
WGCNA Analysis Revealed the Hub Genes Related to Soil Cadmium Stress in Maize Kernel (Zea mays L.)
Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Materials and Experiment Design
2.2. Cd Determination
2.3. Samples Collection and RNA Isolation
2.4. RNA-Seq Data Processing and Analysis
2.5. WGCNA
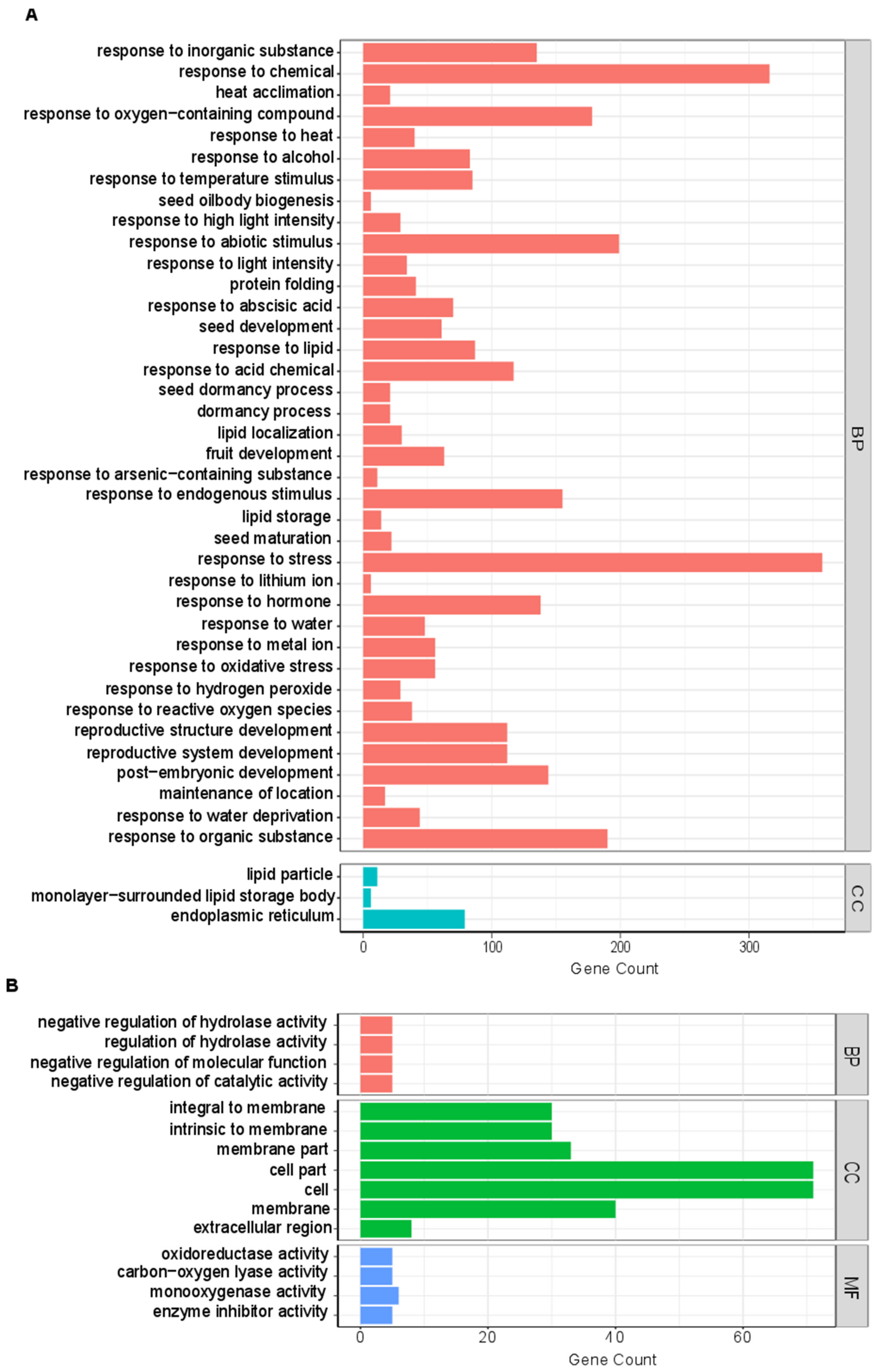
2.6. GO Enrichment Analysis
3. Results
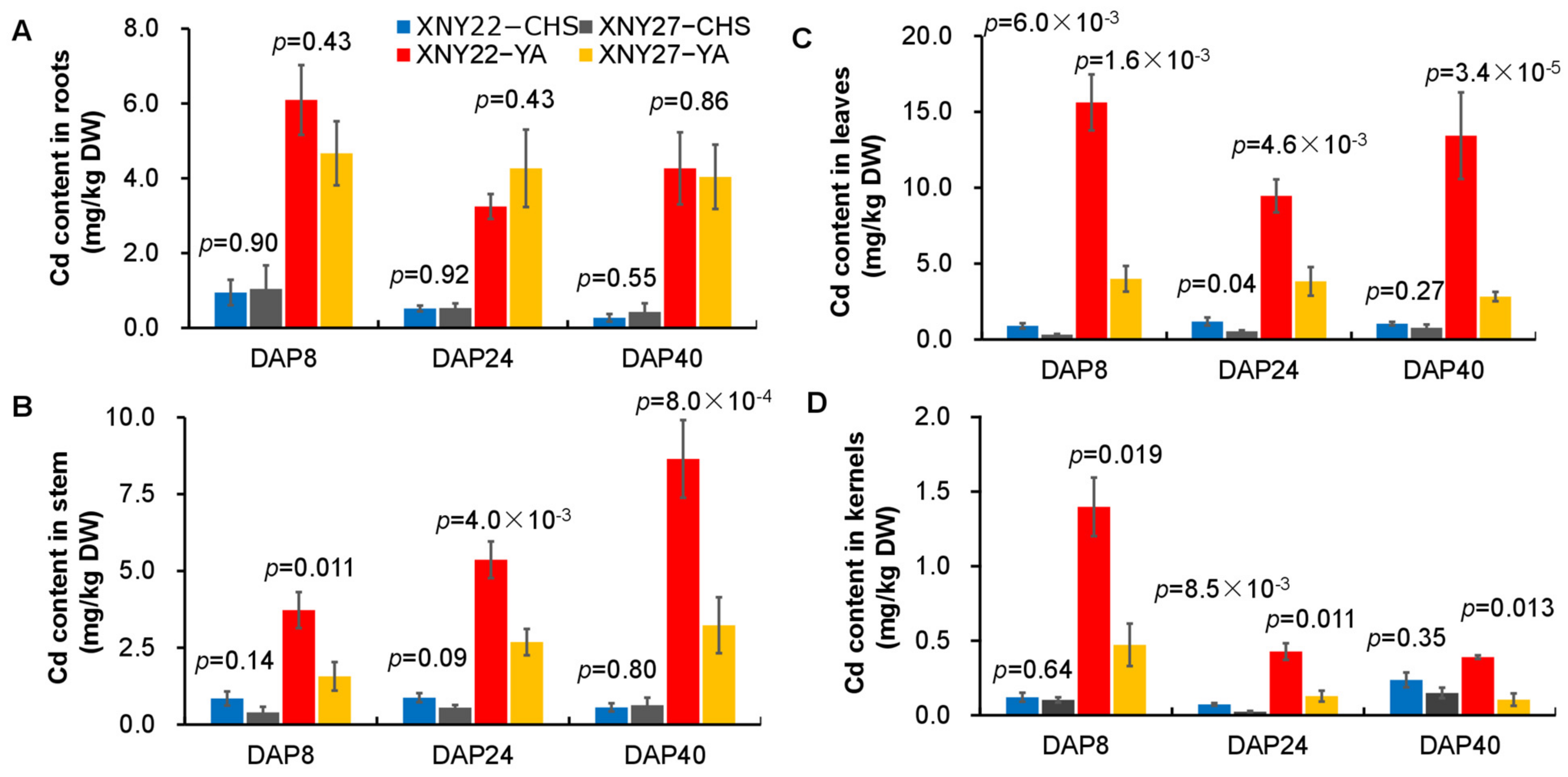
3.1. Cd Accumulation of Hybrid Maize Varieties XNY22 and XNY27 under the Different Cd Content Soil
3.2. Sequencing Quality Statistics
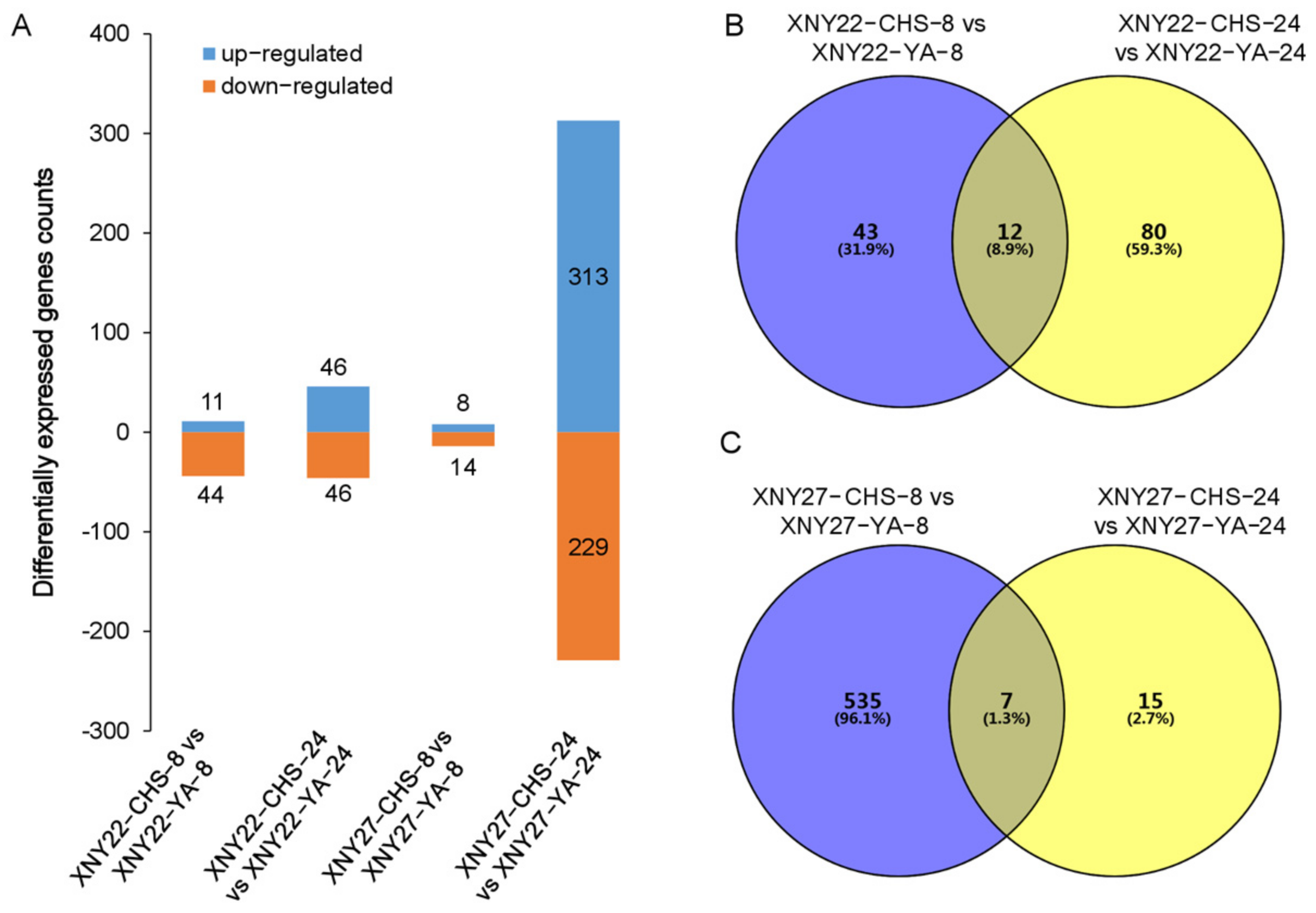
3.3. DEGs Identified between High and Low Cd Accumulation in XNY22 and XNY27
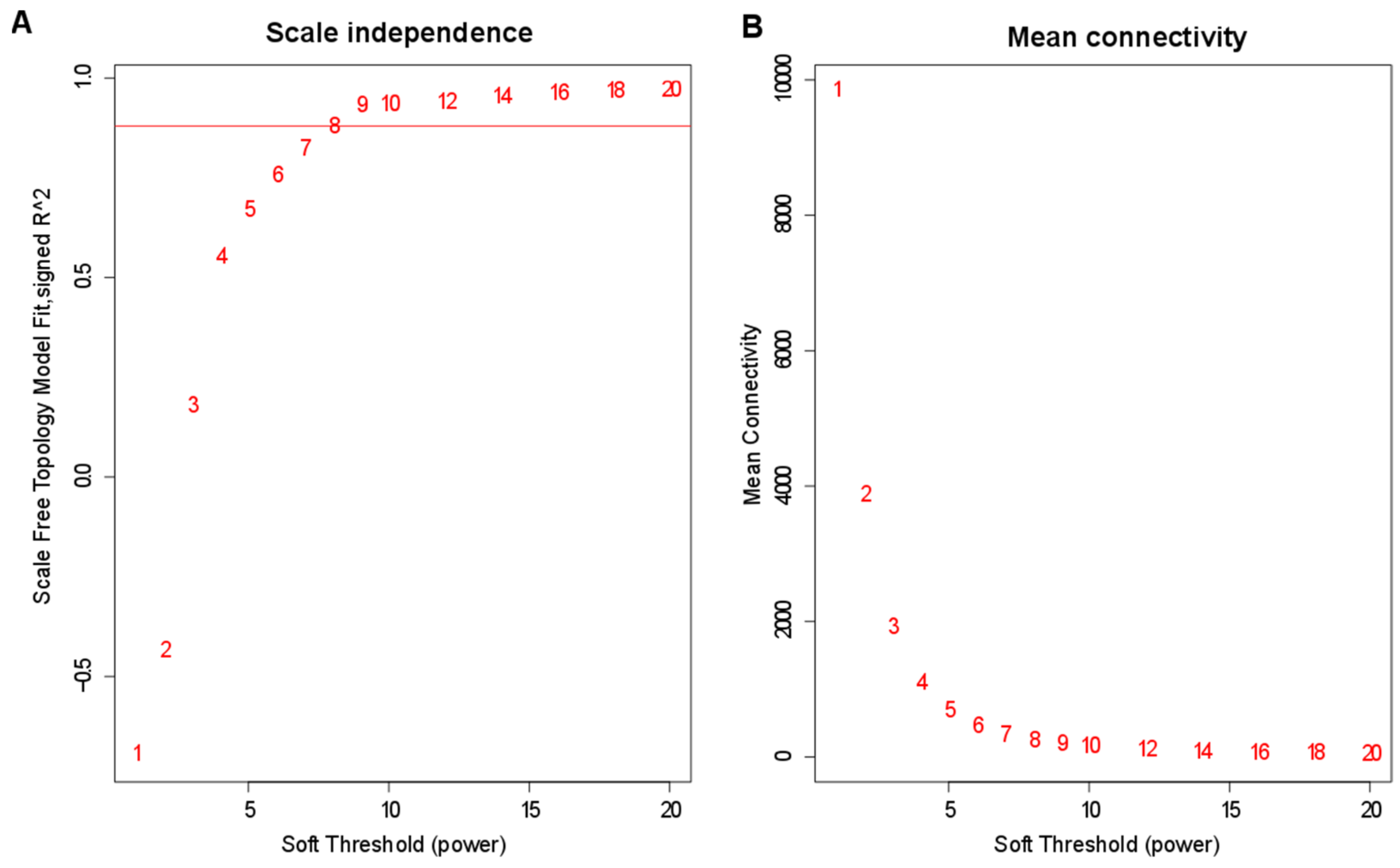
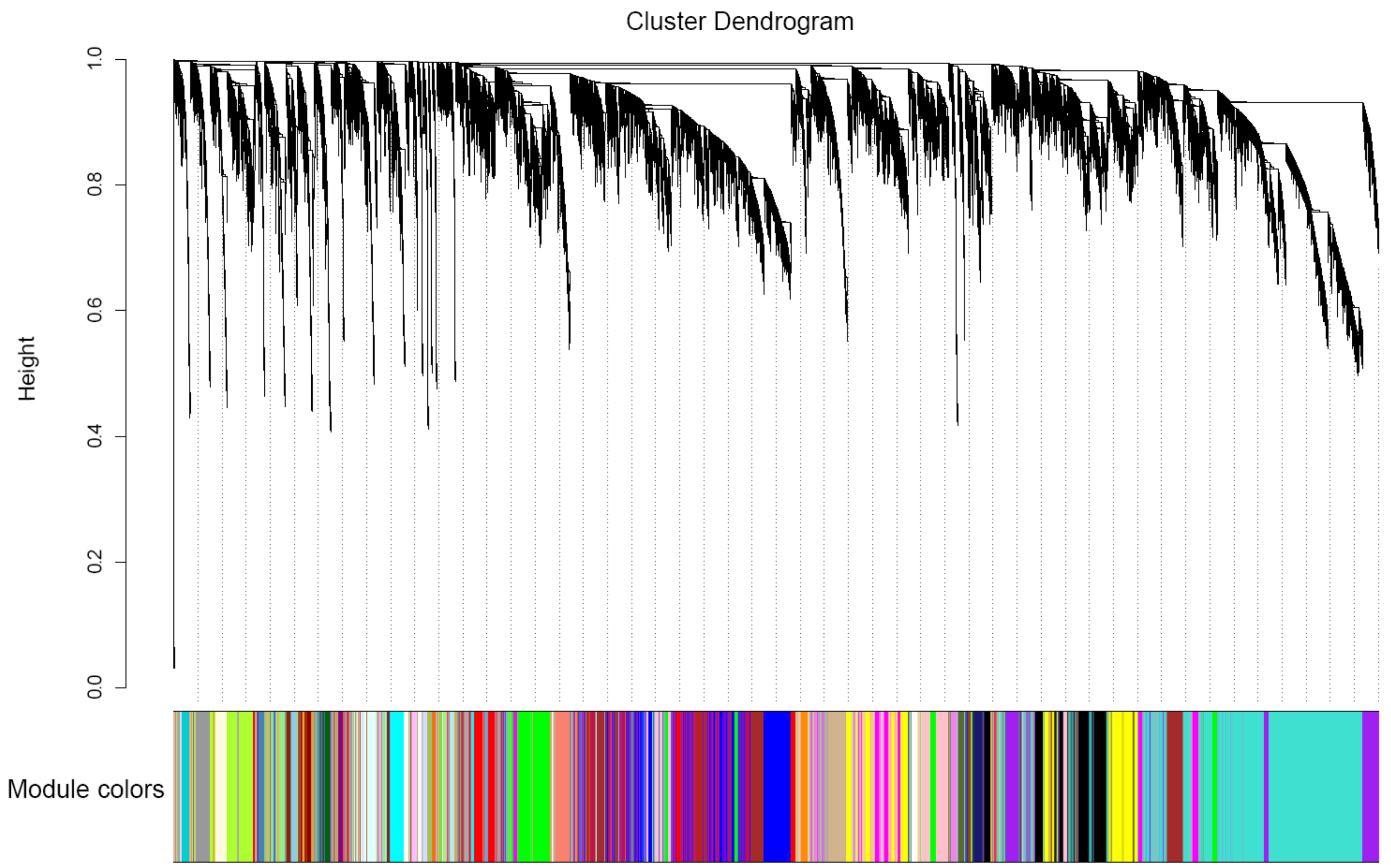
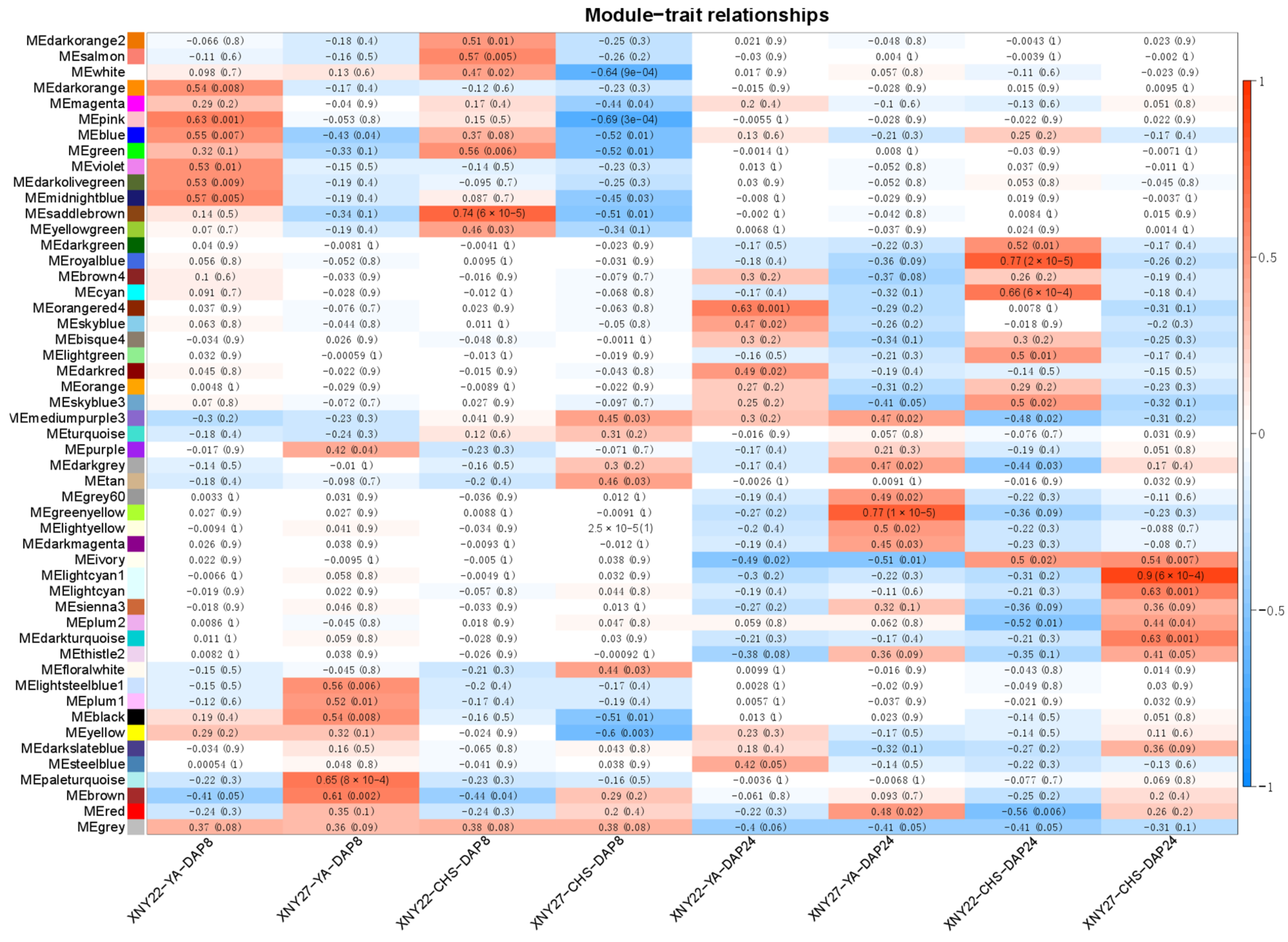
3.4. Construction of Co-Expression Network by WGCNA
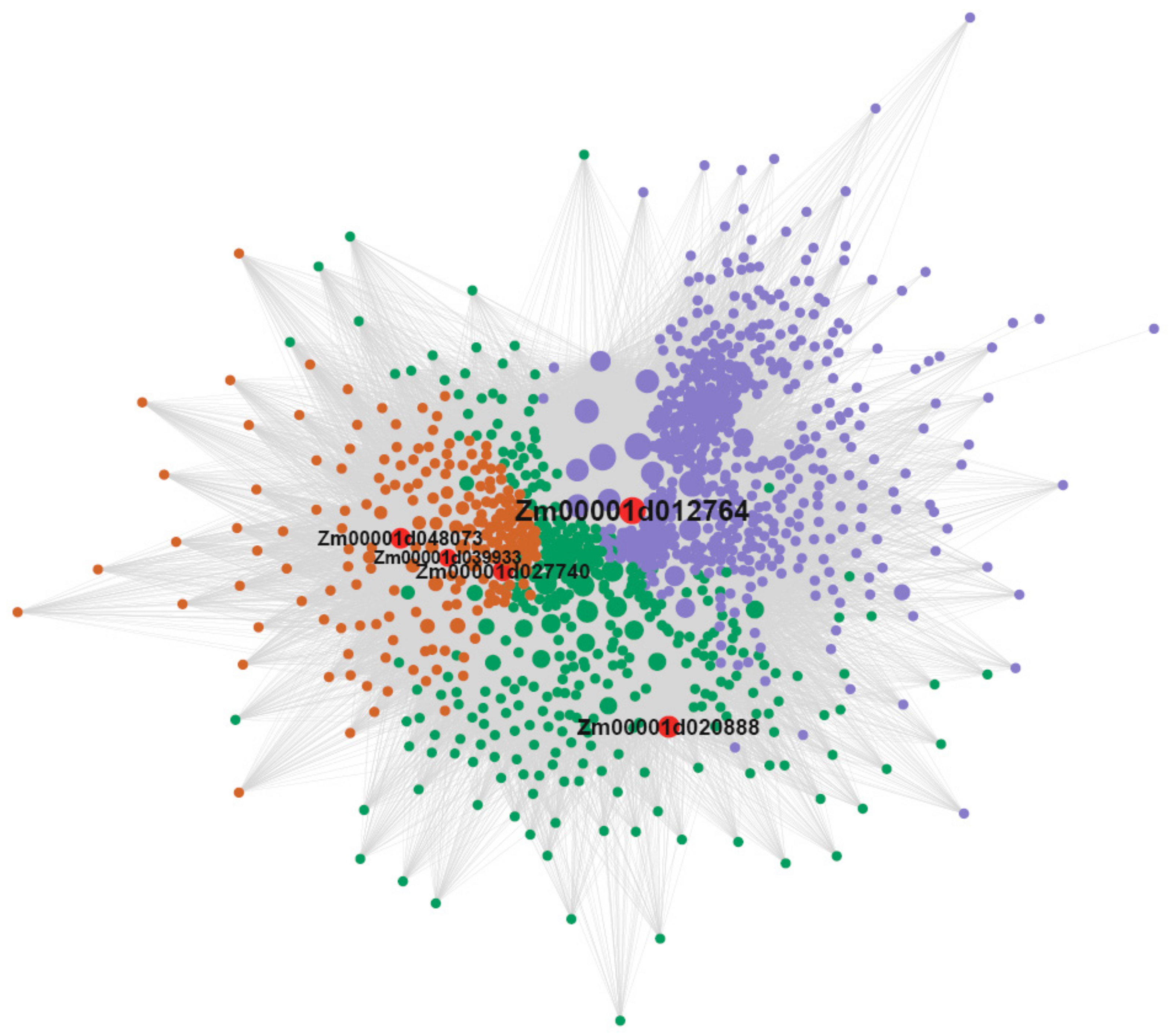
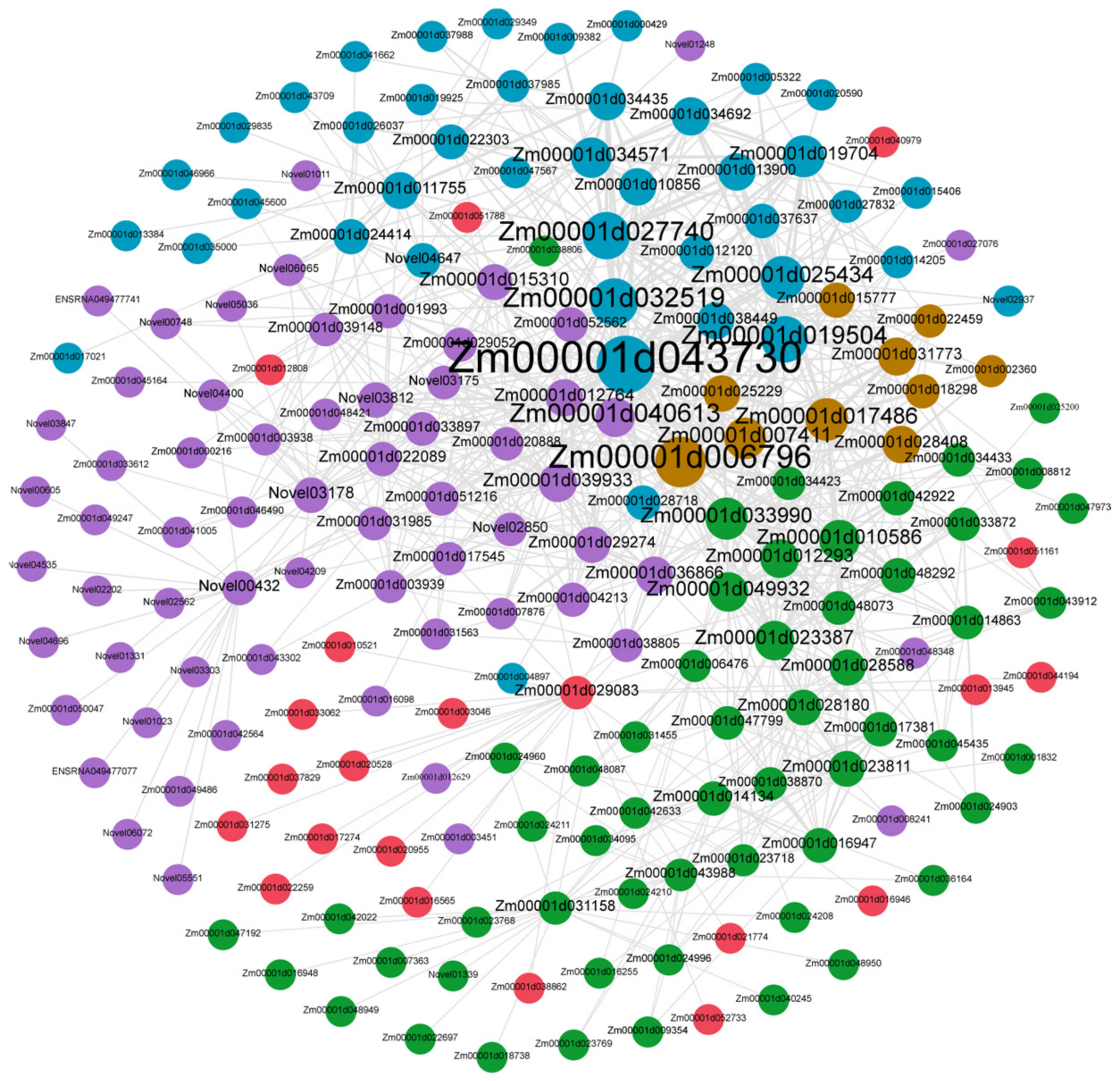
3.5. Candidate-Hub Gene Analysis of XNY22 and XNY27
3.6. GO Enrichment Analysis of Candidate Hub Genes
3.7. Candidate Hub Genes Network Interaction in Phenotypic Significant Enrichment Module
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Valko, M.; Morris, H.; Cronin, M. Metals, toxicity and oxidative stress. Curr. Med. Chem. 2005, 12, 1161–1208. [Google Scholar] [CrossRef] [PubMed]
- Qin, P.; Wang, L.; Liu, K.; Mao, S.; Li, Z.; Gao, S.; Shi, H.; Liu, Y. Genomewide association study of Aegilops tauschii traits under seedling-stage cadmium stress. Crop J. 2015, 3, 405–415. [Google Scholar] [CrossRef]
- Xue, D.; Jiang, H.; Deng, X.; Zhang, X.; Wang, H.; Xu, X.; Hu, J.; Zeng, D.; Guo, L.; Qian, Q. Comparative proteomic analysis provides new insights into cadmium accumulation in rice grain under cadmium stress. J. Hazard. Mater. 2014, 280, 269–278. [Google Scholar] [CrossRef] [PubMed]
- Shukla, U.; Singh, J.; Joshi, P.; Kakkar, P. Effect of bioaccumulation of cadmium on biomass productivity, essential trace elements, chlorophyll biosynthesis, and macromolecules of wheat seedlings. Biol. Trace Elem. Res. 2003, 92, 257–273. [Google Scholar] [CrossRef]
- Wang, X.-K.; Gong, X.; Cao, F.; Wang, Y.; Zhang, G.; Wu, F. HvPAA1 encodes a P-Type ATPase, a novel gene for cadmium accumulation and tolerance in barley (Hordeum vulgare L.). Int. J. Mol. Sci. 2019, 20, 1732. [Google Scholar] [CrossRef] [PubMed]
- Uraguchi, S.; Kamiya, T.; Clemens, S.; Fujiwara, T. Characterization of OsLCT1, a cadmium transporter from indica rice (Oryza sativa). Physiol. Plant. 2014, 151, 339–347. [Google Scholar] [CrossRef]
- Edelstein, M.; Ben-Hur, M. Heavy metals and metalloids: Sources, risks and strategies to reduce their accumulation in horticultural crops. Sci. Hortic. 2018, 234, 431–444. [Google Scholar] [CrossRef]
- Ishikawa, S.; Ae, N.; Sugiyama, M.; Murakami, M.; Arao, T. Genotypic variation in shoot cadmium concentration in rice and soybean in soils with different levels of cadmium contamination. Soil Sci. Plant Nutr. 2005, 51, 101–108. [Google Scholar] [CrossRef]
- Chen, H.; Zhang, W.; Yang, X.; Wang, P.; McGrath, S.P.; Zhao, F.-J. Effective methods to reduce cadmium accumulation in rice grain. Chemosphere 2018, 207, 699–707. [Google Scholar] [CrossRef]
- Wang, Y.; Xu, Y.; Liang, X.; Wang, L.; Sun, Y.; Huang, Q.; Qin, X.; Zhao, L. Soil application of manganese sulfate could reduce wheat Cd accumulation in Cd contaminated soil by the modulation of the key tissues and ionomic of wheat. Sci. Total Environ. 2021, 770, 145328. [Google Scholar] [CrossRef]
- Jia, H.; Wang, X.; Wei, T.; Wang, M.; Liu, X.; Hua, L.; Ren, X.; Guo, J.; Li, J. Exogenous salicylic acid regulates cell wall polysaccharides synthesis and pectin methylation to reduce Cd accumulation of tomato. Ecotoxicol. Environ. Saf. 2021, 207, 111550. [Google Scholar] [CrossRef]
- Yang, Y.; Chen, J.; Huang, Q.; Tang, S.; Wang, J.; Hu, P.; Shao, G. Can liming reduce cadmium (Cd) accumulation in rice (Oryza sativa) in slightly acidic soils? A contradictory dynamic equilibrium between Cd uptake capacity of roots and Cd immobilisation in soils. Chemosphere 2018, 193, 547–556. [Google Scholar] [CrossRef]
- Murakami, M.; Ae, N.; Ishikawa, S. Phytoextraction of cadmium by rice (Oryza sativa L.), soybean (Glycine max (L.) Merr.), and maize (Zea mays L.). Environ. Pollut. 2007, 145, 96–103. [Google Scholar] [CrossRef]
- Juwarkar, A.A.; Yadav, S.K.; Kumar, P.; Singh, S.K. Effect of biosludge and biofertilizer amendment on growth of Jatropha curcas in heavy metal contaminated soils. Environ. Monit. Assess. 2008, 145, 7–15. [Google Scholar] [CrossRef]
- Gnansounou, E.; Dauriat, A.; Wyman, C.E. Refining sweet sorghum to ethanol and sugar: Economic trade-offs in the context of North China. Bioresour. Technol. 2005, 96, 985–1002. [Google Scholar] [CrossRef]
- Yu, J.; Zhang, T.; Zhong, J.; Zhang, X.; Tan, T. Biorefinery of sweet sorghum stem. Biotechnol. Adv. 2012, 30, 811–816. [Google Scholar] [CrossRef]
- Ma, J.F.; Shen, R.F.; Shao, J.F. Transport of cadmium from soil to grain in cereal crops: A review. Pedosphere 2021, 31, 3–10. [Google Scholar] [CrossRef]
- Sasaki, A.; Yamaji, N.; Yokosho, K.; Ma, J.F. Nramp5 Is a Major Transporter Responsible for Manganese and Cadmium Uptake in Rice. Plant Cell 2012, 24, 2155–2167. [Google Scholar] [CrossRef]
- Chang, J.-D.; Huang, S.; Yamaji, N.; Zhang, W.; Ma, J.F.; Zhao, F.-J. OsNRAMP1 transporter contributes to cadmium and manganese uptake in rice. Plant Cell Environ. 2020, 43, 2476–2491. [Google Scholar] [CrossRef]
- Ueno, D.; Yamaji, N.; Kono, I.; Huang, C.F.; Ando, T.; Yano, M.; Ma, J.F. Gene limiting cadmium accumulation in rice. Proc. Natl. Acad. Sci. USA 2010, 107, 16500–16505. [Google Scholar] [CrossRef]
- Uraguchi, S.; Kamiya, T.; Sakamoto, T.; Kasai, K.; Sato, Y.; Nagamura, Y.; Yoshida, A.; Kyozuka, J.; Ishikawa, S.; Fujiwara, T. Low-affinity cation transporter (OsLCT1) regulates cadmium transport into rice grains. Proc. Natl. Acad. Sci. USA 2011, 108, 20959–20964. [Google Scholar] [CrossRef] [PubMed]
- Yamaji, N.; Xia, J.; Mitani-Ueno, N.; Yokosho, K.; Feng Ma, J. Preferential delivery of zinc to developing tissues in rice is mediated by P-type heavy metal ATPase OsHMA2. Plant Physiol. 2013, 162, 927–939. [Google Scholar] [CrossRef] [PubMed]
- Hao, X.; Zeng, M.; Wang, J.; Zeng, Z.; Dai, J.; Xie, Z.; Yang, Y.; Tian, L.; Chen, L.; Li, D. A node-expressed transporter OsCCX2 is involved in grain cadmium accumulation of rice. Front. Plant Sci. 2018, 9, 476. [Google Scholar] [CrossRef] [PubMed]
- Tan, L.; Zhu, Y.; Fan, T.; Peng, C.; Wang, J.; Sun, L.; Chen, C. OsZIP7 functions in xylem loading in roots and inter-vascular transfer in nodes to deliver Zn/Cd to grain in rice. Biochem. Biophys. Res. Commun. 2019, 512, 112–118. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Xu, W.; Xie, J.; Gao, Y.; Wu, L.; Sun, L.; Feng, L.; Chen, X.; Zhang, T.; Dai, C. Variation of a major facilitator superfamily gene contributes to differential cadmium accumulation between rice subspecies. Nat. Commun. 2019, 10, 2562. [Google Scholar] [CrossRef]
- Zheng, H.; Yang, Z.; Wang, W.; Guo, S.; Li, Z.; Liu, K.; Sui, N. Transcriptome analysis of maize inbred lines differing in drought tolerance provides novel insights into the molecular mechanisms of drought responses in roots. Plant Physiol. Biochem. 2020, 149, 11–26. [Google Scholar] [CrossRef]
- Quan, P.-Q.; Li, M.-Z.; Wang, G.-R.; Gu, L.-L.; Liu, X.-D. Comparative transcriptome analysis of the rice leaf folder (Cnaphalocrocis medinalis) to heat acclimation. BMC Genom. 2020, 21, 450. [Google Scholar] [CrossRef]
- Thakur, S.; Choudhary, S.; Bhardwaj, P. Comparative Transcriptome Profiling Under Cadmium Stress Reveals the Uptake and Tolerance Mechanism in Brassica juncea. J. Plant Growth Regul. 2019, 38, 1141–1152. [Google Scholar] [CrossRef]
- Miao, L.; Zhang, L.; Raboanatahiry, N.; Lu, G.; Zhang, X.; Xiang, J.; Gan, J.; Fu, C.; Li, M. Transcriptome analysis of stem and globally comparison with other tissues in Brassica napus. Front. Plant Sci. 2016, 7, 1403. [Google Scholar] [CrossRef]
- Liu, D.; Li, M.; Liu, Y.; Shi, L. Integration of the metabolome and transcriptome reveals the resistance mechanism to low nitrogen in wild soybean seedling roots. Environ. Exp. Bot. 2020, 175, 104043. [Google Scholar] [CrossRef]
- Chang, W.; Zhao, H.; Yu, S.; Yu, J.; Cai, K.; Sun, W.; Liu, X.; Li, X.; Yu, M.; Ali, S. Comparative transcriptome and metabolomic profiling reveal the complex mechanisms underlying the developmental dynamics of tobacco leaves. Genomics 2020, 112, 4009–4022. [Google Scholar] [CrossRef]
- Zhang, B.; Hu, F.; Cai, X.; Cheng, J.; Zhang, Y.; Lin, H.; Hu, K.; Wu, Z. Integrative Analysis of the Metabolome and Transcriptome of a Cultivated Pepper and Its Wild Progenitor Chiltepin (Capsicum annuum L. var. glabriusculum) Revealed the Loss of Pungency During Capsicum Domestication. Front. Plant Sci. 2022, 12, 783496. [Google Scholar] [CrossRef]
- Eckel-Mahan, K.L.; Patel, V.R.; Mohney, R.P.; Vignola, K.S.; Baldi, P.; Sassone-Corsi, P. Coordination of the transcriptome and metabolome by the circadian clock. Proc. Natl. Acad. Sci. USA 2012, 109, 5541–5546. [Google Scholar] [CrossRef]
- Liang, W.; Ni, L.; Carballar-Lejarazú, R.; Zou, X.; Sun, W.; Wu, L.; Yuan, X.; Mao, Y.; Huang, W.; Zou, S. Comparative transcriptome among Euscaphis konishii Hayata tissues and analysis of genes involved in flavonoid biosynthesis and accumulation. BMC Genom. 2019, 20, 24. [Google Scholar] [CrossRef]
- Li, Y.; Fang, J.; Qi, X.; Lin, M.; Zhong, Y.; Sun, L.; Cui, W. Combined Analysis of the Fruit Metabolome and Transcriptome Reveals Candidate Genes Involved in Flavonoid Biosynthesis in Actinidia arguta. Int. J. Mol. Sci. 2018, 19, 1471. [Google Scholar] [CrossRef]
- Zhou, C.; Mei, X.; Rothenberg, D.O.N.; Yang, Z.; Zhang, W.; Wan, S.; Yang, H.; Zhang, L. Metabolome and Transcriptome Analysis Reveals Putative Genes Involved in Anthocyanin Accumulation and Coloration in White and Pink Tea (Camellia sinensis) Flower. Molecules 2020, 25, 190. [Google Scholar] [CrossRef]
- Sun, L.; Wang, J.; Song, K.; Sun, Y.; Qin, Q.; Xue, Y. Transcriptome analysis of rice (Oryza sativa L.) shoots responsive to cadmium stress. Sci. Rep. 2019, 9, 10177. [Google Scholar] [CrossRef]
- Liu, A.; Zhou, Z.; Yi, Y.; Chen, G. Transcriptome analysis reveals the roles of stem nodes in cadmium transport to rice grain. BMC Genom. 2020, 21, 127. [Google Scholar] [CrossRef]
- Tan, M.; Cheng, D.; Yang, Y.; Zhang, G.; Qin, M.; Chen, J.; Chen, Y.; Jiang, M. Co-expression network analysis of the transcriptomes of rice roots exposed to various cadmium stresses reveals universal cadmium-responsive genes. BMC Plant Biol. 2017, 17, 194. [Google Scholar] [CrossRef]
- Su, L.; Xie, Y.; He, Z.; Zhang, J.; Tang, Y.; Zhou, X. Network response of two cherry tomato (Lycopersicon esculentum) cultivars to Cadmium stress as revealed by transcriptome analysis. Ecotoxicol. Environ. Saf. 2021, 222, 112473. [Google Scholar] [CrossRef]
- Zhang, Y.; Chao, J.; Li, X.; Zhang, C.; Khan, R.; Du, S.; Xu, N.; Song, L.; Liu, H.; Shi, Y. Comparative transcriptome combined with biochemical and physiological analyses provide new insights toward cadmium accumulation with two contrasting Nicotiana species. Physiol. Plant. 2021, 173, 369–383. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Zheng, S.; Liu, R.; Lu, J.; Lu, L.; Zhang, C.; Liu, Z.; Luo, C.; Zhang, L.; Wu, Y. Comparative analysis of root transcriptome profiles between low-and high-cadmium-accumulating genotypes of wheat in response to cadmium stress. Funct. Integr. Genom. 2019, 19, 281–294. [Google Scholar] [CrossRef]
- Han, M.; Lu, X.; Yu, J.; Chen, X.; Wang, X.; Malik, W.A.; Wang, J.; Wang, D.; Wang, S.; Guo, L. Transcriptome analysis reveals cotton (Gossypium hirsutum) genes that are differentially expressed in cadmium stress tolerance. Int. J. Mol. Sci. 2019, 20, 1479. [Google Scholar] [CrossRef] [PubMed]
- Cao, F.; Chen, F.; Sun, H.; Zhang, G.; Chen, Z.-H.; Wu, F. Genome-wide transcriptome and functional analysis of two contrasting genotypes reveals key genes for cadmium tolerance in barley. BMC Genom. 2014, 15, 611. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Lu, Q.; Cao, Y.; Wang, M.; Cheng, X.; Yan, Q. Comparative transcriptome analysis of the molecular mechanism of the hairy roots of Brassica campestris L. in response to cadmium stress. Int. J. Mol. Sci. 2019, 21, 180. [Google Scholar] [CrossRef] [PubMed]
- He, Q.; Zhou, T.; Sun, J.; Wang, P.; Yang, C.; Bai, L.; Liu, Z. Transcriptome Profiles of Leaves and Roots of Goldenrain Tree (Koelreuteria paniculata Laxm.) in Response to Cadmium Stress. Int. J. Environ. Res. Public Health 2021, 18, 12046. [Google Scholar] [CrossRef]
- Chen, P.; Chen, T.; Li, Z.; Jia, R.; Luo, D.; Tang, M.; Lu, H.; Hu, Y.; Yue, J.; Huang, Z. Transcriptome analysis revealed key genes and pathways related to cadmium-stress tolerance in Kenaf (Hibiscus cannabinus L.). Ind. Crops Prod. 2020, 158, 112970. [Google Scholar] [CrossRef]
- Hou, F.; Zhou, X.; Liu, P.; Yuan, G.; Zou, C.; Lübberstedt, T.; Pan, G.; Ma, L.; Shen, Y. Genetic dissection of maize seedling traits in an IBM Syn10 DH population under the combined stress of lead and cadmium. Mol. Genet. Genom. 2021, 296, 1057–1070. [Google Scholar] [CrossRef]
- Tang, B.; Luo, M.; Zhang, Y.; Guo, H.; Li, J.; Song, W.; Zhang, R.; Feng, Z.; Kong, M.; Li, H. Natural variations in the P-type ATPase heavy metal transporter gene ZmHMA3 control cadmium accumulation in maize grains. J. Exp. Bot. 2021, 72, 6230–6246. [Google Scholar] [CrossRef]
- Zdunić, Z.; Grljušić, S.; Ledenčan, T.; Duvnjak, T.; Šimić, D. Quantitative trait loci mapping of metal concentrations in leaves of the maize IBM population. Hereditas 2014, 151, 55–60. [Google Scholar] [CrossRef]
- Chen, Y.; Chao, Z.; Jin, M.; Wang, Y.; Li, Y.; Wu, J.; Xiao, Y.; Peng, Y.; Lv, Q.; Gui, S.; et al. A heavy metal transporter gene ZmHMA3a promises safe agricultural production on cadmium-polluted arable land. J. Genet. Genom. 2022. ahead of print. [Google Scholar] [CrossRef]
- Yue, R.; Lu, C.; Qi, J.; Han, X.; Yan, S.; Guo, S.; Liu, L.; Fu, X.; Chen, N.; Yin, H. Transcriptome analysis of cadmium-treated roots in maize (Zea mays L.). Front. Plant Sci. 2016, 7, 1298. [Google Scholar] [CrossRef]
- Cheng, D.; Tan, M.; Yu, H.; Li, L.; Zhu, D.; Chen, Y.; Jiang, M. Comparative analysis of Cd-responsive maize and rice transcriptomes highlights Cd co-modulated orthologs. BMC Genom. 2018, 19, 709. [Google Scholar] [CrossRef]
- Peng, H.; He, X.; Gao, J.; Ma, H.; Zhang, Z.; Shen, Y.; Pan, G.; Lin, H. Transcriptomic changes during maize roots development responsive to Cadmium (Cd) pollution using comparative RNAseq-based approach. Biochem. Biophys. Res. Commun. 2015, 464, 1040–1047. [Google Scholar] [CrossRef]
- Gu, L.; Zhao, M.; Ge, M.; Zhu, S.; Cheng, B.; Li, X. Transcriptome analysis reveals comprehensive responses to cadmium stress in maize inoculated with arbuscular mycorrhizal fungi. Ecotoxicol. Environ. Saf. 2019, 186, 109744. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Luo, M.; Peng, H.; Chen, F.; Li, W. Characterization of cadmium-responsive MicroRNAs and their target genes in maize (Zea mays) roots. BMC Mol. Biol. 2019, 20, 109744. [Google Scholar] [CrossRef]
- Li, S.; Wang, M.; Zhao, Z.; Ma, C.; Chen, S. Adsorption and desorption of Cd by soil amendment: Mechanisms and environmental implications in field-soil remediation. Sustainability 2018, 10, 2337. [Google Scholar] [CrossRef]
- Cao, Y.; Zhao, X.; Liu, Y.; Wang, Y.; Wu, W.; Jiang, Y.; Liao, C.; Xu, X.; Gao, S.; Shen, Y. Genome-wide identification of ZmHMAs and association of natural variation in ZmHMA2 and ZmHMA3 with leaf cadmium accumulation in maize. PeerJ 2019, 7, e7877. [Google Scholar] [CrossRef]
- Kong, J.; Jin, J.; Dong, Q.; Qiu, J.; Li, Y.; Yang, Y.; Shi, Y.; Si, W.; Gu, L.; Yang, F. Maize factors ZmUBP15, ZmUBP16 and ZmUBP19 play important roles for plants to tolerance the cadmium stress and salt stress. Plant Sci. 2019, 280, 77–89. [Google Scholar] [CrossRef]
- Ma, L.; Zhang, M.; Chen, J.; Qing, C.; He, S.; Zou, C.; Yuan, G.; Yang, C.; Peng, H.; Pan, G. GWAS and WGCNA uncover hub genes controlling salt tolerance in maize (Zea mays L.) seedlings. Theor. Appl. Genet. 2021, 134, 3305–3318. [Google Scholar] [CrossRef]
- Zhu, M.; Xie, H.; Wei, X.; Dossa, K.; Yu, Y.; Hui, S.; Tang, G.; Zeng, X.; Yu, Y.; Hu, P.; et al. WGCNA Analysis of Salt-Responsive Core Transcriptome Identifies Novel Hub Genes in Rice. Genes 2019, 10, 719. [Google Scholar] [CrossRef]
- Amrine, K.C.; Blanco-Ulate, B.; Cantu, D. Discovery of core biotic stress responsive genes in Arabidopsis by weighted gene co-expression network analysis. PLoS ONE 2015, 10, e0118731. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, Y.; Sadhukhan, A.; Tazib, T.; Nakano, Y.; Kusunoki, K.; Kamara, M.; Chaffai, R.; Iuchi, S.; Sahoo, L.; Kobayashi, M.; et al. Joint genetic and network analyses identify loci associated with root growth under NaCl stress in Arabidopsis thaliana. Plant Cell Environ. 2016, 39, 918–934. [Google Scholar] [CrossRef] [PubMed]
- Lou, Q.; Chen, L.; Mei, H.; Xu, K.; Wei, H.; Feng, F.; Li, T.; Pang, X.; Shi, C.; Luo, L. Root transcriptomic analysis revealing the importance of energy metabolism to the development of deep roots in rice (Oryza sativa L.). Front. Plant Sci. 2017, 8, 1314. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Kim, D.; Pertea, G.; Trapnell, C.; Pimentel, H.; Kelley, R.; Salzberg, S.L. TopHat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013, 14, R36. [Google Scholar] [CrossRef]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Nat. Preced. 2010, 11, R106. [Google Scholar]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef]
- Luo, J.-S.; Huang, J.; Zeng, D.-L.; Peng, J.-S.; Zhang, G.-B.; Ma, H.-L.; Guan, Y.; Yi, H.-Y.; Fu, Y.-L.; Han, B. A defensin-like protein drives cadmium efflux and allocation in rice. Nat. Commun. 2018, 9, 645. [Google Scholar] [CrossRef]
- Khanam, R.; Kumar, A.; Nayak, A.K.; Shahid, M.; Tripathi, R.; Vijayakumar, S.; Bhaduri, D.; Kumar, U.; Mohanty, S.; Panneerselvam, P.; et al. Metal(loid)s (As, Hg, Se, Pb and Cd) in paddy soil: Bioavailability and potential risk to human health. Sci. Total Environ. 2020, 699, 134330. [Google Scholar] [CrossRef]
- Ishikawa, S. Mechanisms of cadmium accumulation in rice grains and molecular breeding for its reduction. Soil Sci. Plant Nutr. 2020, 66, 28–33. [Google Scholar] [CrossRef]
- Zhang, J.; Zhu, Y.; Yu, L.; Yang, M.; Zou, X.; Yin, C.; Lin, Y. Research Advances in Cadmium Uptake, Transport and Resistance in Rice (Oryza sativa L.). Cells 2022, 11, 569. [Google Scholar] [CrossRef] [PubMed]
- NaziaTahir; Ullah, A.; Tahir, A.; Rashid, H.U.; Rehman, T.u.; Danish, S.; Hussain, B.; Akca, H. Strategies for reducing Cd concentration in paddy soil for rice safety. J. Clean. Prod. 2021, 316, 128116. [Google Scholar] [CrossRef]
- Dos Reis, R.A.; Keunen, E.; Mourato, M.P.; Martins, L.L.; Vangronsveld, J.; Cuypers, A. Accession-specific life strategies affect responses in leaves of Arabidopsis thaliana plants exposed to excess Cu and Cd. J. Plant Physiol. 2018, 223, 37–46. [Google Scholar] [CrossRef]
- Wang, A.; Wang, M.; Liao, Q.; He, X. Characterization of Cd translocation and accumulation in 19 maize cultivars grown on Cd-contaminated soil: Implication of maize cultivar selection for minimal risk to human health and for phytoremediation. Environ. Sci. Pollut. Res. 2016, 23, 5410–5419. [Google Scholar] [CrossRef]
- Xu, W.; Lu, G.; Dang, Z.; Liao, C.; Chen, Q.; Yi, X. Uptake and distribution of Cd in sweet maize grown on contaminated soils: A field-scale study. Bioinorg. Chem. Appl. 2013, 2013, 959764. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Name | Total Raw Reads | Total Clean Reads | Clean Bases | Q20 | Q30 | GC Content |
|---|---|---|---|---|---|---|
| XNY22-YA-8-1 | 61,002,492 | 58,995,782 | 4.42G | 95.89% | 90.00% | 55.10% |
| XNY22-YA-8-2 | 49,000,066 | 47,314,434 | 3.55G | 95.86% | 89.94% | 55.16% |
| XNY22-YA-8-3 | 50,773,422 | 48,946,308 | 3.67G | 95.90% | 90.03% | 55.30% |
| XNY22-YA-24-1 | 57,585,600 | 55,670,330 | 4.18G | 95.94% | 90.06% | 57.05% |
| XNY22-YA-24-2 | 71,045,424 | 68,906,076 | 5.17G | 96.11% | 90.55% | 55.47% |
| XNY22-YA-24-3 | 74,261,462 | 71,988,508 | 5.4G | 96.38% | 91.05% | 56.07% |
| XNY27-YA-8-1 | 49,047,692 | 47,515,340 | 3.56G | 96.21% | 90.64% | 54.99% |
| XNY27-YA-8-2 | 54,774,602 | 53,113,822 | 3.98G | 96.06% | 90.31% | 55.35% |
| XNY27-YA-8-3 | 50,077,846 | 48,608,644 | 3.65G | 96.05% | 90.29% | 54.80% |
| XNY27-YA-24-1 | 65,085,908 | 62,577,096 | 4.69G | 96.30% | 90.86% | 56.86% |
| XNY27-YA-24-2 | 61,293,978 | 59,407,268 | 4.46G | 96.26% | 90.83% | 56.71% |
| XNY27-YA-24-3 | 62,471,238 | 60,300,228 | 4.52G | 96.17% | 90.63% | 57.02% |
| XNY22-CHS-8-1 | 45,920,496 | 44,668,098 | 3.35G | 96.16% | 90.61% | 55.41% |
| XNY22-CHS-8-2 | 50,754,324 | 49,405,462 | 3.71G | 96.35% | 91.00% | 55.62% |
| XNY22-CHS-8-3 | 46,733,440 | 45,233,552 | 3.39G | 96.18% | 90.64% | 55.65% |
| XNY22-CHS-24-1 | 51,689,096 | 49,994,612 | 3.75G | 96.13% | 90.45% | 55.07% |
| XNY22-CHS-24-2 | 44,128,234 | 42,698,694 | 3.2G | 96.09% | 90.35% | 56.72% |
| XNY22-CHS-24-3 | 55,802,774 | 53,912,512 | 4.04G | 96.03% | 90.22% | 56.36% |
| XNY27-CHS-8-1 | 45,568,500 | 44,342,446 | 3.33G | 96.13% | 90.52% | 54.78% |
| XNY27-CHS-8-2 | 47,235,914 | 45,699,000 | 3.43G | 95.95% | 90.06% | 55.69% |
| XNY27-CHS-8-3 | 49,151,816 | 47,584,118 | 3.57G | 96.03% | 90.22% | 56.59% |
| XNY27-CHS-24-1 | 52,646,388 | 50,889,064 | 3.82G | 96.14% | 90.47% | 56.59% |
| XNY27-CHS-24-2 | 57,725,532 | 55,861,930 | 4.19G | 96.07% | 90.31% | 57.71% |
| XNY27-CHS-24-3 | 44,060,200 | 42,491,670 | 3.19G | 95.95% | 90.09% | 55.09% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Y.; Zhang, Y.; Luo, H.; Lv, D.; Yi, Z.; Duan, M.; Deng, M. WGCNA Analysis Revealed the Hub Genes Related to Soil Cadmium Stress in Maize Kernel (Zea mays L.). Genes 2022, 13, 2130. https://doi.org/10.3390/genes13112130
Li Y, Zhang Y, Luo H, Lv D, Yi Z, Duan M, Deng M. WGCNA Analysis Revealed the Hub Genes Related to Soil Cadmium Stress in Maize Kernel (Zea mays L.). Genes. 2022; 13(11):2130. https://doi.org/10.3390/genes13112130
Chicago/Turabian StyleLi, Yongjin, Ying Zhang, Hongbing Luo, Dan Lv, Zhenxie Yi, Meijuan Duan, and Min Deng. 2022. "WGCNA Analysis Revealed the Hub Genes Related to Soil Cadmium Stress in Maize Kernel (Zea mays L.)" Genes 13, no. 11: 2130. https://doi.org/10.3390/genes13112130
APA StyleLi, Y., Zhang, Y., Luo, H., Lv, D., Yi, Z., Duan, M., & Deng, M. (2022). WGCNA Analysis Revealed the Hub Genes Related to Soil Cadmium Stress in Maize Kernel (Zea mays L.). Genes, 13(11), 2130. https://doi.org/10.3390/genes13112130