
Prenatal Genetic Testing in the Era of Next Generation Sequencing: A One-Center Canadian Experience
 , , ,
, , ,
Abstract
1. Introduction
2. Materials and Methods
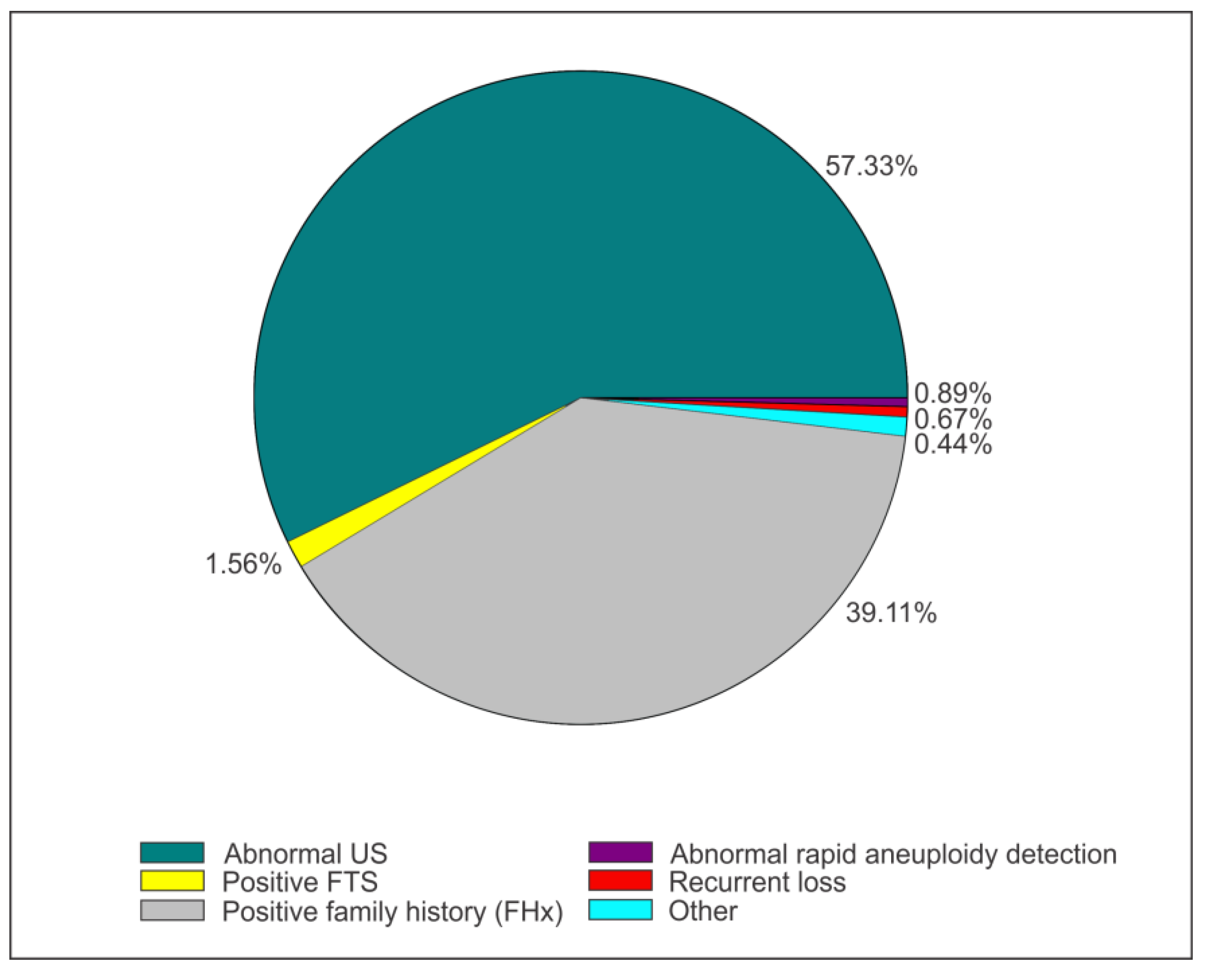
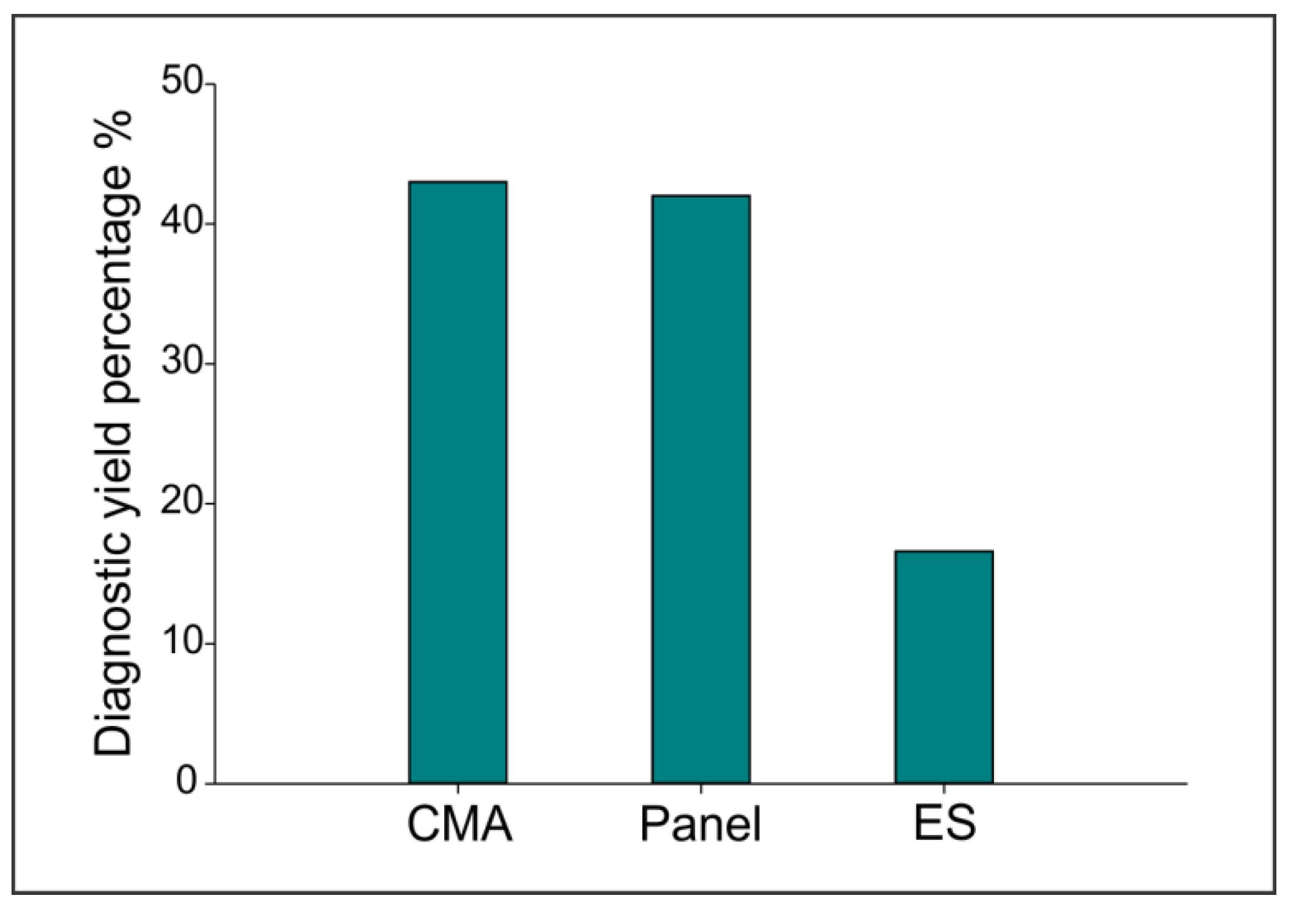
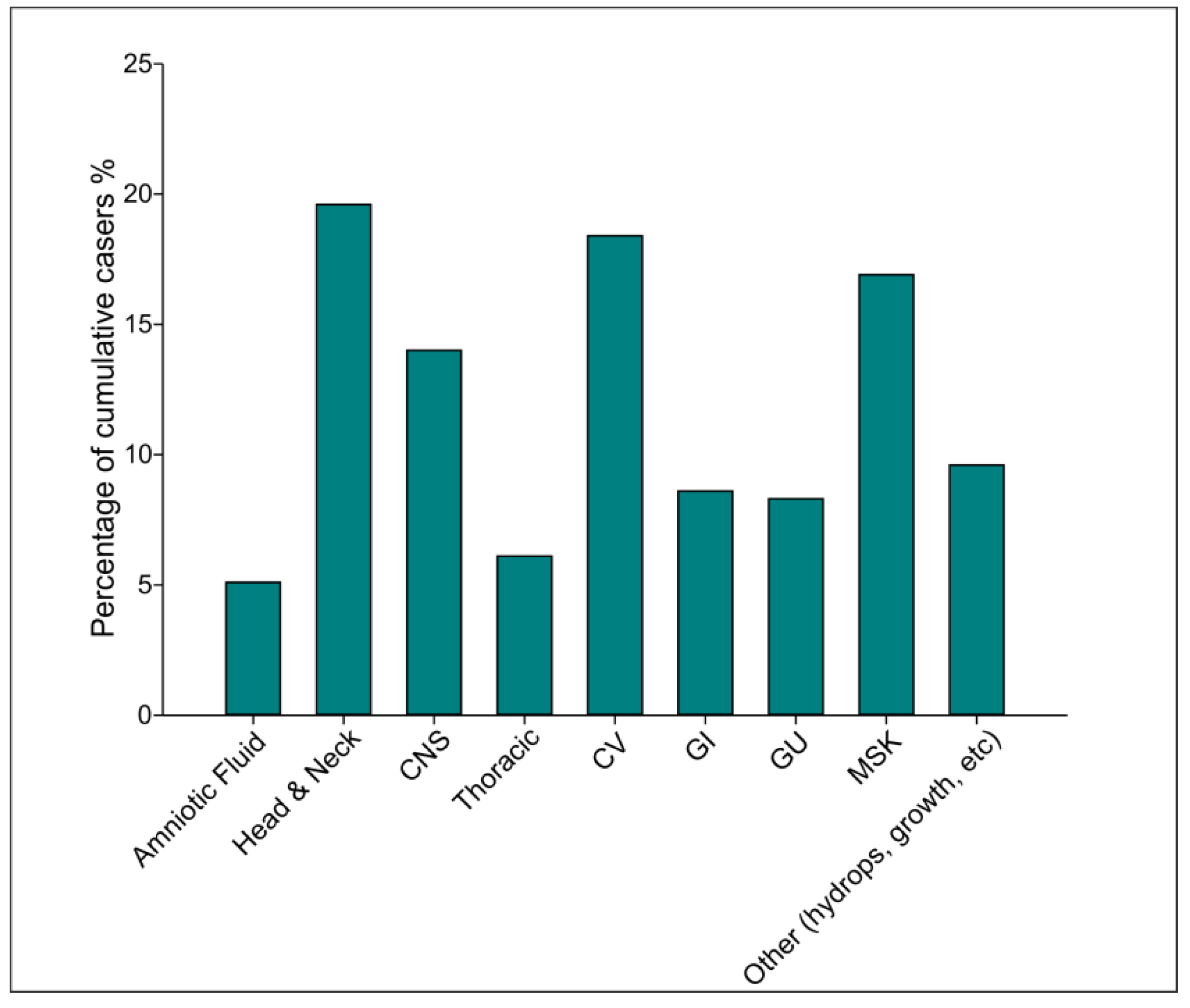
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| CMA | Chromosomal microarray |
| CNS | Central nervous system |
| CV | Cardiovascular |
| ES | Exome sequencing |
| FHx | Positive family history |
| FTS | First trimester screen |
| GA | Gestational age |
| GI | Gastrointestinal |
| MRI | Magnetic resonance imaging |
| MSK | Musculoskeletal |
| NGS | Next generation sequencing |
| PP | Previous pregnancy |
| PC | Previous child |
| QF-PCR | Quantitative Fluorescent-Polymerase chain reaction |
| WES | Whole exome sequencing |
| VUS | Variants of unknown significance |
| ? | Variants of unknown significance |
References
- Bardi, F.; Bergman, J.E.H.; Siemensma-Mühlenberg, N.; Elvan-Taşpınar, A.; de Walle, H.E.K.; Bakker, M.K. Prenatal diagnosis and pregnancy outcome of major structural anomalies detectable in the first trimester: A population-based cohort study in the Netherlands. Paediatr. Perinat. Epidemiol. 2022, 36, 804–814. [Google Scholar] [CrossRef] [PubMed]
- Dolk, H.; Loane, M.; Garne, E. The prevalence of congenital anomalies in Europe. Adv. Exp. Med. Biol. 2010, 686, 349–364. [Google Scholar] [CrossRef] [PubMed]
- Greenbaum, G.; Rubin, A.; Templeton, A.R.; Rosenberg, N.A. Network-based hierarchical population structure analysis for large genomic data sets. Genome. Res. 2019, 29, 2020–2033. [Google Scholar] [CrossRef]
- Monaghan, K.G.; Leach, N.T.; Pekarek, D.; Prasad, P.; Rose, N.C. The use of fetal exome sequencing in prenatal diagnosis: A points to consider document of the American College of Medical Genetics and Genomics (ACMG). Genet. Med. 2020, 22, 675–680. [Google Scholar] [CrossRef]
- Zhang, B.; Lu, B.Y.; Yu, B.; Zheng, F.X.; Zhou, Q.; Chen, Y.P.; Zhang, X.Q. Noninvasive prenatal screening for fetal common sex chromosome aneuploidies from maternal blood. J. Int. Med. Res. 2017, 45, 621–630. [Google Scholar] [CrossRef] [PubMed]
- Hillman, S.C.; McMullan, D.J.; Hall, G.; Togneri, F.S.; James, N.; Maher, E.J.; Meller, C.H.; Williams, D.; Wapner, R.J.; Maher, E.R.; et al. Use of prenatal chromosomal microarray: Prospective cohort study and systematic review and meta-analysis. Ultrasound Obs. Gynecol. 2013, 41, 610–620. [Google Scholar] [CrossRef]
- Wapner, R.J.; Martin, C.L.; Levy, B.; Ballif, B.C.; Eng, C.M.; Zachary, J.M.; Savage, M.; Platt, L.D.; Saltzman, D.; Grobman, W.A.; et al. Chromosomal microarray versus karyotyping for prenatal diagnosis. N. Engl. J. Med. 2012, 367, 2175–2184. [Google Scholar] [CrossRef]
- Yang, Y.; Muzny, D.M.; Reid, J.G.; Bainbridge, M.N.; Willis, A.; Ward, P.A.; Braxton, A.; Beuten, J.; Xia, F.; Niu, Z.; et al. Clinical whole-exome sequencing for the diagnosis of mendelian disorders. N. Engl. J. Med. 2013, 369, 1502–1511. [Google Scholar] [CrossRef]
- Sawyer, S.L.; Hartley, T.; Dyment, D.A.; Beaulieu, C.L.; Schwartzentruber, J.; Smith, A.; Bedford, H.M.; Bernard, G.; Bernier, F.P.; Brais, B.; et al. Utility of whole-exome sequencing for those near the end of the diagnostic odyssey: Time to address gaps in care. Clin. Genet. 2016, 89, 275–284. [Google Scholar] [CrossRef]
- Mone, F.; McMullan, D.; Williams, D.; Chitty, L.; Maher, E.; Kilby, M. Evidence to Support the Clinical Utility of Prenatal Exome Sequencing in Evaluation of the Fetus with Congenital Anomalies. BJOG Int. J. Obstet. Amp. Gynaecol. 2021, 128, e39–e50. [Google Scholar] [CrossRef]
- Lazier, J.; Hartley, T.; Brock, J.A.; Caluseriu, O.; Chitayat, D.; Laberge, A.M.; Langlois, S.; Lauzon, J.; Nelson, T.N.; Parboosingh, J.; et al. Clinical application of fetal genome-wide sequencing during pregnancy: Position statement of the Canadian College of Medical Geneticists. J. Med. Genet. 2021, 59, 931–937. [Google Scholar] [CrossRef] [PubMed]
- Van den Veyver, I.B.; Chandler, N.; Wilkins-Haug, L.E.; Wapner, R.J.; Chitty, L.S. International Society for Prenatal Diagnosis Updated Position Statement on the use of genome-wide sequencing for prenatal diagnosis. Prenat. Diagn. 2022, 42, 796–803. [Google Scholar] [CrossRef] [PubMed]
- Lord, J.; McMullan, D.J.; Eberhardt, R.Y.; Rinck, G.; Hamilton, S.J.; Quinlan-Jones, E.; Prigmore, E.; Keelagher, R.; Best, S.K.; Carey, G.K.; et al. Prenatal exome sequencing analysis in fetal structural anomalies detected by ultrasonography (PAGE): A cohort study. Lancet 2019, 393, 747–757. [Google Scholar] [CrossRef]
- Han, J.; Yang, Y.D.; He, Y.; Liu, W.J.; Zhen, L.; Pan, M.; Yang, X.; Zhang, V.W.; Liao, C.; Li, D.Z. Rapid prenatal diagnosis of skeletal dysplasia using medical trio exome sequencing: Benefit for prenatal counseling and pregnancy management. Prenat. Diagn. 2020, 40, 577–584. [Google Scholar] [CrossRef]
- Tolusso, L.K.; Hazelton, P.; Wong, B.; Swarr, D.T. Beyond diagnostic yield: Prenatal exome sequencing results in maternal, neonatal, and familial clinical management changes. Genet. Med. 2021, 23, 909–917. [Google Scholar] [CrossRef] [PubMed]
- Mellis, R.; Oprych, K.; Scotchman, E.; Hill, M.; Chitty, L.S. Diagnostic yield of exome sequencing for prenatal diagnosis of fetal structural anomalies: A systematic review and meta-analysis. Prenat. Diagn. 2022, 42, 662–685. [Google Scholar] [CrossRef]
- Government of Alberta. Population Statistics. Available online: https://www.alberta.ca/population-statistics.aspx#population-projections (accessed on 16 September 2022).
- Government of Alberta. Alberta Health Care Insurance Plan (AHCIP). Available online: https://www.alberta.ca/ahcip.aspx (accessed on 16 September 2022).
- Committee Opinion No. 682: Microarrays and Next-Generation Sequencing Technology: The Use of Advanced Genetic Diagnostic Tools in Obstetrics and Gynecology. Obs. Gynecol. 2016, 128, e262–e268. [CrossRef]
- Srebniak, M.I.; Joosten, M.; Knapen, M.; Arends, L.R.; Polak, M.; van Veen, S.; Go, A.; Van Opstal, D. Frequency of submicroscopic chromosomal aberrations in pregnancies without increased risk for structural chromosomal aberrations: Systematic review and meta-analysis. Ultrasound Obs. Gynecol. 2018, 51, 445–452. [Google Scholar] [CrossRef]
- Normand, E.A.; Braxton, A.; Nassef, S.; Ward, P.A.; Vetrini, F.; He, W.; Patel, V.; Qu, C.; Westerfield, L.E.; Stover, S.; et al. Clinical exome sequencing for fetuses with ultrasound abnormalities and a suspected Mendelian disorder. Genome. Med. 2018, 10, 74. [Google Scholar] [CrossRef]
- Liu, P.; Meng, L.; Normand, E.A.; Xia, F.; Song, X.; Ghazi, A.; Rosenfeld, J.; Magoulas, P.L.; Braxton, A.; Ward, P.; et al. Reanalysis of Clinical Exome Sequencing Data. N. Engl. J. Med. 2019, 380, 2478–2480. [Google Scholar] [CrossRef]
- Sobreira, N.L.M.; Arachchi, H.; Buske, O.J.; Chong, J.X.; Hutton, B.; Foreman, J.; Schiettecatte, F.; Groza, T.; Jacobsen, J.O.B.; Haendel, M.A.; et al. Matchmaker Exchange. Curr. Protoc. Hum. Genet. 2017, 95, 9.31.1–9.31.15. [Google Scholar] [CrossRef]
- Au, P.Y.B.; You, J.; Caluseriu, O.; Schwartzentruber, J.; Majewski, J.; Bernier, F.P.; Ferguson, M.; Valle, D.; Parboosingh, J.S.; Sobreira, N.; et al. GeneMatcher aids in the identification of a new malformation syndrome with intellectual disability, unique facial dysmorphisms, and skeletal and connective tissue abnormalities caused by de novo variants in HNRNPK. Hum. Mutat. 2015, 36, 1009–1014. [Google Scholar] [CrossRef]
- de Koning, M.A.; Haak, M.C.; Adama van Scheltema, P.N.; Peeters-Scholte, C.; Koopmann, T.T.; Nibbeling, E.A.R.; Aten, E.; den Hollander, N.S.; Ruivenkamp, C.A.L.; Hoffer, M.J.V.; et al. From diagnostic yield to clinical impact: A pilot study on the implementation of prenatal exome sequencing in routine care. Genet. Med. 2019, 21, 2303–2310. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| a. Prenatally Assessed Cases for Which Pre- and Postnatal Positive Results of Gene Panels Were Obtained *. | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Stud-y ID | Reason for Referral | Anomalies Identified | Prenatal Genetic Testing | Pregnancy Outcomes | Postnatal Genetic Testing | Diagnosis | Gene | Management Implications (RR%) | |
| Prenatal Detection | Postnatal Detection | ||||||||
| Prenatal testing | |||||||||
| 67573 | AbN US | 9 | 9 | (-) RAD, CMA (+) SD panel | TOP | nil | Hypochondrogenesis | COL2A1 | <1% |
| 66234 | AbN US | 9 | 9 | (-) RAD, CMA (+) SD panel | Stillbirth | nil | Opsismodysplasia | INPPL1 | <1% |
| 66328 | AbN US | 3, 4, 5, 6, 7 | 3, 4, 5, 6, 7, 11 | (-) RAD, CMA (+) Noonan panel | TOP | nil | Noonan syndrome | PTPN11 | <1% |
| 66330 | AbN US | 1, 5, 9 | 1, 5, 9 | (-) RAD, CMA (+) Arthrogryposis panel | Nn demise | nil | Nemaline myopathy | NEB | 25% |
| 66883 | AbN US | 7, 8 | 3, 7, 8 | (-) RAD, CMA (+) BW panel | Living | nil | Beckwith-Wiedemann syndrome | KvDMR2 loss of methylation | <1% |
| 66944 | FHx | nil | 11 | (-) RAD, CMA (+) SCID panel | Living | nil | SCID | IL2RG | 25% |
| 67196 | AbN US | 3 | 1, 3, 10 | (-) RAD, CMA (+) Noonan panel | Living | nil | Noonan syndrome (MIM163950) | PTPN11 | <1% |
| 67478 | FHx | 10 | (-) CMA (+) Targeted panel | Living | nil | EBS (MIM131800) | KRT14 | 50% | |
| 67497 | AbN US | 9 | 9 | (-) RAD, CMA (+) SD panel | Living | nil | Hypophosphatasia | ALPL | 25% |
| 67797 | AbN US | 1, 2, 6 | 1, 3, 5, 6, 7 | (-) RAD, CMA (+) Noonan panel | Stillbirth | nil | Noonan syndrome | PTPN11 | <1% |
| 67981 | AbN US | 3, 4 | 3, 4 | (-) RAD, CMA (+) Brain malformation panel | TOP | nil | L1CAM-related disorders | L1CAM | 25% |
| 68743 | AbN US | 7 | 7 | (-) RAD, CMA (+) Congenital diarrhea panel | Living | nil | Congenital diarrhea | SLC26A3 | 25% |
| 58884 | AbN US | 2, 5, 9 | 5, 9 | (-) RAD, CMA (+) SD panel | stillbirth | nil | Thanatophoric dysplasia | FGFR3 | <1% |
| Postnatal testing | |||||||||
| 67259 | AbN US | 9 | 3, 9 | nil | TOP | (-) CMA (+) SD panel | OI2 | COL2A1 | <1% |
| 68120 | AbN US | 4, 8 | 4, 8 | nil | TOP | (-) CMA (+) Ciliopathy Panel | Joubert Syndrome | RPGRIP1L | 25% |
| 68445 | FHx | 9 | 9 | nil | TOP | (-) CMA (+) SD panel | Thanatophoric dysplasia | FGFR3 | <1% |
| 69205 | AbN US | 4, 9 | 3, 4, 8, 9 | nil | Infantile demise | (-) CMA (+) Brain malformation panel | Zellweger syndrome | PEX1 | 25% |
| 66125 | AbN US | 2, 6 | 1, 2, 3, 4, 6, 9 | (-) RAD, CMA | Living | (+) Craniosynostosis panel | Acromelic fronto-nasal dysostosis | ZSWIM6 | <1% |
| 67274 | AbN US | 6, 7 | 1, 3, 4, 6, 7 | (-) RAD, CMA | Nn demise | (+) Aicardi-Goutieres panel | Aicardi-Goutieres syndrome | RNASEH2C | 25% |
| 67502 | AbN US | 3 | 1, 3, 9 | (-) RAD, CMA | Living | (+) Stickler syndrome panel | Stickler syndrome | COL11A1 | 50% |
| 7009 | FHx | nil | nil | Living | (+) MH panel | Malignant Hyperthermia | RYR1 | 50% | |
| 17890 | FHx | nil | 1, 3 | nil | Living | (+) CDL panel | Cornelia de Lange syndrome | NIPBL | 50% |
| 63327 | AbN US | 3, 9 | 3, 9 | nil | Nn demise | (+) Craniosynostosis panel | Beare-Stevenson syndrome | FGFR2 | <1% |
| 64860 | FHx | nil | 1, 3, 9 | nil | Living | (+) OI panel | Bruck Syndrome | FKBP10 | 25% |
| 66134 | FHx | nil | 11 | nil | Living | (+) DSPD panel | Delta Storage pool disease | RUNX1 | 50% |
| 66206 | FHx | nil | 1, 9 | nil | Living | (+) Hposhosphatemia panel | Hypophosphatemia | PHEX | 50% |
| 66428 | AbN US | 6 | 3, 6 | nil | Living | (+) Brain malfomation panel | Tuberous Sclerosis | TSC1 | 1% |
| 67255 | AbN US | 9 | 1, 3, 9 | nil | Nn demise | (+) SD panel | Campomelic dysplasia | SOX9 | <1% |
| 67739 | AbN US | 1, 3, 9 | 1, 3, 9 | (-) RAD | TOP | (+) SD panel | Thanatophoric dysplasia | FGFR3 | <1% |
| 68014 | FTS+ | 1, 2, 3, 5, 7 | 3, 5, 10 | nil | Living | (+) Vascular malformation panel | RASA1-related disorder | RASA1 | <1% |
| 69624 | FHx | 3, 9 | 3 | nil | Living | (+) Craniosynostosis panel | Saethre-Chotzen syndrome | TWIST | 50% |
| 67669 | FHx | 2, 9 | 3, 4, 9 | nil | Living | (+) Congenital muscular dystrophy panel | Myotonic dystrophy type 1 | DMPK | 50% |
| b. Prenatally assessed cases for which pre- and postnatal variants of unknown significance (VUS) results of gene panels were obtained. | |||||||||
| 59041 | FTS+ | 1, 3, 4, 6, 9 | (-) RAD, CMA (?) Panels (hydrops, brain malformations) | TOP | (?) WES | AR combined oxidative phosphorylation deficiency 27/Alpers-Huttenlocher syndrome | CARS2 (NM_ 024537.4): c.563C>T,p.Thr188Met | 25%? | |
| 67291 | FTS+ | 3, 8 | 1, 3, 4, 9 | (-) RAD, CMA (?) Noonan Panel | Living | nil | Noonan syndrome? Lost to follow up | LZTR1 (NM_006767.3):c.2063G>A,p.(Arg688His) | ? |
| 67347 | FHx | nil | nil | (-) RAD, CMA (?) Bleeding disorder panel | Living | nil | Bleeding disorder | ABCG5(NM_022436.2):c.235G>A,p.(Gly79Arg); FREM1(NM_144966.5):c.987A>T,p)Lys329Asn); MPL(NM_005373.2):c.655C>G,p(Gln219Glu) | ? |
| 67980 | AbN US | 6 | 6, 8, 11 | (-) RAD, CMA (?) Noonan syndrome/Rasopathies | Living | nil | Complex syndromal presentation | KAT6B(NM_0123303.3): c.1217G>A, p.(Arg406Gln)mat | ? |
| 68077 | AbN US | 9 | 1, 9 | (-) RAD, CMA (?) SD Panel | Living | nil | Pseudohypoparathyroidism | GNAS(NM_000516.4):c.349G>A,p.(Val117Met),mat | 50%? |
| 67042 | AbN US | 4, 6 | 4, 6 | (-) RAD, CMA | Living | (-) CMA (?) Brain malformation panel | Joubert syndrome | TUBA8(NM_018943.2):c.1118G>A,p.(Arg373Gln), het VLDLR(NM003383.3):c.836G>A,p(Arg279Gln),het | ? |
| 66890 | FHx | 3, 9 | 3, 9 | nil | TOP | (-) CMA (?) Arthrogryposis panel | Beals syndrome | FBN2(NM_001999.3):c.8363C>T,p.(Ala2788Val), het | ? |
| 67846 | AbN US | 2, 8 | 8 | nil | Stillbirth | (-) CMA (?) CAKUT Panel | Bilateral Renal agenesis | FREM2(NM_207361.6):c.9038C>T,p.(Thr3013Met), het | ? |
| 68247 | AbN US | 3, 8, 9 | 3, 8, 9 | (-) RAD | TOP | (-) CMA (?) Arthrogryposis panel | Rothmund-Thmpson syndrome | RECQL4(NM_004260.3): c.551G>A,p.(Gly184Asp), hom | 25%? |
| 66794 | AbN US | 1, 2 | (-) RAD, CMA | Infantile demise | (?) Hydrops Panel | Feingold syndrome | MYCN(NM_005378.4):c.718G>A,p.(Gly240Ser) | ? | |
| 66824 | AbN US | 1, 2, 9 | nil | TOP | (-) CMA, (?) Hydrops Panel | Desbuquois dysplasia | CANT1(NM_138793.3):c.872C>T,p(Thr291Met), het | ? | |
| a. Prenatal Cases in Which a Diagnosis Was Achieved via Whole Exome Sequencing | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Study ID | Reason for Referral | Anomalies Identified | Prenatal Genetic Testing | Pregnancy Outcome | Postnatal Testing | Diagnosis | Genetic Finding | Management Implications | |
| Prenatal Detection | Postnatal Detection | ||||||||
| 64702001 | AbN US | 3, 4, 6, 7, 8 | 3, 4, 6, 7, 8 | nil | TOP | (-) RAD (-) CMA (+) WES | Developmental delay with or without dysmorphic facies and autism (MIM618454) AD | TRRAP (NM_003496.3), c.3127G>A, p.(Ala1043Thr), heterozygous, de novo | RR < 1% |
| 71280001 | AbN US | 3, 4, 6 | 3, 4, 6, 7, 8 | (-) RAD, CMA, Panel | TOP | (+) WES | Neurooculocardio-genitourinary syndrome (MIM618652), AD | WDR37 (NM_014023.3), c.389C>T, p.(Thr130Ile), heterozygous, de novo | RR < 1% |
| 71354001 | AbN US | 5, 6 | 3, 5, 6, 7, 8, 9 | (-) RAD, CMA, Panel | Neonatal Demise | (+) WES | Osteopathia striata with cranial sclerosis (MIM 300373), XL | AMER1 (NM_152424.4), c.19G>&, p.Glu7 *; hemizygous, maternal | RR25% |
| 67893001 | AbN US | 6 | 6 | (-) RAD, CMA in Alberta (+) WES in another province | Living | nil | 1. RASA1-related disorder (MIM 608354), AD 2. Kleefstra sdr. type 2 | 1.RASA1 (NM_002890.2), c.2131C>T,p.Arg711X; heterozygous, maternal 2.KMT2C (NM_170606.2), c.9773A>C, p.His3258Pro heterozygous, de novo | Surveillance 50% RR |
| 66175001 | AbN US | 3 | 3, 9 | (-) RAD, CMA | Living | (-) CLP Panel (+) WES | Hartsfield syndrome (MIM615465), AD | FGFR1 (NM_023110.2), c.1474G>A,p.(Val492Met), heterozygous, de novo | Anticipatory management RR < 1% |
| 67295001 | AbN US | 9 | 3, 9 | nil | Living | (+) CMA (+) WES | 1. Prader-Willi Syndrome (MIM176270), imprinting 2. Ullrich Muscular Dystrophy (MIM254090), AD | 1.Del 15q11, paternal 2.COL6A2(NM_001849.3), c.848G > A,p.Gly283Glu, heterozygous, maternal | Treatment Anticipatory management RR 50% |
| 69829001 | AbN US | 1, 4, 5, 9 | 1, 3, 4, 5, 9 | nil | Living | (-) CMA (+) WES | Escobar Syndrome (MIM265000), AR | CHRNG (NM_005199.4), c.202C>T, p.Arg68 *; c.459dup, p.Val154Serfs*24, combined heterozygous, parents carriers | Anticipatory management RR25% |
| 69573001 | AbN US | 1, 2, 3 | 1, 2, 3, 6 | (-) RAD, CMA | Living | (+) WES | 1. Costello Syndrome (MIM218040),AD 2. ERF-related disorder (MIM600775),AD | 1.HRAS (NM_005343.3), c.34G>A, p.(Gly12Ser), heterozygous, de novo 2.ERF (NM_006494.3), c.891_892del, p.(Gly299Argfs * 9), heterozygous, de novo | Anticipatory management RR < 1% |
| 68143001 | AbN US | 6 | 3, 6, 7 | (-) RAD, CMA | Demise at 4 mo | (-) Panel (+) WES | Zhu-Tokita-Takenouchi-Kim syndrome (ZTTKS),AD | SON (NM_032195.2), c.5867G>A, p.(Arg1956His), heterozygous, de novo | RR < 1% |
| b. Prenatal Cases in which WES identified a Variant of Unknown Significance | |||||||||
| 67663001 | AbN US | 3 | 3, 4 | nil | Living | (+) CMA (-) CLP Panel (?) WES | 1. 15q13 dup syndrome; 2. Intellectual Developmental disorder, auto-somal dominant 52 | 1. Paternal 2. ASH1L (NM_018489.2): c.614A>C,p.(Lys205Thr), pat | 50% |
| 68424001 | FHx | 2, 6, 7, 8 | 2, 3, 6, 7, 8, 9 | nil | TOP | (-) RAD, (-) CMA (?) WES | CDX2 variant, gene not associated with human phenotype | CDX (NM_001265.4): c.592C>T,p.(Arg198Trp), de novo | <1% |
| 67572001 | AbN US | 4, 9 | 3, 4, 9 | nil | TOP | (-) CMA (?) WES | RYR3 variants, not associated with human phenotype | RYR3 (NM_001036.4): c.90T>A,p.(His30Gln), pat C,11730G>C,p.Leu391Phe),mat | 25% |
| 69260001 | AbN US | 6, 7, 9 | 6, 7, 9 | (-) RAD, CMA, Panel | TOP | (?) WES | Mandibulofacial dysostosis, Guyon Almeida type (MIM610536) | EFTUD2(NM_004247.3):c.658C>T,p.(Arg220Cys), de novo | <1% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Almubarak, A.; Zhang, D.; Kosak, M.; Rathwell, S.; Doonanco, J.; Eaton, A.J.; Kannu, P.; Lazier, J.; Lui, M.; Niederhoffer, K.Y.; et al. Prenatal Genetic Testing in the Era of Next Generation Sequencing: A One-Center Canadian Experience. Genes 2022, 13, 2019. https://doi.org/10.3390/genes13112019
Almubarak A, Zhang D, Kosak M, Rathwell S, Doonanco J, Eaton AJ, Kannu P, Lazier J, Lui M, Niederhoffer KY, et al. Prenatal Genetic Testing in the Era of Next Generation Sequencing: A One-Center Canadian Experience. Genes. 2022; 13(11):2019. https://doi.org/10.3390/genes13112019
Chicago/Turabian StyleAlmubarak, Asra, Dan Zhang, Mackenzie Kosak, Sarah Rathwell, Jasmine Doonanco, Alison J. Eaton, Peter Kannu, Joanna Lazier, Monique Lui, Karen Y. Niederhoffer, and et al. 2022. "Prenatal Genetic Testing in the Era of Next Generation Sequencing: A One-Center Canadian Experience" Genes 13, no. 11: 2019. https://doi.org/10.3390/genes13112019
APA StyleAlmubarak, A., Zhang, D., Kosak, M., Rathwell, S., Doonanco, J., Eaton, A. J., Kannu, P., Lazier, J., Lui, M., Niederhoffer, K. Y., MacPherson, M. J., Sorsdahl, M., & Caluseriu, O. (2022). Prenatal Genetic Testing in the Era of Next Generation Sequencing: A One-Center Canadian Experience. Genes, 13(11), 2019. https://doi.org/10.3390/genes13112019