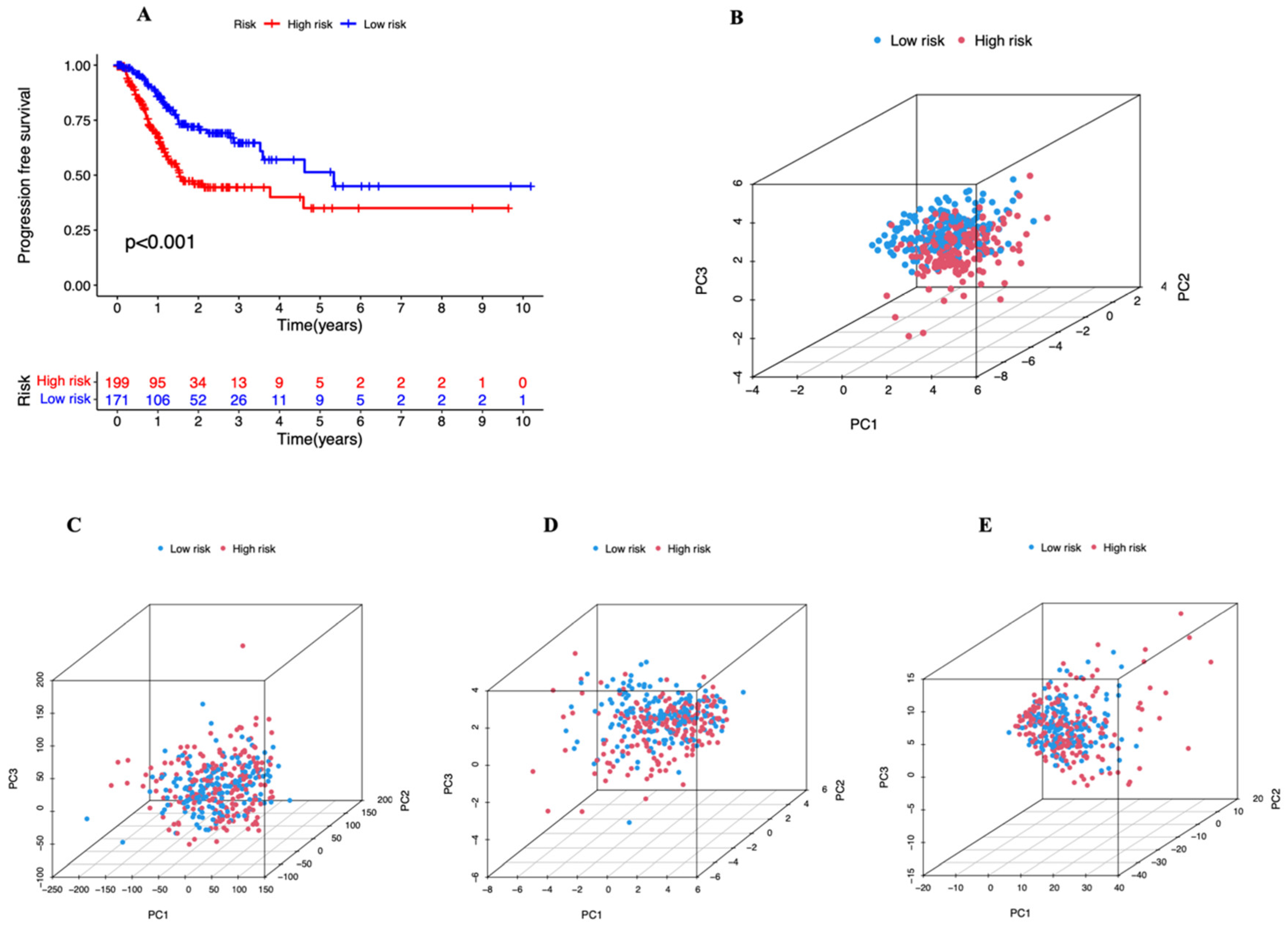
3.2. A Cuproptosis-Related Long Noncoding RNAs Prognostic Marker for STAD: Development and Validation
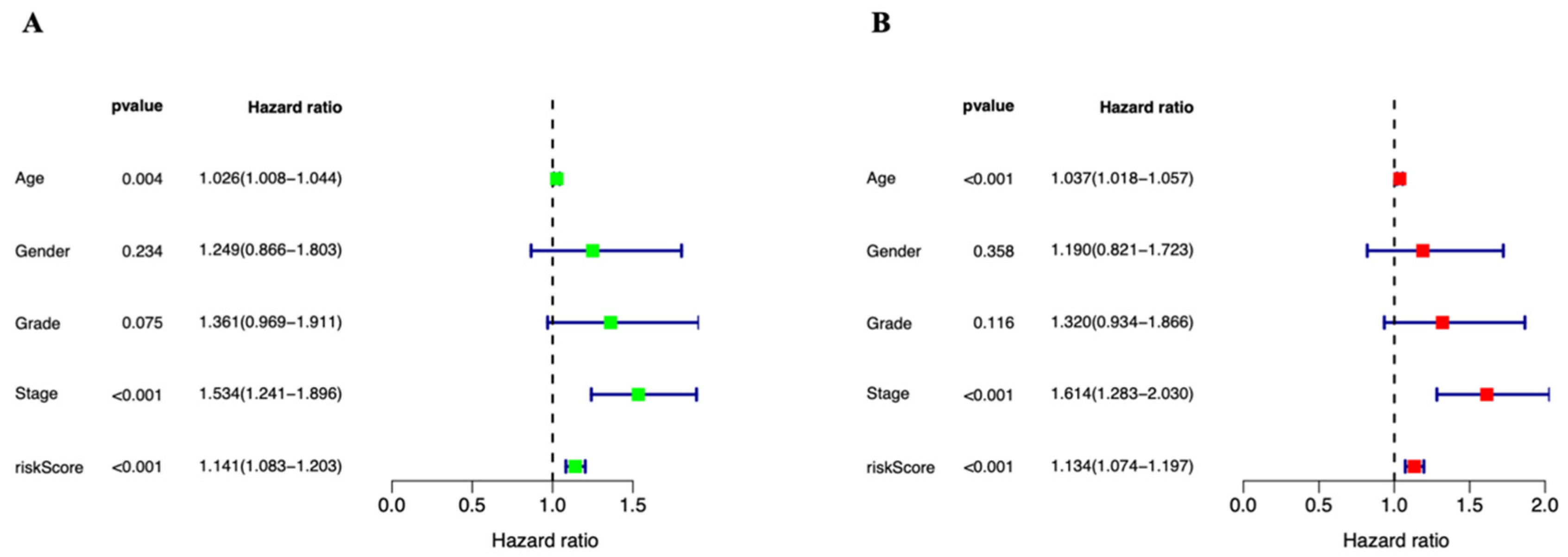
3.3. Independent Prognostic Marker for Cuproptosis-Related lncRNAs in Predicting Overall Survival
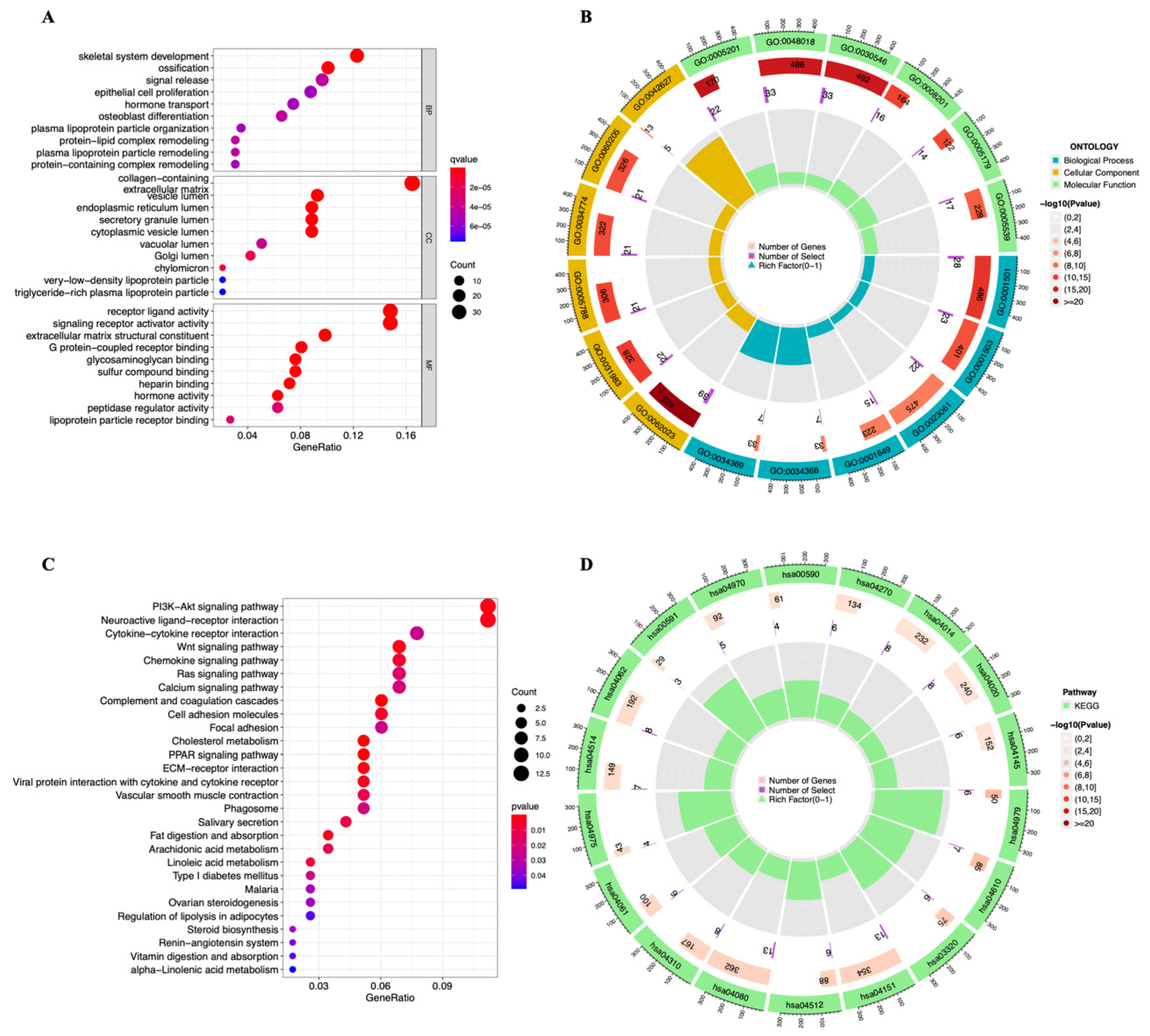
3.4. Functional Enrichment Analysis
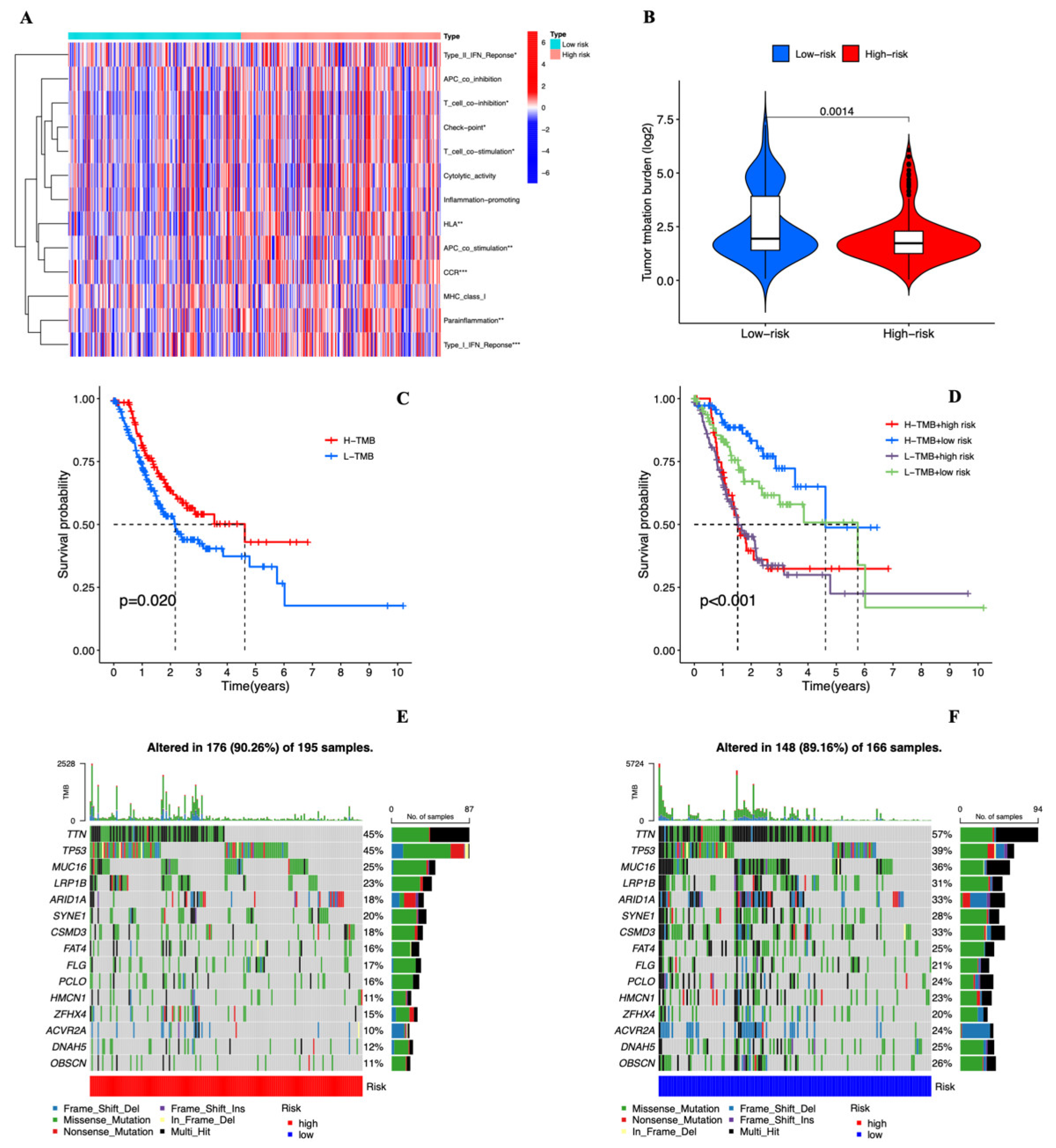
3.5. Estimation of Intratumoral Immune Cell Infiltration and Immunotherapy
3.6. Tumor Mutational Burden of the Cuproptosis-related lncRNAs Prognostic Marker in STAD Samples
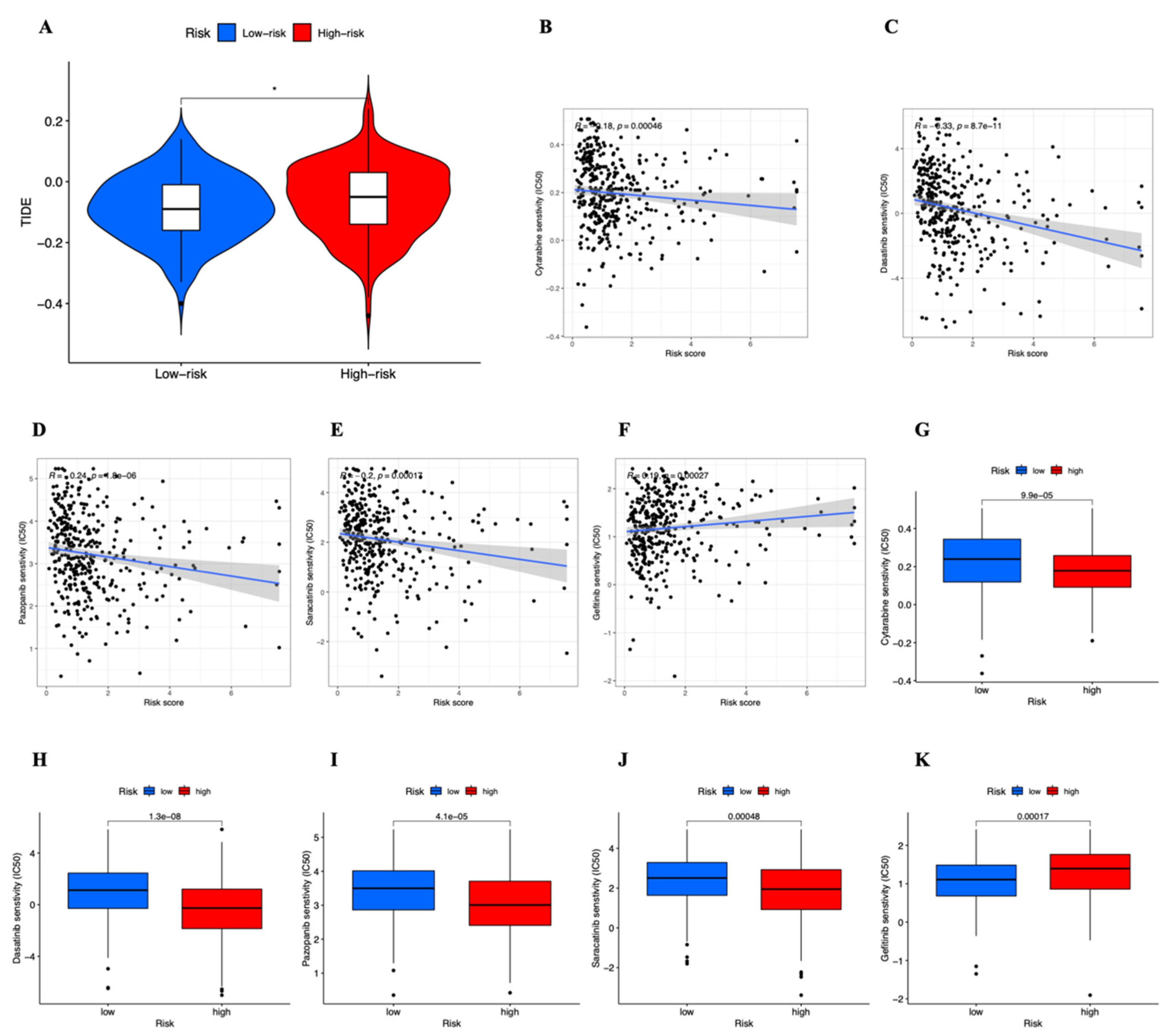
3.7. Drug Sensitivity
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Joshi, S.S.; Badgwell, B.D. Current treatment and recent progress in gastric cancer. CA Cancer J. Clin. 2021, 71, 264–279. [Google Scholar] [CrossRef] [PubMed]
- Seeneevassen, L.; Bessède, E.; Mégraud, F.; Lehours, P.; Dubus, P.; Varon, C. Gastric Cancer: Advances in Carcinogenesis Research and New Therapeutic Strategies. Int. J. Mol. Sci. 2021, 22, 3418. [Google Scholar] [CrossRef] [PubMed]
- Li, C.Y.; Liang, G.Y.; Yao, W.Z.; Sui, J.; Shen, X.; Zhang, Y.Q.; Peng, H.; Hong, W.W.; Ye, Y.C.; Zhang, Z.Y.; et al. Integrated analysis of long non-coding RNA competing interactions reveals the potential role in progression of human gastric cancer. Int. J. Oncol. 2016, 48, 1965–1976. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.C.; Zhu, Y.; Chen, W.B.; Lin, L.W.; Chen, D.H.; Huang, J.R.; Pan, K.; Lin, Y.; Wu, B.T.; Dai, Y.; et al. Integrated analysis of long non-coding RNAs and mRNA expression profiles reveals the potential role of lncRNAs in gastric cancer pathogenesis. Int. J. Oncol. 2014, 45, 619–628. [Google Scholar] [CrossRef]
- Li, F.; Huang, C.; Li, Q.; Wu, X. Construction and Comprehensive Analysis for Dysregulated Long Non-Coding RNA (lncRNA)-Associated Competing Endogenous RNA (ceRNA) Network in Gastric Cancer. Med. Sci. Monit. 2018, 24, 37–49. [Google Scholar] [CrossRef]
- Tsvetkov, P.; Coy, S.; Petrova, B.; Dreishpoon, M.; Verma, A.; Abdusamad, M.; Rossen, J.; Joesch-Cohen, L.; Humeidi, R.; Spangler, R.D.; et al. Copper induces cell death by targeting lipoylated TCA cycle proteins. Science 2022, 375, 1254–1261. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, Y.; Ye, S.; Feng, H.; Ma, L. Development and validation of cuproptosis-related gene signature in the prognostic prediction of liver cancer. Front. Oncol. 2022, 12, 985484. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Mu, J.; Wang, J.; Zhou, Q.; Wang, J. Construction and validation of a cuproptosis-related lncRNA signature as a novel and robust prognostic model for colon adenocarcinoma. Front. Oncol. 2022, 12, 961213. [Google Scholar] [CrossRef]
- Jiang, R.; Huan, Y.; Li, Y.; Gao, X.; Sun, Q.; Zhang, F.; Jiang, T. Transcriptional and genetic alterations of cuproptosis-related genes correlated to malignancy and immune-infiltrate of esophageal carcinoma. Cell Death Discov. 2022, 8, 370. [Google Scholar] [CrossRef]
- Li, P.; Li, J.; Wen, F.; Cao, Y.; Luo, Z.; Zuo, J.; Wu, F.; Li, Z.; Li, W.; Wang, F. A novel cuproptosis-related LncRNA signature: Prognostic and therapeutic value for acute myeloid leukemia. Front. Oncol. 2022, 12, 966920. [Google Scholar] [CrossRef] [PubMed]
- Ye, L.; Jin, W. Identification of lncRNA-associated competing endogenous RNA networks for occurrence and prognosis of gastric carcinoma. J. Clin. Lab. Anal. 2021, 35, e24028. [Google Scholar] [CrossRef]
- Yang, L.; Yu, J.; Tao, L.; Huang, H.; Gao, Y.; Yao, J.; Liu, Z. Cuproptosis-Related lncRNAs are Biomarkers of Prognosis and Immune Microenvironment in Head and Neck Squamous Cell Carcinoma. Front. Genet. 2022, 13, 947551. [Google Scholar] [CrossRef] [PubMed]
- Hu, Q.; Wang, R.; Ma, H.; Zhang, Z.; Xue, Q. Cuproptosis predicts the risk and clinical outcomes of lung adenocarcinoma. Front. Oncol. 2022, 12, 922332. [Google Scholar] [CrossRef] [PubMed]
- Mi, J.X.; Zhang, Y.N.; Lai, Z.; Li, W.; Zhou, L.; Zhong, F. Principal Component Analysis based on Nuclear norm Minimization. Neural. Netw. 2019, 118, 1–16. [Google Scholar] [CrossRef]
- Tang, S.; Zhao, L.; Wu, X.B.; Wang, Z.; Cai, L.Y.; Pan, D.; Li, Y.; Zhou, Y.; Shen, Y. Identification of a Novel Cuproptosis-Related Gene Signature for Prognostic Implication in Head and Neck Squamous Carcinomas. Cancers 2022, 14, 3986. [Google Scholar] [CrossRef]
- Xu, S.; Liu, D.; Chang, T.; Wen, X.; Ma, S.; Sun, G.; Wang, L.; Chen, S.; Xu, Y.; Zhang, H. Cuproptosis-Associated lncRNA Establishes New Prognostic Profile and Predicts Immunotherapy Response in Clear Cell Renal Cell Carcinoma. Front. Genet. 2022, 13, 938259. [Google Scholar] [CrossRef]
- Fernández, A.; Ordóñez, R.; Reiter, R.J.; González-Gallego, J.; Mauriz, J.L. Melatonin and endoplasmic reticulum stress: Relation to autophagy and apoptosis. J. Pineal Res. 2015, 59, 292–307. [Google Scholar] [CrossRef]
- Ye, L.; Jin, F.; Kumar, S.K.; Dai, Y. The mechanisms and therapeutic targets of ferroptosis in cancer. Expert Opin. Ther. Targets 2021, 25, 965–986. [Google Scholar] [CrossRef]
- Chen, X.; Kang, R.; Kroemer, G.; Tang, D. Broadening horizons: The role of ferroptosis in cancer. Nat. Rev. Clin. Oncol. 2021, 18, 280–296. [Google Scholar] [CrossRef]
- Li, D.; Li, Y. The interaction between ferroptosis and lipid metabolism in cancer. Signal Transduct. Target Ther. 2020, 5, 108. [Google Scholar] [CrossRef] [PubMed]
- Tsang, T.; Posimo, J.M.; Gudiel, A.A.; Cicchini, M.; Feldser, D.M.; Brady, D.C. Copper is an essential regulator of the autophagic kinases ULK1/2 to drive lung adenocarcinoma. Nat. Cell Biol. 2020, 22, 412–424. [Google Scholar] [CrossRef]
- Hwang, J.J.; Park, M.H.; Koh, J.Y. Copper activates TrkB in cortical neurons in a metalloproteinase-dependent manner. J. Neurosci. Res. 2007, 85, 2160–2166. [Google Scholar] [CrossRef]
- He, F.; Chang, C.; Liu, B.; Li, Z.; Li, H.; Cai, N.; Wang, H.H. Copper (II) Ions Activate Ligand-Independent Receptor Tyrosine Kinase (RTK) Signaling Pathway. Biomed. Res. Int. 2019, 2019, 4158415. [Google Scholar] [CrossRef] [PubMed]
- DaRosa, P.A.; Wang, Z.; Jiang, X.; Pruneda, J.N.; Cong, F.; Klevit, R.E.; Xu, W. Allosteric activation of the RNF146 ubiquitin ligase by a poly(ADP-ribosyl)ation signal. Nature 2015, 517, 223–226. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics | Type | Total (%) | Test Group (%) | Train Group (%) | p-Value |
|---|---|---|---|---|---|
| Age | ≤65 | 163 (43.94%) | 86 (46.49%) | 77 (41.4%) | 0.3511 |
| >65 | 205 (55.26%) | 97 (52.43%) | 108 (58.06%) | ||
| unknow | 3 (0.81%) | 2 (1.08%) | 1 (0.54%) | ||
| Gender | FEMALE | 133 (35.85%) | 69 (37.3%) | 64 (34.41%) | 0.637 |
| MALE | 238 (64.15%) | 116 (62.7%) | 122 (65.59%) | ||
| Grade | G1 | 10 (2.7%) | 3 (1.62%) | 7 (3.76%) | 0.2236 |
| G2 | 134 (36.12%) | 62 (33.51%) | 72 (38.71%) | ||
| G3 | 218 (58.76%) | 115 (62.16%) | 103 (55.38%) | ||
| unknown | 9 (2.43%) | 5 (2.7%) | 4 (2.15%) | ||
| Stage | Stage I | 50 (13.48%) | 23 (12.43%) | 27 (14.52%) | 0.1095 |
| Stage II | 111 (29.92%) | 50 (27.03%) | 61 (32.8%) | ||
| Stage III | 149 (40.16%) | 85 (45.95%) | 64 (34.41%) | ||
| Stage IV | 38 (10.24%) | 15 (8.11%) | 23 (12.37%) | ||
| unknown | 23 (6.2%) | 12 (6.49%) | 11 (5.91%) | ||
| T Stage | T1 | 18 (4.85%) | 7 (3.78%) | 11 (5.91%) | 0.8158 |
| T2 | 78 (21.02%) | 39 (21.08%) | 39 (20.97%) | ||
| T3 | 167 (45.01%) | 85 (45.95%) | 82 (44.09%) | ||
| T4 | 100 (26.95%) | 50 (27.03%) | 50 (26.88%) | ||
| unknown | 8 (2.16%) | 4 (2.16%) | 4(2.15%) | ||
| M Stage | M0 | 328 (88.41%) | 165 (89.19%) | 163(87.63%) | 1 |
| M1 | 25 (6.74%) | 13(7.03%) | 12 (6.45%) | ||
| unknown | 18 (4.85%) | 7 (3.78%) | 11 (5.91%) | ||
| N Stage | N0 | 108 (29.11%) | 55 (29.73%) | 53 (28.49%) | 0.8005 |
| N1 | 97 (26.15%) | 52 (28.11%) | 45 (24.19%) | ||
| N2 | 74 (19.95%) | 37 (20%) | 37 (19.89%) | ||
| N3 | 74 (19.95%) | 34 (18.38%) | 40 (21.51%) | ||
| unknown | 18 (4.85%) | 7 (3.78%) | 11 (5.91%) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tu, H.; Zhang, Q.; Xue, L.; Bao, J. Cuproptosis-Related lncRNA Gene Signature Establishes a Prognostic Model of Gastric Adenocarcinoma and Evaluate the Effect of Antineoplastic Drugs. Genes 2022, 13, 2214. https://doi.org/10.3390/genes13122214
Tu H, Zhang Q, Xue L, Bao J. Cuproptosis-Related lncRNA Gene Signature Establishes a Prognostic Model of Gastric Adenocarcinoma and Evaluate the Effect of Antineoplastic Drugs. Genes. 2022; 13(12):2214. https://doi.org/10.3390/genes13122214
Chicago/Turabian StyleTu, Hengjia, Qingling Zhang, Lingna Xue, and Junrong Bao. 2022. "Cuproptosis-Related lncRNA Gene Signature Establishes a Prognostic Model of Gastric Adenocarcinoma and Evaluate the Effect of Antineoplastic Drugs" Genes 13, no. 12: 2214. https://doi.org/10.3390/genes13122214
APA StyleTu, H., Zhang, Q., Xue, L., & Bao, J. (2022). Cuproptosis-Related lncRNA Gene Signature Establishes a Prognostic Model of Gastric Adenocarcinoma and Evaluate the Effect of Antineoplastic Drugs. Genes, 13(12), 2214. https://doi.org/10.3390/genes13122214