

A TSHZ3 Frame-Shift Variant Causes Neurodevelopmental and Renal Disorder Consistent with Previously Described Proximal Chromosome 19q13.11 Deletion Syndrome

 , , and
, , and 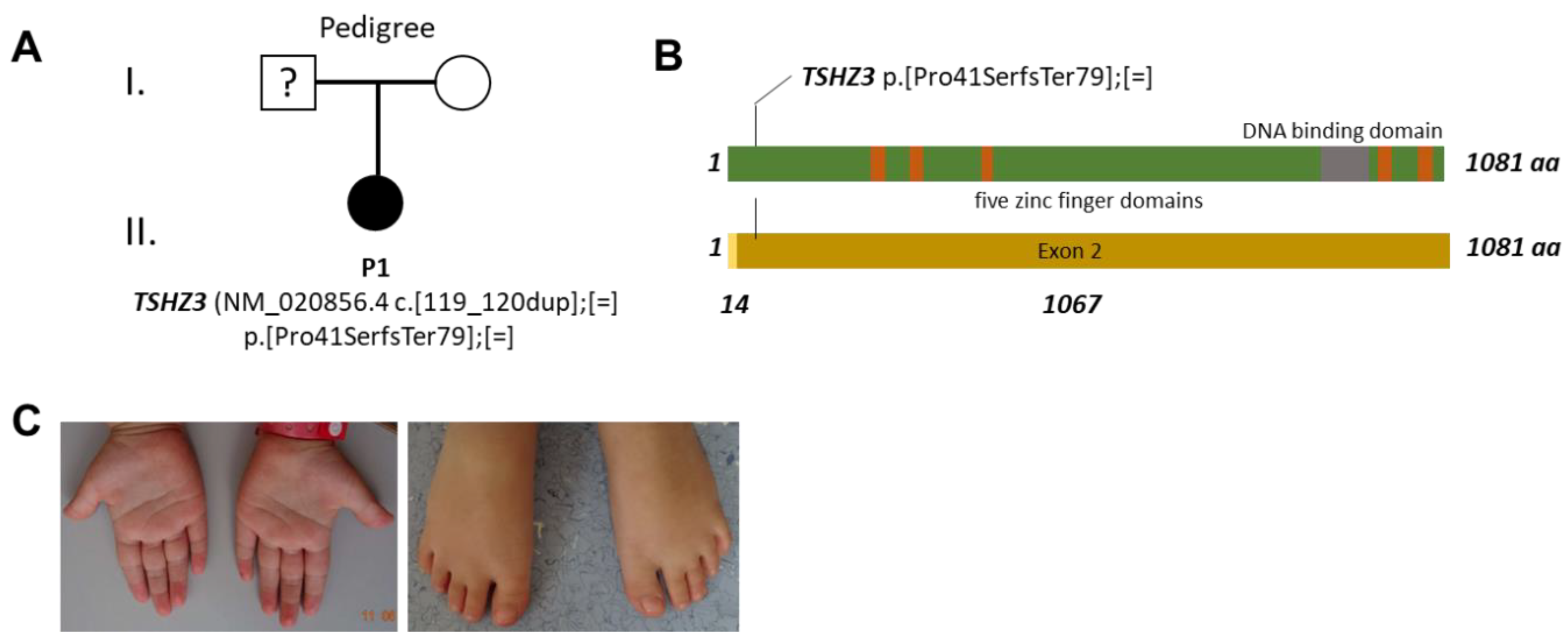{kind=link}
Abstract
1. Introduction
2. Materials and Methods
3. Results
3.1. Clinical Description
3.2. Genetic Testing
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Caubit, X.; Gubellini, P.; Andrieux, J.; Roubertoux, P.L.; Metwaly, M.; Jacq, B.; Fatmi, A.; Had-Aissouni, L.; Kwan, K.Y.; Salin, P.; et al. TSHZ3 deletion causes an autism syndrome and defects in cortical projection neurons. Nat. Genet. 2016, 48, 1359–1369. [Google Scholar] [CrossRef]
- Jenkins, D.; Caubit, X.; Dimovski, A.; Matevska, N.; Lye, C.M.; Cabuk, F.; Gucev, Z.; Tasic, V.; Fasano, L.; Woolf, A.S. Analysis of TSHZ2 and TSHZ3 genes in congenital pelvi-ureteric junction obstruction. Nephrol. Dial. Transplant. 2010, 25, 54–60. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kulharya, A.S.; Michaelis, R.C.; Norris, K.S.; Taylor, H.A.; Garcia-Heras, J. Constitutional del(19)(q12q13.1) in a three-year-old girl with severe phenotypic abnormalities affecting multiple organ systems. Am. J. Med. Genet. 1998, 77, 391–394. [Google Scholar] [CrossRef]
- Adalat, S.; Bockenhauer, D.; Ledermann, S.E.; Hennekam, R.C.; Woolf, A.S. Renal malformations associated with mutations of developmental genes: Messages from the clinic. Pediatr. Nephrol. 2010, 25, 2247–2255. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, S.; Bandholz, A.M.; Parkash, S.; Dyack, S.; Rideout, A.L.; Leppig, K.A.; Thiese, H.; Wheeler, P.G.; Tsang, M.; Ballif, B.C.; et al. Phenotypic and molecular characterization of 19q12q13.1 deletions: A report of five patients. Am. J. Med. Genet. A 2014, 164, 62–69. [Google Scholar] [CrossRef] [PubMed]
- Malan, V.; Raoul, O.; Firth, H.V.; Royer, G.; Turleau, C.; Bernheim, A.; Willatt, L.; Munnich, A.; Vekemans, M.; Lyonnet, S.; et al. 19q13.11 deletion syndrome: A novel clinically recognisable genetic condition identified by array comparative genomic hybridisation. J. Med. Genet. 2009, 46, 635–640. [Google Scholar] [CrossRef] [PubMed]
- Roubertoux, P.L.; Tordjman, S.; Caubit, X.; di Cristopharo, J.; Ghata, A.; Fasano, L.; Kerkerian-Le Goff, L.; Gubellini, P.; Carlier, M. Construct Validity and Cross Validity of a Test Battery Modeling Autism Spectrum Disorder (ASD) in Mice. Behav. Genet. 2020, 50, 26–40. [Google Scholar] [CrossRef] [PubMed]
- Chabbert, D.; Caubit, X.; Roubertoux, P.L.; Carlier, M.; Habermann, B.; Jacq, B.; Salin, P.; Metwaly, M.; Frahm, C.; Fatmi, A.; et al. Postnatal Tshz3 Deletion Drives Altered Corticostriatal Function and Autism Spectrum Disorder-like Behavior. Biol. Psychiatry 2019, 86, 274–285. [Google Scholar] [CrossRef] [PubMed]
- Wagner, M.; Berutti, R.; Lorenz-Depiereux, B.; Graf, E.; Eckstein, G.; Mayr, J.A.; Meitinger, T.; Ahting, U.; Prokisch, H.; Strom, T.M.; et al. Mitochondrial DNA mutation analysis from exome sequencing-A more holistic approach in diagnostics of suspected mitochondrial disease. J. Inherit. Metab. Dis. 2019, 42, 909–917. [Google Scholar] [CrossRef] [PubMed]
- Nicolaou, N.; Pulit, S.L.; Nijman, I.J.; Monroe, G.R.; Feitz, W.F.J.; Schreuder, M.F.; van Eerde, A.M.; de Jong, T.P.V.M.; Giltay, J.; van der Zwaag, B.; et al. Prioritization and burden analysis of rare variants in 208 candidate genes suggest they do not play a major role in CAKUT. Kidney Int. 2016, 89, 476–486. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Feichtinger, R.G.; Preisel, M.; Steinbrücker, K.; Brugger, K.; Radda, A.; Wortmann, S.B.; Mayr, J.A. A TSHZ3 Frame-Shift Variant Causes Neurodevelopmental and Renal Disorder Consistent with Previously Described Proximal Chromosome 19q13.11 Deletion Syndrome. Genes 2022, 13, 2191. https://doi.org/10.3390/genes13122191
Feichtinger RG, Preisel M, Steinbrücker K, Brugger K, Radda A, Wortmann SB, Mayr JA. A TSHZ3 Frame-Shift Variant Causes Neurodevelopmental and Renal Disorder Consistent with Previously Described Proximal Chromosome 19q13.11 Deletion Syndrome. Genes. 2022; 13(12):2191. https://doi.org/10.3390/genes13122191
Chicago/Turabian StyleFeichtinger, René G., Martin Preisel, Katja Steinbrücker, Karin Brugger, Alexandra Radda, Saskia B. Wortmann, and Johannes A. Mayr. 2022. "A TSHZ3 Frame-Shift Variant Causes Neurodevelopmental and Renal Disorder Consistent with Previously Described Proximal Chromosome 19q13.11 Deletion Syndrome" Genes 13, no. 12: 2191. https://doi.org/10.3390/genes13122191
APA StyleFeichtinger, R. G., Preisel, M., Steinbrücker, K., Brugger, K., Radda, A., Wortmann, S. B., & Mayr, J. A. (2022). A TSHZ3 Frame-Shift Variant Causes Neurodevelopmental and Renal Disorder Consistent with Previously Described Proximal Chromosome 19q13.11 Deletion Syndrome. Genes, 13(12), 2191. https://doi.org/10.3390/genes13122191