
SNP-by-CpG Site Interactions in ABCA7 Are Associated with Cognition in Older African Americans
 , , and
, , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample
2.2. Measures
2.2.1. General Cognitive Function
- The Weschler Adult Intelligence Scale-Revised: Digit Symbol Substitution Test (DSST) measured complex visual attention, sustained and focused concentration, response speed, and visuomotor coordination. The DSST is related to the executive function of working memory in cognition [34]. In this test, participants matched symbols to numbers according to a key located at the top of the page. The DSST score comprised the number of symbols correctly matched within 90 s.
- The Controlled Oral Word Association Test (COWA-FAS) tested for verbal fluency (phonetic association) and language. This required participants to generate as many words as possible that start with F, A, and S in 1 min. The score consisted of the total number of admissible words generated.
- The Rey Auditory Verbal Learning Test (RAVLT) measured delayed recall, relating to the cognitive functions of new learning, immediate memory span, and vulnerability to learning interference, and recognition memory. Scores were determined by the number of words recalled after a 30-min delay. Scores ranged from 0 to 15.
- The Stroop Color–Word Test (SCWT) assessed concentration effectiveness by requiring participants to state the color of a word, rather than the word written. The score sums the number of color words that were correctly stated in 45 s. Specifically, the ability to shift perceptual sets in response to novel stimuli was tested.
- The Trail Making Test A (TMTA) evaluated visual conceptual tracking, as participants are required to connect a set of 25 circles quickly and accurately. TMTA provided information on the cognitive functions of visual search, scanning, processing speed, and executive functions. The TMTA score was measured as the amount of time (seconds) the participants took to complete the task. The maximum time allowed was 240 s. Prior to analysis, TMTA scores were natural log-transformed and recoded so that higher scores indicated better cognitive function.
2.2.2. Demographic Data
2.2.3. Genetic Data
2.2.4. DNA Methylation Data
2.2.5. Gene Expression Data
2.3. Statistical Analysis
2.3.1. Genetic Analysis
2.3.2. Epigenetic Analysis
2.3.3. Genetic-Epigenetic Interaction Analysis
2.3.4. Gene Expression Analysis
2.3.5. Multiple Testing Correction
3. Results
3.1. Sample Characteristics
3.2. Correlation among Six Cognitive Outcomes
3.3. Correlation among ABCA7 SNPs
3.4. Genetic Associations
3.5. Epigenetic Associations
3.6. Genetic-Epigenetic Interactions
3.7. Gene Expression Associations
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hampel, H.; Hardy, J.; Blennow, K.; Chen, C.; Perry, G.; Kim, S.H.; Villemagne, V.L.; Aisen, P.; Vendruscolo, M.; Iwatsubo, T.; et al. The Amyloid-β Pathway in Alzheimer’s Disease. Mol. Psychiatry 2021, 26, 5481–5503. [Google Scholar] [CrossRef] [PubMed]
- Grundke-Iqbal, I.; Iqbal, K.; Tung, Y.C.; Quinlan, M.; Wisniewski, H.M.; Binder, L.I. Abnormal Phosphorylation of the Microtubule-Associated Protein Tau (Tau) in Alzheimer Cytoskeletal Pathology. Proc. Natl. Acad. Sci. USA 1986, 83, 4913–4917. [Google Scholar] [CrossRef] [PubMed]
- 2021 Alzheimer’s Disease Facts and Figures. Alzheimers Dement. 2021, 17, 327–406. [CrossRef]
- Barnes, L.L.; Bennett, D.A. Alzheimer’s Disease in African Americans: Risk Factors and Challenges for the Future. Health Aff. 2014, 33, 580–586. [Google Scholar] [CrossRef] [PubMed]
- Tang, M.X.; Cross, P.; Andrews, H.; Jacobs, D.M.; Small, S.; Bell, K.; Merchant, C.; Lantigua, R.; Costa, R.; Stern, Y.; et al. Incidence of AD in African-Americans, Caribbean Hispanics, and Caucasians in Northern Manhattan. Neurology 2001, 56, 49–56. [Google Scholar] [CrossRef]
- Ridge, P.G.; Mukherjee, S.; Crane, P.K.; Kauwe, J.S.K.; Consortium, A.D.G. Alzheimer’s Disease: Analyzing the Missing Heritability. PLoS ONE 2013, 8, e79771. [Google Scholar] [CrossRef]
- Migliore, L.; Coppedè, F. Genetics, Environmental Factors and the Emerging Role of Epigenetics in Neurodegenerative Diseases. Mutat. Res. 2009, 667, 82–97. [Google Scholar] [CrossRef]
- Reitz, C.; Jun, G.; Naj, A.; Rajbhandary, R.; Vardarajan, B.N.; Wang, L.-S.; Valladares, O.; Lin, C.-F.; Larson, E.B.; Graff-Radford, N.R.; et al. Variants in the ATP-Binding Cassette Transporter (ABCA7), Apolipoprotein E Ε4,and the Risk of Late-Onset Alzheimer Disease in African Americans. JAMA 2013, 309, 1483–1492. [Google Scholar] [CrossRef]
- Strittmatter, W.J.; Saunders, A.M.; Schmechel, D.; Pericak-Vance, M.; Enghild, J.; Salvesen, G.S.; Roses, A.D. Apolipoprotein E: High-Avidity Binding to Beta-Amyloid and Increased Frequency of Type 4 Allele in Late-Onset Familial Alzheimer Disease. Proc. Natl. Acad. Sci. USA 1993, 90, 1977–1981. [Google Scholar] [CrossRef]
- Strittmatter, W.J.; Roses, A.D. Apolipoprotein E and Alzheimer Disease. Proc. Natl. Acad. Sci. USA 1995, 92, 4725–4727. [Google Scholar] [CrossRef]
- Corder, E.H.; Saunders, A.M.; Strittmatter, W.J.; Schmechel, D.E.; Gaskell, P.C.; Small, G.W.; Roses, A.D.; Haines, J.L.; Pericak-Vance, M.A. Gene Dose of Apolipoprotein E Type 4 Allele and the Risk of Alzheimer’s Disease in Late Onset Families. Science 1993, 261, 921–923. [Google Scholar] [CrossRef]
- Lamartinière, Y.; Boucau, M.-C.; Dehouck, L.; Krohn, M.; Pahnke, J.; Candela, P.; Gosselet, F.; Fenart, L. ABCA7 Downregulation Modifies Cellular Cholesterol Homeostasis and Decreases Amyloid-β Peptide Efflux in an In Vitro Model of the Blood-Brain Barrier. J. Alzheimers Dis. 2018, 64, 1195–1211. [Google Scholar] [CrossRef]
- Aikawa, T.; Holm, M.-L.; Kanekiyo, T. ABCA7 and Pathogenic Pathways of Alzheimer’s Disease. Brain Sci. 2018, 8, 27. [Google Scholar] [CrossRef]
- Zhao, Q.-F.; Yu, J.-T.; Tan, M.-S.; Tan, L. ABCA7 in Alzheimer’s Disease. Mol. Neurobiol. 2015, 51, 1008–1016. [Google Scholar] [CrossRef]
- Wang, N.; Lan, D.; Gerbod-Giannone, M.; Linsel-Nitschke, P.; Jehle, A.W.; Chen, W.; Martinez, L.O.; Tall, A.R. ATP-Binding Cassette Transporter A7 (ABCA7) Binds Apolipoprotein A-I and Mediates Cellular Phospholipid but Not Cholesterol Efflux. J. Biol. Chem. 2003, 278, 42906–42912. [Google Scholar] [CrossRef]
- Logue, M.W.; Lancour, D.; Farrell, J.; Simkina, I.; Fallin, M.D.; Lunetta, K.L.; Farrer, L.A. Targeted Sequencing of Alzheimer Disease Genes in African Americans Implicates Novel Risk Variants. Front. Neurosci. 2018, 12, 592. [Google Scholar] [CrossRef]
- N’Songo, A.; Carrasquillo, M.M.; Wang, X.; Burgess, J.D.; Nguyen, T.; Asmann, Y.W.; Serie, D.J.; Younkin, S.G.; Allen, M.; Pedraza, O.; et al. African American Exome Sequencing Identifies Potential Risk Variants at Alzheimer Disease Loci. Neurol. Genet. 2017, 3, e141. [Google Scholar] [CrossRef]
- Jaenisch, R.; Bird, A. Epigenetic Regulation of Gene Expression: How the Genome Integrates Intrinsic and Environmental Signals. Nat. Genet. 2003, 33, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Cain, J.A.; Montibus, B.; Oakey, R.J. Intragenic CpG Islands and Their Impact on Gene Regulation. Front. Cell Dev. Biol. 2022, 10, 832348. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.S.; Sun, Y.; Wang, T. Epigenome-Wide Association Study of Alzheimer’s Disease Replicates 22 Differentially Methylated Positions and 30 Differentially Methylated Regions. Clin. Epigenet. 2020, 12, 149. [Google Scholar] [CrossRef]
- Yu, L.; Chibnik, L.B.; Srivastava, G.P.; Pochet, N.; Yang, J.; Xu, J.; Kozubek, J.; Obholzer, N.; Leurgans, S.E.; Schneider, J.A.; et al. Association of Brain DNA Methylation in SORL1, ABCA7, HLA-DRB5, SLC24A4, and BIN1 with Pathological Diagnosis of Alzheimer Disease. JAMA Neurol. 2015, 72, 15–24. [Google Scholar] [CrossRef] [PubMed]
- De Jager, P.L.; Srivastava, G.; Lunnon, K.; Burgess, J.; Schalkwyk, L.C.; Yu, L.; Eaton, M.L.; Keenan, B.T.; Ernst, J.; McCabe, C.; et al. Alzheimer’s Disease: Early Alterations in Brain DNA Methylation at ANK1, BIN1, RHBDF2 and Other Loci. Nat. Neurosci. 2014, 17, 1156–1163. [Google Scholar] [CrossRef]
- Lunnon, K.; Smith, R.; Hannon, E.; De Jager, P.L.; Srivastava, G.; Volta, M.; Troakes, C.; Al-Sarraj, S.; Burrage, J.; Macdonald, R.; et al. Methylomic Profiling Implicates Cortical Deregulation of ANK1 in Alzheimer’s Disease. Nat. Neurosci. 2014, 17, 1164–1170. [Google Scholar] [CrossRef]
- Yamazaki, K.; Yoshino, Y.; Mori, T.; Yoshida, T.; Ozaki, Y.; Sao, T.; Mori, Y.; Ochi, S.; Iga, J.; Ueno, S. Gene Expression and Methylation Analysis of ABCA7 in Patients with Alzheimer’s Disease. J. Alzheimers Dis. 2017, 57, 171–181. [Google Scholar] [CrossRef]
- Apostolova, L.G.; Risacher, S.L.; Duran, T.; Stage, E.C.; Goukasian, N.; West, J.D.; Do, T.M.; Grotts, J.; Wilhalme, H.; Nho, K.; et al. Associations of the Top 20 Alzheimer Disease Risk Variants with Brain Amyloidosis. JAMA Neurol. 2018, 75, 328–341. [Google Scholar] [CrossRef]
- De Roeck, A.; Van Broeckhoven, C.; Sleegers, K. The Role of ABCA7 in Alzheimer’s Disease: Evidence from Genomics, Transcriptomics and Methylomics. Acta Neuropathol. 2019, 138, 201–220. [Google Scholar] [CrossRef]
- Nettiksimmons, J.; Tranah, G.; Evans, D.S.; Yokoyama, J.S.; Yaffe, K. Gene-Based Aggregate SNP Associations between Candidate AD Genes and Cognitive Decline. Age 2016, 38, 41. [Google Scholar] [CrossRef]
- Engelman, C.D.; Koscik, R.L.; Jonaitis, E.M.; Okonkwo, O.C.; Hermann, B.P.; La Rue, A.; Sager, M.A. Interaction between Two Cholesterol Metabolism Genes Influences Memory: Findings from the Wisconsin Registry for Alzheimer’s Prevention. J. Alzheimers Dis. 2013, 36, 749–757. [Google Scholar] [CrossRef]
- Carrasquillo, M.M.; Crook, J.E.; Pedraza, O.; Thomas, C.S.; Pankratz, V.S.; Allen, M.; Nguyen, T.; Malphrus, K.G.; Ma, L.; Bisceglio, G.D.; et al. Late-Onset Alzheimer’s Risk Variants in Memory Decline, Incident Mild Cognitive Impairment, and Alzheimer’s Disease. Neurobiol. Aging 2015, 36, 60–67. [Google Scholar] [CrossRef]
- Cacabelos, R.; Torrellas, C. Epigenetics of Aging and Alzheimer’s Disease: Implications for Pharmacogenomics and Drug Response. Int. J. Mol. Sci. 2015, 16, 30483–30543. [Google Scholar] [CrossRef]
- Daniels, P.R.; Kardia, S.L.R.; Hanis, C.L.; Brown, C.A.; Hutchinson, R.; Boerwinkle, E.; Turner, S.T.; Genetic Epidemiology Network of Arteriopathy study. Familial Aggregation of Hypertension Treatment and Control in the Genetic Epidemiology Network of Arteriopathy (GENOA) Study. Am. J. Med. 2004, 116, 676–681. [Google Scholar] [CrossRef] [PubMed]
- Lezak, M.D.; Howieson, D.B.; Loring, D.W.; Hannay, H.; Fischer, J. Neuropsychological Assessment; Oxford University Press: New York, NY, USA, 2004; ISBN 978-0-19-511121-7. [Google Scholar]
- Smith, J.A.; Mosley, T.H., Jr.; Turner, S.T.; Kardia, S.L. Shared Genetic Effects among Measures of Cognitive Function and Leukoaraiosis. In Brain Injury–Pathogenesis, Monitoring, Recovery and Management; Agrawal, A., Ed.; Intechopen: London, UK, 2012; p. 39. [Google Scholar]
- Jaeger, J. Digit Symbol Substitution Test. J. Clin. Psychopharmacol. 2018, 38, 513–519. [Google Scholar] [CrossRef] [PubMed]
- Davies, G.; Armstrong, N.; Bis, J.C.; Bressler, J.; Chouraki, V.; Giddaluru, S.; Hofer, E.; Ibrahim-Verbaas, C.A.; Kirin, M.; Lahti, J.; et al. Genetic Contributions to Variation in General Cognitive Function: A Meta-Analysis of Genome-Wide Association Studies in the CHARGE Consortium (N = 53 949). Mol. Psychiatry 2015, 20, 183–192. [Google Scholar] [CrossRef]
- Gao, X.; Jia, M.; Zhang, Y.; Breitling, L.P.; Brenner, H. DNA Methylation Changes of Whole Blood Cells in Response to Active Smoking Exposure in Adults: A Systematic Review of DNA Methylation Studies. Clin. Epigenet. 2015, 7, 113. [Google Scholar] [CrossRef] [PubMed]
- Fortin, J.-P.; Fertig, E.; Hansen, K. ShinyMethyl: Interactive Quality Control of Illumina 450k DNA Methylation Arrays in R. F1000 Res. 2014, 3, 175. [Google Scholar] [CrossRef]
- Xu, Z.; Niu, L.; Li, L.; Taylor, J.A. ENmix: A Novel Background Correction Method for Illumina HumanMethylation450 BeadChip. Nucleic Acids Res. 2016, 44, e20. [Google Scholar] [CrossRef]
- Fortin, J.-P.; Triche, T.J., Jr.; Hansen, K.D. Preprocessing, Normalization and Integration of the Illumina HumanMethylationEPIC Array with Minfi. Bioinformatics 2017, 33, 558–560. [Google Scholar] [CrossRef]
- Aryee, M.J.; Jaffe, A.E.; Corrada-Bravo, H.; Ladd-Acosta, C.; Feinberg, A.P.; Hansen, K.D.; Irizarry, R.A. Minfi: A Flexible and Comprehensive Bioconductor Package for the Analysis of Infinium DNA Methylation Microarrays. Bioinformatics 2014, 30, 1363–1369. [Google Scholar] [CrossRef]
- Niu, L.; Xu, Z.; Taylor, J.A. RCP: A Novel Probe Design Bias Correction Method for Illumina Methylation BeadChip. Bioinformatics 2016, 32, 2659–2663. [Google Scholar] [CrossRef]
- Lehne, B.; Drong, A.W.; Loh, M.; Zhang, W.; Scott, W.R.; Tan, S.-T.; Afzal, U.; Scott, J.; Jarvelin, M.-R.; Elliott, P.; et al. A Coherent Approach for Analysis of the Illumina HumanMethylation450 BeadChip Improves Data Quality and Performance in Epigenome-Wide Association Studies. Genome Biol. 2015, 16, 37. [Google Scholar] [CrossRef]
- Hansen, K. IlluminaHumanMethylationEPICanno.ilm10b2.hg19: Annotation for Illumina’s EPIC Methylation Arrays, R package version 0.6.0; BioConductor: Boston, MA, USA, 2016. [Google Scholar]
- Houseman, E.A.; Accomando, W.P.; Koestler, D.C.; Christensen, B.C.; Marsit, C.J.; Nelson, H.H.; Wiencke, J.K.; Kelsey, K.T. DNA Methylation Arrays as Surrogate Measures of Cell Mixture Distribution. BMC Bioinform. 2012, 13, 86. [Google Scholar] [CrossRef]
- Du, P.; Zhang, X.; Huang, C.-C.; Jafari, N.; Kibbe, W.A.; Hou, L.; Lin, S.M. Comparison of Beta-Value and M-Value Methods for Quantifying Methylation Levels by Microarray Analysis. BMC Bioinform. 2010, 11, 587. [Google Scholar] [CrossRef]
- Weisenberger, C.D.J.; Van den Berg, D.; Pan, F.; Berman, B.P.; Laird, P.W. Comprehensive DNA Methylation Analysis on the Illumina® Infinium® Assay Platform; Illumina: San Diego, CA, USA, 2008; p. 4. [Google Scholar]
- Lockstone, H.E. Exon Array Data Analysis Using Affymetrix Power Tools and R Statistical Software. Brief. Bioinform. 2011, 12, 634–644. [Google Scholar] [CrossRef]
- Irizarry, R.A.; Bolstad, B.M.; Collin, F.; Cope, L.M.; Hobbs, B.; Speed, T.P. Summaries of Affymetrix GeneChip Probe Level Data. Nucleic Acids Res. 2003, 31, e15. [Google Scholar] [CrossRef]
- Dai, M.; Wang, P.; Boyd, A.D.; Kostov, G.; Athey, B.; Jones, E.G.; Bunney, W.E.; Myers, R.M.; Speed, T.P.; Akil, H.; et al. Evolving Gene/Transcript Definitions Significantly Alter the Interpretation of GeneChip Data. Nucleic Acids Res. 2005, 33, e175. [Google Scholar] [CrossRef]
- Saha, A.; Battle, A. False Positives in Trans-EQTL and Co-Expression Analyses Arising from RNA-Sequencing Alignment Errors. F1000Research 2018, 7, 1860. [Google Scholar] [CrossRef]
- Johnson, W.E.; Li, C.; Rabinovic, A. Adjusting Batch Effects in Microarray Expression Data Using Empirical Bayes Methods. Biostatistics 2007, 8, 118–127. [Google Scholar] [CrossRef]
- Martin, T.C.; Hardiman, T.; Yet, I.; Tsai, P.-C.; Bell, J.T. CoMET: Visualisation of Regional Epigenome-Wide Association Scan (EWAS) Results and DNA Co-Methylation Patterns. BMC Bioinform. 2015, 16, 131. [Google Scholar] [CrossRef]
- BioRender. Available online: https://app.biorender.com/illustrations/634485ef72e6ef474cc65999 (accessed on 10 October 2022).
- Lenth, R.V.; Buerkner, P.; Herve, M.; Love, J.; Riebl, H.; Singmann, H. Emmeans: Estimated Marginal Means, Aka Least-Squares Means, R package version: 1.8.2; CRAN: Stamford, CT, USA, 2022. [Google Scholar]
- Fox, J.; Weisberg, S.; Price, B.; Friendly, M.; Hong, J.; Andersen, R.; Firth, D.; Taylor, S. ; R Core Team. Effects: Effect Displays for Linear, Generalized Linear, and Other Models, R package version4.2-2; CRAN: Stamford, CT, USA, 2022. [Google Scholar]
- Ggplot2 Package—RDocumentation. Available online: https://www.rdocumentation.org/packages/ggplot2/versions/3.3.5 (accessed on 17 December 2021).
- The Gtex Consortium. The GTEx Consortium Atlas of Genetic Regulatory Effects across Human Tissues. Science 2020, 369, 1318–1330. [Google Scholar] [CrossRef]
- Gene: ABCA7 (ENSG00000064687)-Summary–Homo_sapiens-Ensembl Genome Browser 107. Available online: https://useast.ensembl.org/Homo_sapiens/Gene/Summary?db=core;g=ENSG00000064687;r=19:1039997-1065572 (accessed on 14 October 2022).
- Komaki, S.; Shiwa, Y.; Furukawa, R.; Hachiya, T.; Ohmomo, H.; Otomo, R.; Satoh, M.; Hitomi, J.; Sobue, K.; Sasaki, M.; et al. IMETHYL: An Integrative Database of Human DNA Methylation, Gene Expression, and Genomic Variation. Hum. Genome Var. 2018, 5, 18008. [Google Scholar] [CrossRef]
- Hachiya, T.; Furukawa, R.; Shiwa, Y.; Ohmomo, H.; Ono, K.; Katsuoka, F.; Nagasaki, M.; Yasuda, J.; Fuse, N.; Kinoshita, K.; et al. Genome-Wide Identification of Inter-Individually Variable DNA Methylation Sites Improves the Efficacy of Epigenetic Association Studies. NPJ Genom. Med. 2017, 2, 11. [Google Scholar] [CrossRef] [PubMed]
- The R Project for Statistical Computing. Available online: https://www.r-project.org/ (accessed on 14 June 2022).
- Naj, A.C.; Jun, G.; Beecham, G.W.; Wang, L.-S.; Vardarajan, B.N.; Buros, J.; Gallins, P.J.; Buxbaum, J.D.; Jarvik, G.P.; Crane, P.K.; et al. Common Variants in MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 Are Associated with Late-Onset Alzheimer’s Disease. Nat. Genet. 2011, 43, 436–441. [Google Scholar] [CrossRef] [PubMed]
- Vasquez, J.B.; Fardo, D.W.; Estus, S. ABCA7 Expression Is Associated with Alzheimer’s Disease Polymorphism and Disease Status. Neurosci. Lett. 2013, 556, 58–62. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, S.; Gene, D.; Stefansson, H.; Jonsson, T.; Johannsdottir, H.; Ingason, A.; Helgason, H.; Sulem, P.; Magnusson, O.T.; Gudjonsson, S.A.; et al. Loss-of-Function Variants in ABCA7 Confer Risk of Alzheimer’s Disease. Nat. Genet. 2015, 47, 445–447. [Google Scholar] [CrossRef] [PubMed]
- De Roeck, A.; Duchateau, L.; Van Dongen, J.; Cacace, R.; Bjerke, M.; Van den Bossche, T.; Cras, P.; Vandenberghe, R.; De Deyn, P.P.; Engelborghs, S.; et al. An Intronic VNTR Affects Splicing of ABCA7 and Increases Risk of Alzheimer’s Disease. Acta Neuropathol. 2018, 135, 827–837. [Google Scholar] [CrossRef]
- De Roeck, A.; Van den Bossche, T.; van der Zee, J.; Verheijen, J.; De Coster, W.; Van Dongen, J.; Dillen, L.; Baradaran-Heravi, Y.; Heeman, B.; Sanchez-Valle, R.; et al. Deleterious ABCA7 Mutations and Transcript Rescue Mechanisms in Early Onset Alzheimer’s Disease. Acta Neuropathol. 2017, 134, 475–487. [Google Scholar] [CrossRef]
- Hof, P.R.; Glannakopoulos, P.; Bouras, C. The Neuropathological Changes Associated with Normal Brain Aging. Histol. Histopathol. 1996, 11, 1075–1088. [Google Scholar]
- Perl, D.P. Neuropathology of Alzheimer’s Disease. Mt. Sinai J. Med. 2010, 77, 32–42. [Google Scholar] [CrossRef]
- Vivot, A.; Glymour, M.M.; Tzourio, C.; Amouyel, P.; Chêne, G.; Dufouil, C. Association of Alzheimer’s Related Genotypes with Cognitive Decline in Multiple Domains: Results from the Three-City Dijon Study. Mol. Psychiatry 2015, 20, 1173–1178. [Google Scholar] [CrossRef]
- Smith, J.A.; Kho, M.; Zhao, W.; Yu, M.; Mitchell, C.; Faul, J.D. Genetic Effects and Gene-by-Education Interactions on Episodic Memory Performance and Decline in an Aging Population. Soc. Sci. Med. 2021, 271, 112039. [Google Scholar] [CrossRef]
- Davies, G.; Lam, M.; Harris, S.E.; Trampush, J.W.; Luciano, M.; Hill, W.D.; Hagenaars, S.P.; Ritchie, S.J.; Marioni, R.E.; Fawns-Ritchie, C.; et al. Study of 300,486 Individuals Identifies 148 Independent Genetic Loci Influencing General Cognitive Function. Nat. Commun. 2018, 9, 2098. [Google Scholar] [CrossRef]
- Dumitrescu, L.; Mahoney, E.R.; Mukherjee, S.; Lee, M.L.; Bush, W.S.; Engelman, C.D.; Lu, Q.; Fardo, D.W.; Trittschuh, E.H.; Mez, J.; et al. Genetic Variants and Functional Pathways Associated with Resilience to Alzheimer’s Disease. Brain 2020, 143, 2561–2575. [Google Scholar] [CrossRef]
- Marioni, R.E.; McRae, A.F.; Bressler, J.; Colicino, E.; Hannon, E.; Li, S.; Prada, D.; Smith, J.A.; Trevisi, L.; Tsai, P.-C.; et al. Meta-Analysis of Epigenome-Wide Association Studies of Cognitive Abilities. Mol. Psychiatry 2018, 23, 2133–2144. [Google Scholar] [CrossRef]
- Yu, L.; Chibnik, L.B.; Yang, J.; McCabe, C.; Xu, J.; Schneider, J.A.; De Jager, P.L.; Bennett, D.A. Methylation Profiles in Peripheral Blood CD4+ Lymphocytes versus Brain: The Relation to Alzheimer’s Disease Pathology. Alzheimers Dement. 2016, 12, 942–951. [Google Scholar] [CrossRef]
- Ashraf-ganjouei, A.; Moradi, K.; Bagheri, S.; Aarabi, M.H. The Association between Systemic Inflammation and Cognitive Performance in Healthy Adults. J. Neuroimmunol. 2020, 345, 577272. [Google Scholar] [CrossRef]
- Weinstein, G.; Lutski, M.; Goldbourt, U.; Tanne, D. C-Reactive Protein Is Related to Future Cognitive Impairment and Decline in Elderly Individuals with Cardiovascular Disease. Arch. Gerontol. Geriatr. 2017, 69, 31–37. [Google Scholar] [CrossRef]
- Cukier, H.N.; Kunkle, B.W.; Vardarajan, B.N.; Rolati, S.; Hamilton-Nelson, K.L.; Kohli, M.A.; Whitehead, P.L.; Dombroski, B.A.; Van Booven, D.; Lang, R.; et al. ABCA7 Frameshift Deletion Associated with Alzheimer Disease in African Americans. Neurol. Genet. 2016, 2, e79. [Google Scholar] [CrossRef]
- Raj, T.; Li, Y.I.; Wong, G.; Humphrey, J.; Wang, M.; Ramdhani, S.; Wang, Y.-C.; Ng, B.; Gupta, I.; Haroutunian, V.; et al. Integrative Transcriptome Analyses of the Aging Brain Implicate Altered Splicing in Alzheimer’s Disease Susceptibility. Nat. Genet. 2018, 50, 1584–1592. [Google Scholar] [CrossRef]
- Humphries, C.; Kohli, M.A.; Whitehead, P.; Mash, D.C.; Pericak-Vance, M.A.; Gilbert, J. Alzheimer Disease (AD) Specific Transcription, DNA Methylation and Splicing in Twenty AD Associated Loci. Mol. Cell. Neurosci. 2015, 67, 37–45. [Google Scholar] [CrossRef]
- Tsuboi, K.; Nagatomo, T.; Gohno, T.; Higuchi, T.; Sasaki, S.; Fujiki, N.; Kurosumi, M.; Takei, H.; Yamaguchi, Y.; Niwa, T.; et al. Single CpG Site Methylation Controls Estrogen Receptor Gene Transcription and Correlates with Hormone Therapy Resistance. J. Steroid Biochem. Mol. Biol. 2017, 171, 209–217. [Google Scholar] [CrossRef]
- Qiu, C.; Shen, H.; Fu, X.; Xu, C.; Deng, H. Meta-Analysis of Genome-Wide Association Studies Identifies Novel Functional CpG-SNPs Associated with Bone Mineral Density at Lumbar Spine. Int. J. Genom. 2018, 2018, 6407257. [Google Scholar] [CrossRef] [PubMed]
- Gertz, J.; Varley, K.E.; Reddy, T.E.; Bowling, K.M.; Pauli, F.; Parker, S.L.; Kucera, K.S.; Willard, H.F.; Myers, R.M. Analysis of DNA Methylation in a Three-Generation Family Reveals Widespread Genetic Influence on Epigenetic Regulation. PLoS Genet. 2011, 7, e1002228. [Google Scholar] [CrossRef] [PubMed]
- Harlid, S.; Ivarsson, M.I.L.; Butt, S.; Hussain, S.; Grzybowska, E.; Eyfjörd, J.E.; Lenner, P.; Försti, A.; Hemminki, K.; Manjer, J.; et al. A Candidate CpG SNP Approach Identifies a Breast Cancer Associated ESR1-SNP. Int. J. Cancer 2011, 129, 1689–1698. [Google Scholar] [CrossRef] [PubMed]
- Dayeh, T.A.; Olsson, A.H.; Volkov, P.; Almgren, P.; Rönn, T.; Ling, C. Identification of CpG-SNPs Associated with Type 2 Diabetes and Differential DNA Methylation in Human Pancreatic Islets. Diabetologia 2013, 56, 1036–1046. [Google Scholar] [CrossRef]
- Taqi, M.M.; Bazov, I.; Watanabe, H.; Sheedy, D.; Harper, C.; Alkass, K.; Druid, H.; Wentzel, P.; Nyberg, F.; Yakovleva, T.; et al. Prodynorphin CpG-SNPs Associated with Alcohol Dependence: Elevated Methylation in the Brain of Human Alcoholics. Addict. Biol. 2011, 16, 499–509. [Google Scholar] [CrossRef]
- Bani-Fatemi, A.; Gonçalves, V.F.; Zai, C.; de Souza, R.; Le Foll, B.; Kennedy, J.L.; Wong, A.H.; De Luca, V. Analysis of CpG SNPs in 34 Genes: Association Test with Suicide Attempt in Schizophrenia. Schizophr. Res. 2013, 147, 262–268. [Google Scholar] [CrossRef]
- Fridman, A.L.; Tainsky, M.A. Critical Pathways in Cellular Senescence and Immortalization Revealed by Gene Expression Profiling. Oncogene 2008, 27, 5975–5987. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Mean (SD) or n% | |
|---|---|
| Age at cognition measurement (years) | 63.31 (8.08) |
| Age difference between methylation and cognition measurements (years) a | 6.03 (1.29) |
| Sex | |
| Female | 475 (74.90%) |
| Male | 159 (25.10%) |
| Educational attainment | |
| At least some college | 300 (47.32%) |
| High school degree/GED | 169 (26.66%) |
| Less than high School degree/GED | 165 (26.03%) |
| Smoking status | |
| Current smoker | 105 (16.56%) |
| Former smoker | 146 (23.03%) |
| Never smoker | 383 (60.41%) |
| General cognitive function | 0.00 (1.00) |
| Delayed recall (RAVLT, number of words recalled) | 7.05 (3.34) |
| Processing speed (DSST, number of symbols) | 34.44 (12.62) |
| Word fluency (COWA-FAS, number of words) | 29.73 (11.61) |
| Concentration effectiveness (SCWT, number of items) | 22.53 (9.83) |
| Visual conceptual tracking (TMTA, seconds to test completion) | 61.63 (31.96) |
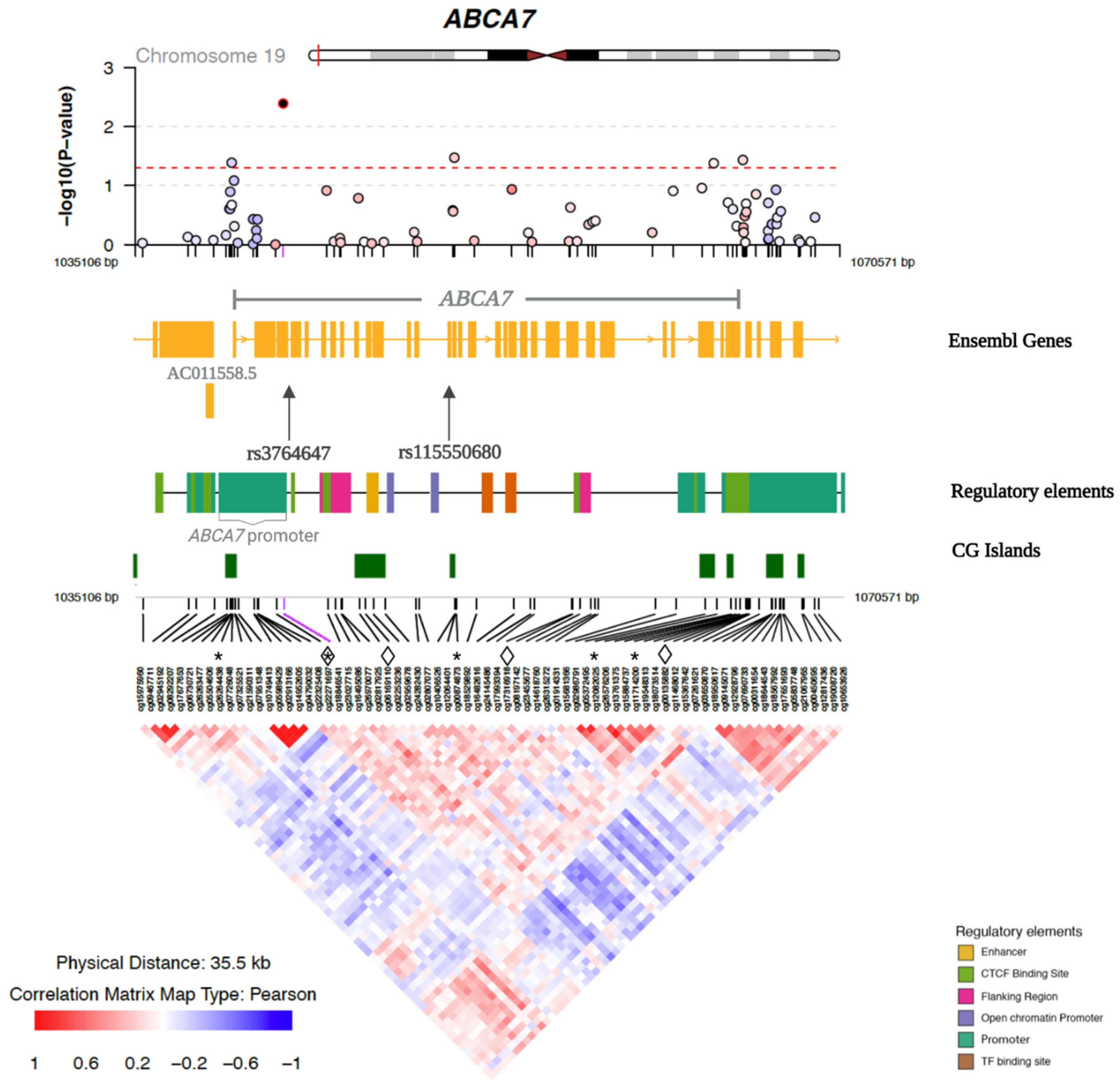
| SNP Annotation | CpG Site Annotation | Main Effects | Interaction | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SNP*CpG Site Interaction | SNP | Position | Risk Allele | RAF | Cpg Site | Position | Site Type | Relation to CGI | βSNP | p-Value | βCpG | p-Value | βinteraction | p-Value |
| rs3764647*cg00135882 | rs3764647 | 1044712 | G | 0.20 | cg00135882 | 1065783 | Promoter | North Shore | −0.01 | 0.875 | 0.24 | 0.086 | −0.80 | 1.46 × 10−4 ** |
| rs3764647*cg22271697 | rs3764647 | 1044712 | G | 0.20 | cg22271697 | 1042537 | Promoter | North Shelf | −0.07 | 0.319 | 0.16 | 7.23 × 10−6 * | −0.18 | 5.77 × 10−4 ** |
| rs115550680*cg06169110 | rs115550680 | 1050420 | G | 0.06 | cg06169110 | 1046615 | Gene Body | CG Island | −0.23 | 0.045 * | 0.06 | 0.143 | −0.38 | 2.18 × 10−4 ** |
| rs115550680*cg17316918 | rs115550680 | 1050420 | G | 0.06 | cg17316918 | 1056930 | Gene Body | Open Sea | −0.05 | 0.661 | −0.06 | 0.164 | 0.41 | 4.84 × 10−4 ** |
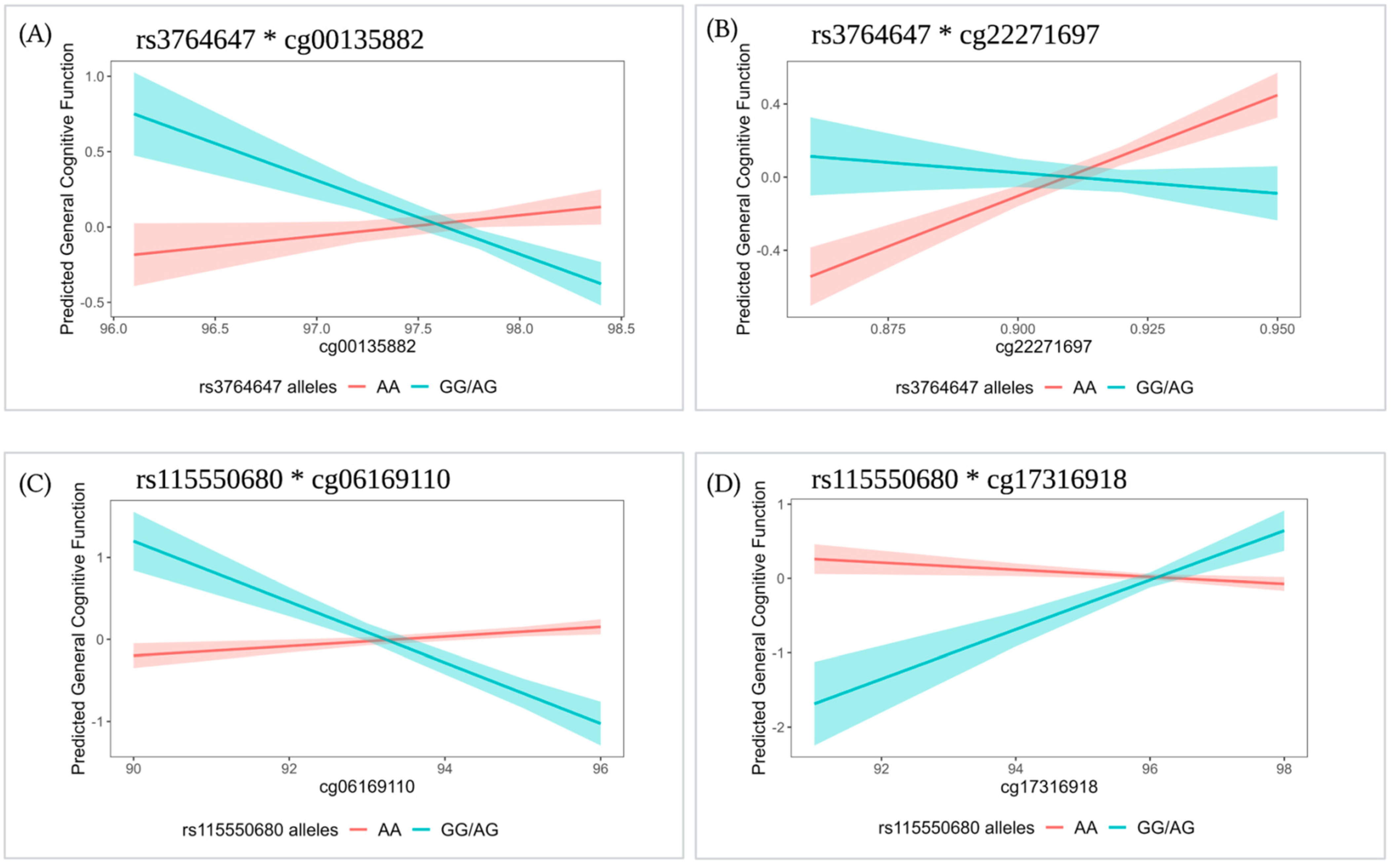
| SNP | Cpg Site | Genotype | βCpG | p-Value |
|---|---|---|---|---|
| rs3764647 a | cg00135882 | AA | 0.09 | 0.566 |
| GG/AG | −0.68 | 0.004 * | ||
| rs3764647 a | cg22271697 | AA | 0.14 | 2.00 × 10−4 * |
| GG/AG | −0.02 | 0.719 | ||
| rs115550680 b | cg06169110 | AA | 0.05 | 0.221 |
| GG/AG | −0.37 | 2.00 × 10−4 * | ||
| rs115550680 b | cg17316918 | AA | −0.06 | 0.202 |
| GG/AG | 0.33 | 0.004 * |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chaar, D.L.; Nguyen, K.; Wang, Y.-Z.; Ratliff, S.M.; Mosley, T.H.; Kardia, S.L.R.; Smith, J.A.; Zhao, W. SNP-by-CpG Site Interactions in ABCA7 Are Associated with Cognition in Older African Americans. Genes 2022, 13, 2150. https://doi.org/10.3390/genes13112150
Chaar DL, Nguyen K, Wang Y-Z, Ratliff SM, Mosley TH, Kardia SLR, Smith JA, Zhao W. SNP-by-CpG Site Interactions in ABCA7 Are Associated with Cognition in Older African Americans. Genes. 2022; 13(11):2150. https://doi.org/10.3390/genes13112150
Chicago/Turabian StyleChaar, Dima L., Kim Nguyen, Yi-Zhe Wang, Scott M. Ratliff, Thomas H. Mosley, Sharon L. R. Kardia, Jennifer A. Smith, and Wei Zhao. 2022. "SNP-by-CpG Site Interactions in ABCA7 Are Associated with Cognition in Older African Americans" Genes 13, no. 11: 2150. https://doi.org/10.3390/genes13112150
APA StyleChaar, D. L., Nguyen, K., Wang, Y.-Z., Ratliff, S. M., Mosley, T. H., Kardia, S. L. R., Smith, J. A., & Zhao, W. (2022). SNP-by-CpG Site Interactions in ABCA7 Are Associated with Cognition in Older African Americans. Genes, 13(11), 2150. https://doi.org/10.3390/genes13112150