
Next Generation Sequencing after Invasive Prenatal Testing in Fetuses with Congenital Malformations: Prenatal or Neonatal Investigation


Abstract
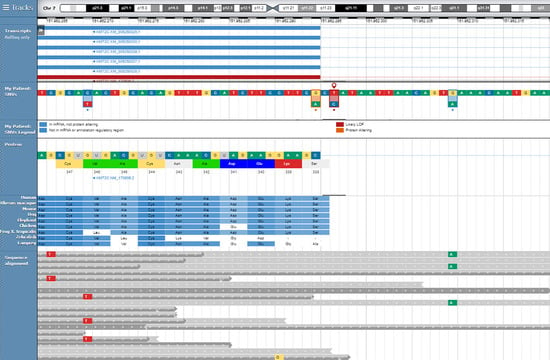
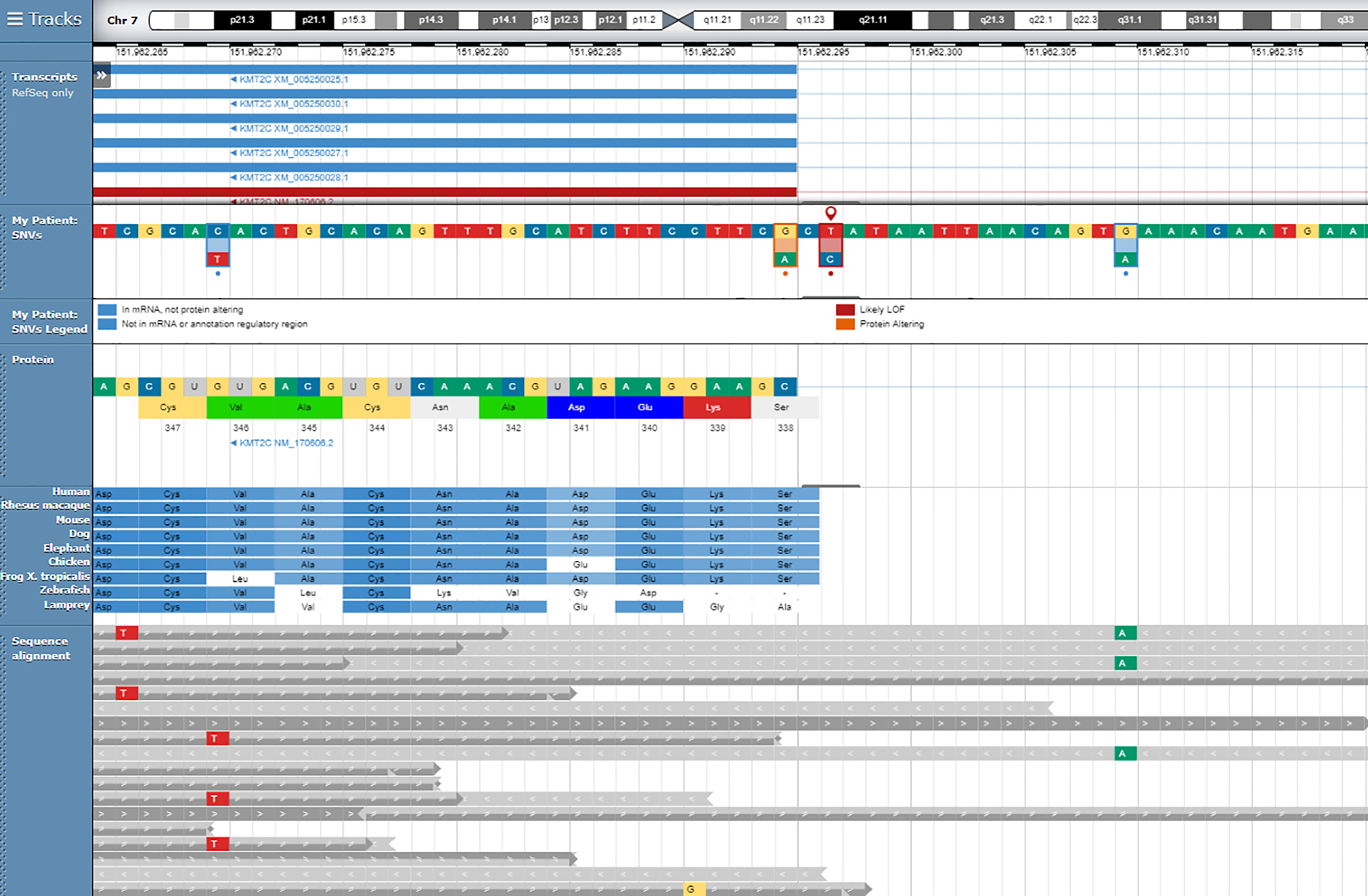
:
{kind=link}
{kind=link}
1. Introduction
2. Prenatal Imaging for Diagnosis of Fetal Structural Differences
3. Clinical Genetics
4. Cell Free Fetal DNA (cffDNA) Based Testing
5. Next Generation Sequencing: Techniques Used for Prenatal Diagnosis
5.1. Exome Sequencing
5.2. Whole Genome Sequencing
6. Variant Classification and Challenges Faced
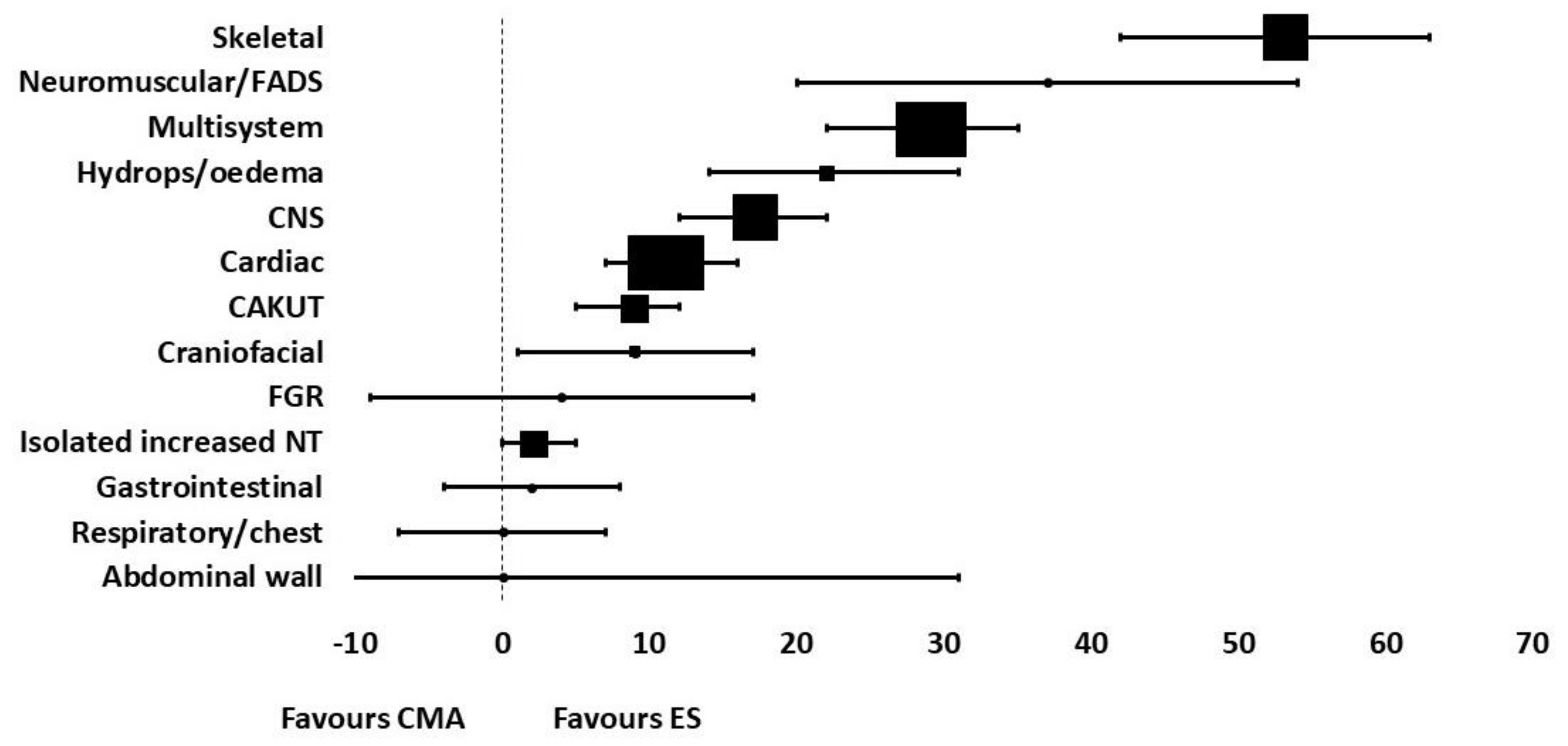
7. Diagnostic Yield of Next Generation Sequencing in Single System and Multisystem Fetal Disease
7.1. Cardiac
7.2. Neurological
7.3. Increased Nuchal Translucency
7.4. Hydrops
7.5. Renal and Urinary Tract
7.6. Skeletal Dysplasias
7.7. Overall Diagnostic Yield of NGS in Single System Congenital Abnormalities
8. Genetic and Genomic Investigation in the Neonatal Period
9. Postmortem Clinico-Pathological Correlation
10. Planning a Future Pregnancy
11. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mone, F.; McMullan, D.J.; Williams, D.; Chitty, L.S.; Maher, E.R.; Kilby, M.D. Fetal Genomics Steering Group of the British Society for Genetic Medicine; on behalf of the Royal College of Obstetricians and Gynaecologists. Evidence to Support the Clinical Utility of Prenatal Exome Sequencing in Evaluation of the Fetus with Congenital Anomalies. Scientific Impact Paper No. 64. BJOG 2021, 128, e39–e50. [Google Scholar] [CrossRef] [PubMed]
- Lord, J.; McMullan, D.J.; Eberhardt, R.Y.; Rinck, G.; Hamilton, S.J.; Quinlan-Jones, E.; Prigmore, E.; Keelagher, R.; Best, S.K.; Carey, G.K.; et al. Prenatal Assessment of Genomes and Exomes Consortium. Prenatal exome sequencing analysis in fetal structural anomalies detected by ultrasonography (PAGE): A cohort study. Lancet 2019, 393, 747–757. [Google Scholar] [CrossRef] [Green Version]
- Petrovski, S.; Aggarwal, V.; Giordano, J.L.; Stosic, M.; Wou, K.; Bier, L.; Spiegel, E.; Brennan, K.; Stong, N.; Jobanputra, V.; et al. Whole-exome sequencing in the evaluation of fetal structural anomalies: A prospective cohort study. Lancet 2019, 393, 758–767. [Google Scholar] [CrossRef]
- Mone, F.; Abu Subieh, H.; Doyle, S.; Hamilton, S.; Mcmullan, D.J.; Allen, S.; Marton, T.; Williams, D.; Kilby, M.D. Evolving fetal phenotypes and clinical impact of progressive prenatal exome sequencing pathways: Cohort study. Ultrasound Obs. Gynecol. 2022, 59, 723–730. [Google Scholar] [CrossRef] [PubMed]
- Mellis, R.; Oprych, K.; Scotchman, E.; Hill, M.; Chitty, L.S. Diagnostic yield of exome sequencing for prenatal diagnosis of fetal structural anomalies: A systematic review and meta-analysis. Prenat. Diagn. 2022, 42, 662–685. [Google Scholar] [CrossRef]
- Chandler, N.J.; Scotchman, E.; Mellis, R.; Ramachandran, V.; Roberts, R.; Chitty, L.S. Lessons learnt from prenatal exome sequencing. Prenat. Diagn. 2022, 42, 831–844. [Google Scholar] [CrossRef]
- NHS England. National Genomic Test Directories. February 2022. Available online: https://www.england.nhs.uk/publication/national-genomic-test-directories/ (accessed on 16 March 2022).
- NHS England. Rapid Exome Sequencing Service for Fetal Anomalies Testing. July 2021. Available online: https://www.england.nhs.uk/wp-content/uploads/2021/07/B0179_Guidance-rapid-exome-sequencing-service-for-fetal-anomalies_July21.pdf (accessed on 16 March 2022).
- Meng, L.; Pammi, M.; Saronwala, A.; Magoulas, P.; Ghazi, A.R.; Vetrini, F.; Zhang, J.; He, W.; Dharmadhikari, A.V.; Qu, C.; et al. Use of Exome Sequencing for Infants in Intensive Care Units: Ascertainment of Severe Single-Gene Disorders and Effect on Medical Management. JAMA Pediatr. 2017, 171, e173438. [Google Scholar] [CrossRef]
- Salomon, L.J.; Alfirevic, Z.; Berghella, V.; Bilardo, C.; Hernandez-Andrade, E.; Johnsen, S.L.; Kalache, K.; Leung, K.Y.; Malinger, G.; Munoz, H.; et al. Practice guidelines for performance of the routine mid-trimester fetal ultrasound scan. Ultrasound Obstet. Gynecol. 2011, 37, 116–126. [Google Scholar] [CrossRef]
- Public Health England. NHS Fetal Anomaly Screening Programme. July 2021. NHS Fetal Anomaly Screening Programme (FASP): Programme Overview—GOV.UK. Available online: www.gov.uk (accessed on 1 July 2022).
- Dolk, H.; Loane, M.; Garne, E. The prevalence of congenital anomalies in Europe. Adv. Exp. Med. Biol. 2010, 686, 349–364. [Google Scholar]
- Fryer, A. Genetics of fetal anomalies. In Fetal Medicine (Royal College of Obstetricians and Gynaecologists Advanced Skills); Kumar, B., Alfirevic, Z., Eds.; Cambridge University Press: Cambridge, MA, USA, 2016. [Google Scholar]
- Griffiths, P.D.; Bradburn, M.; Campbell, M.J.; Cooper, C.L.; Graham, R.; Jarvis, D.; Kilby, M.D.; Mason, G.; Mooney, C.; Robson, S.C.; et al. Use of MRI in the diagnosis of fetal brain abnormalities in utero (MERIDIAN): A multicentre, prospective cohort study. Lancet 2017, 389, 538–546. [Google Scholar] [CrossRef] [Green Version]
- Mone, F.; Doyle, S.; Ahmad, A.; Abu Subieh, H.; Hamilton, S.; Allen, S.; Marton, T.; Williams, D.; Kilby, M.D. Diagnostic and perinatal outcomes in consanguineous couples with a structural fetal anomaly: A cohort study. Acta Obstet. Gynecol. Scand. 2021, 100, 418–424. [Google Scholar] [CrossRef] [PubMed]
- Mone, F.; O’Connor, C.; Hamilton, S.; Quinlan-Jones, E.; Allen, S.; Marton, T.; Williams, D.; Kilby, M.D. Evolution of a prenatal genetic clinic-A 10-year cohort study. Prenat. Diagn. 2020, 40, 618–625. [Google Scholar] [CrossRef] [PubMed]
- Quinlan-Jones, E.; Hillman, S.C.; Kilby, M.D.; Greenfield, S.M. Parental experiences of prenatal whole exome sequencing (WES) in cases of ultrasound diagnosed fetal structural anomaly. Prenat. Diagn. 2017, 37, 1225–1231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hillman, S.C.; Skelton, J.; Quinlan-Jones, E.; Wilson, A.; Kilby, M.D. “If it helps...” the use of microarray technology in prenatal testing: Patient and partners reflections. Am. J. Med. Genet. A 2013, 161A, 1619–1627. [Google Scholar] [CrossRef] [PubMed]
- Horn, R.; Parker, M. Health professionals’ and researchers’ perspectives on prenatal whole genome and exome sequencing: “We can’t shut the door now, the genie’s out, we need to refine it”. PLoS ONE 2018, 13, e0204158. [Google Scholar] [CrossRef] [Green Version]
- Kilby, M.D. The role of next generation sequencing in the investigation of ultrasound identified fetal structural anomalies. BJOG Int. J. Obstet. Gynaecol. 2021, 128, 420. [Google Scholar] [CrossRef]
- Hillman, S.C.; McMullan, D.J.; Hall, G.; Togneri, F.S.; James, N.; Maher, E.J.; Meller, C.H.; Williams, D.; Wapner, R.J.; Maher, E.R.; et al. Use of prenatal chromosomal microarray: Prospective cohort study and systematic review and meta-analysis. Ultrasound Obstet. Gynecol. 2013, 41, 610–620. [Google Scholar] [CrossRef]
- Chong, H.P.; Hamilton, S.; Mone, F.; Cheung, K.W.; Togneri, F.S.; Morris, R.K.; Quinlan-Jones, E.; Williams, D.; Allen, S.; McMullan, D.J.; et al. Prenatal chromosomal microarray testing of fetuses with ultrasound structural anomalies: A prospective cohort study of over 1000 consecutive cases. Prenat. Diagn. 2019, 39, 1064–1069. [Google Scholar] [CrossRef]
- Everett, T.R.; Chitty, L.S. Cell-free fetal DNA: The new tool in fetal medicine. Ultrasound Obstet. Gynecol. 2015, 45, 499–507. [Google Scholar] [CrossRef]
- Young, E.; Bowns, B.; Gerrish, A.; Parks, M.; Court, S.; Clokie, S.; Mashayamombe-Wolfgarten, C.; Hewitt, J.; Williams, D.; Cole, T.; et al. Clinical Service Delivery of Noninvasive Prenatal Diagnosis by Relative Haplotype Dosage for Single-Gene Disorders. J. Mol. Diagn. 2020, 22, 1151–1161. [Google Scholar] [CrossRef]
- Allen, S.; Young, E.; Bowns, B. Noninvasive prenatal diagnosis for single gene disorders. Curr. Opin. Obstet. Gynecol. 2017, 29, 73–79. Available online: https://journals.lww.com/co-obgyn/Fulltext/2017/04000/Noninvasive_prenatal_diagnosis_for_single_gene.5.aspx (accessed on 1 July 2022).
- Parks, M.; Court, S.; Bowns, B.; Cleary, S.; Clokie, S.; Hewitt, J.; Williams, D.; Cole, T.; MacDonald, F.; Griffiths, M.; et al. Non-invasive prenatal diagnosis of spinal muscular atrophy by relative haplotype dosage. Eur. J. Hum. Genet. 2017, 25, 416–422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parks, M.; Court, S.; Cleary, S.; Clokie, S.; Hewitt, J.; Williams, D.; Cole, T.; MacDonald, F.; Griffiths, M.; Allen, S. Non-invasive prenatal diagnosis of Duchenne and Becker muscular dystrophies by relative haplotype dosage. Prenat. Diagn. 2016, 36, 312–320. [Google Scholar] [CrossRef] [PubMed]
- Cayrefourcq, L.; Vincent, M.-C.; Pierredon, S.; Moutou, C.; Imbert-Bouteille, M.; Haquet, E.; Puechberty, J.; Willems, M.; Liautard-Haag, C.; Molinari, N.; et al. Single Circulating Fetal Trophoblastic Cells Eligible for Non Invasive Prenatal Diagnosis: The Exception Rather than the Rule. Sci. Rep. 2020, 10, 9861. [Google Scholar] [CrossRef] [PubMed]
- Sabbatinelli, G.; Fantasia, D.; Palka, C.; Morizio, E.; Alfonsi, M.; Calabrese, G. Isolation and Enrichment of Circulating Fetal Cells for NIPD: An Overview. Diagnostics 2021, 11, 2239. [Google Scholar] [CrossRef] [PubMed]
- Castleman, J.S.; Wall, E.; Allen, S.; Williams, D.; Doyle, S.; Kilby, M.D. The prenatal exome—A door to prenatal diagnostics? Expert Rev. Mol. Diagn. 2021, 21, 465–474. [Google Scholar] [CrossRef]
- Meienberg, J.; Bruggmann, R.; Oexle, K.; Matyas, G. Clinical sequencing: Is WGS the better WES? Hum. Genet. 2016, 135, 359–362. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Greenfeld, E.; Watkins, N.; Belesiotis, P.; Zaidi, S.H.; Marshall, C.; Thiruvahindrapuram, B.; Shannon, P.; Roifman, M.; Chong, K.; et al. Diagnostic yield of genome sequencing for prenatal diagnosis of fetal structural anomalies. Prenat. Diagn. 2022, 42, 822–830. [Google Scholar] [CrossRef]
- Van den Veyver, I.B.; Chandler, N.; Wilkins-Haug, L.E.; Wapner, R.J.; Chitty, L.S. International Society for Prenatal Diagnosis Updated Position Statement on the use of genome-wide sequencing for prenatal diagnosis. Prenat. Diagn. 2022, 42, 796–803. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. Off. J. Am. Coll. Med. Genet. 2015, 17, 405–424. [Google Scholar] [CrossRef] [Green Version]
- Biesecker, L.; Harrison, S.; Azzariti, D. Sequence Variation Interpretation Working Group. Sequence Variant Interpretation. [Internet] Clinical Genome Resource (US). 2022. Available online: https://clinicalgenome.org/working-groups/sequence-variant-interpretation/ (accessed on 18 July 2022).
- Ellard, S.; Baple, E.; Callaway, A.; Berry, I.; Forrester, N.; Turnbull, C.; Owens, M.; Eccles, D.M.; Abbs, S.; Scott, R.; et al. ACGS Best Practice Guidelines for Variant Classification in Rare Disease 2020. Available online: https://www.acgs.uk.com/media/11631/uk-practice-guidelines-for-variant-classification-v4-01-2020.pdf (accessed on 14 July 2022).
- Mone, F.; Eberhardt, R.Y.; Morris, R.K.; Hurles, M.E.; McMullan, D.J.; Maher, E.R.; Lord, J.; Chitty, L.S.; Giordano, J.L.; Wapner, R.J.; et al. COngenital heart disease and the Diagnostic yield with Exome sequencing (CODE) study: Prospective cohort study and systematic review. Ultrasound Obstet. Gynecol. 2021, 57, 43–51. [Google Scholar] [CrossRef]
- Qiao, F.; Wang, Y.; Zhang, C.; Zhou, R.; Wu, Y.; Wang, C.; Meng, L.; Mao, P.; Cheng, Q.; Luo, C.; et al. Comprehensive evaluation of genetic variants using chromosomal microarray analysis and exome sequencing in fetuses with congenital heart defect. Ultrasound Obstet. Gynecol. 2021, 58, 377–387. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Fu, F.; Yu, Q.; Wang, D.; Jing, X.; Zhang, Y.; Li, F.; Li, F.; Han, J.; Pan, M.; et al. Prenatal exome sequencing in fetuses with congenital heart defects. Clin. Genet. 2020, 98, 215–230. [Google Scholar] [CrossRef] [PubMed]
- Baptiste, C.; Mellis, R.; Aggarwal, V.; Lord, J.; Eberhardt, R.; Kilby, M.D.; Maher, E.R.; Wapner, R.; Giordano, J.; Chitty, L. Fetal central nervous system anomalies: When should we offer exome sequencing? Prenat. Diagn. 2022, 42, 736–743. [Google Scholar] [CrossRef] [PubMed]
- Mellis, R.; Eberhardt, R.Y.; Hamilton, S.J.; McMullan, D.J.; Kilby, M.D.; Maher, E.R.; Hurles, M.E.; Giordano, J.L.; Aggarwal, V.; Goldstein, D.B.; et al. Fetal exome sequencing for isolated increased nuchal translucency: Should we be doing it? BJOG 2022, 129, 52–61. [Google Scholar] [CrossRef] [PubMed]
- Bilardo, C.M.; Timmerman, E.; Pajkrt, E.; van Maarle, M. Increased nuchal translucency in euploid fetuses—what should we be telling the parents? Prenat. Diagn. 2010, 30, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Bilardo, C.M.; Müller, M.A.; Pajkrt, E.; Clur, S.A.; van Zalen, M.M.; Bijlsma, E.K. Increased nuchal translucency thickness and normal karyotype: Time for parental reassurance. Ultrasound Obstet. Gynecol. 2007, 30, 11–18. [Google Scholar] [CrossRef]
- Mone, F.; Eberhardt, R.Y.; Hurles, M.E.; Mcmullan, D.J.; Maher, E.R.; Lord, J.; Chitty, L.S.; Dempsey, E.; Homfray, T.; Giordano, J.L.; et al. Fetal hydrops and the Incremental yield of Next-generation sequencing over standard prenatal Diagnostic testing (FIND) study: Prospective cohort study and meta-analysis. Ultrasound Obstet. Gynecol. 2021, 58, 509–518. [Google Scholar] [CrossRef]
- Zhou, X.; Wang, Y.; Shao, B.; Wang, C.; Hu, P.; Qiao, F.; Xu, Z. Molecular diagnostic in fetuses with isolated congenital anomalies of the kidney and urinary tract by whole-exome sequencing. J. Clin. Lab. Anal. 2020, 34, e23480. [Google Scholar] [CrossRef]
- Lei, T.-Y.; Fu, F.; Li, R.; Wang, D.; Wang, R.-Y.; Jing, X.-Y.; Deng, Q.; Li, Z.Z.; Liu, Z.Q.; Yang, X.; et al. Whole-exome sequencing for prenatal diagnosis of fetuses with congenital anomalies of the kidney and urinary tract. Nephrol. Dial Transpl. 2017, 32, 1665–1675. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Pan, L.; Teng, Y.; Liang, D.; Li, Z.; Wu, L. Molecular diagnosis for 55 fetuses with skeletal dysplasias by whole-exome sequencing: A retrospective cohort study. Clin Genet. 2021, 100, 219–226. [Google Scholar] [CrossRef]
- Kucińska-Chahwan, A.; Roszkowski, T.; Nowakowska, B.; Geremek, M.; Paczkowska, M.; Bijok, J.; Massalska, D. Genetic causes of the skeletal system abnormalities diagnosed by prenatal sonography with the use of exome sequencing: Single institution experience. Ultrasound Obstet. Gynecol. 2021. Epub ahead of print. [Google Scholar] [CrossRef]
- Peng, Y.; Yang, S.; Huang, X.; Pang, J.; Liu, J.; Hu, J.; Shen, X.; Tang, C.; Wang, H. Whole Exome Sequencing Analysis in Fetal Skeletal Dysplasia Detected by Ultrasonography: An Analysis of 38 Cases. Front. Genet. 2021, 12, 728544. [Google Scholar] [CrossRef] [PubMed]
- Holm, I.A.; Agrawal, P.B.; Ceyhan-Birsoy, O.; Christensen, K.D.; Fayer, S.; Frankel, L.A.; Genetti, C.A.; Krier, J.B.; LaMay, R.C.; Levy, H.L.; et al. The BabySeq project: Implementing genomic sequencing in newborns. BMC Pediatr. 2018, 18, 225. [Google Scholar] [CrossRef] [Green Version]
- Steele, B.; Mackie, A.; Wigley, C. UK National Screening Committee and Genomics England. Implications for Whole Genome Sequencing for Newborn Screening. A Public Dialogue. July 2021. UK: UK National Screening Committee and Genomics England; 2021. Available online: https://assets.publishing.service.gov.uk/government/uploads/system/uploads/attachment_data/file/999931/WGS_for_newborn_screening_FINAL_ACCESSIBLE.pdf (accessed on 18 July 2022).
- Quinlan-Jones, E.; Lord, J.; Williams, D.; Hamilton, S.; Marton, T.; Eberhardt, R.Y.; Rinck, G.; Prigmore, E.; Keelagher, R.; McMullan, D.J.; et al. Molecular autopsy by trio exome sequencing (ES) and postmortem examination in fetuses and neonates with prenatally identified structural anomalies. Genet. Med. 2019, 21, 1065–1073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yates, C.L.; Monaghan, K.G.; Copenheaver, D.; Retterer, K.; Scuffins, J.; Kucera, C.R.; Friedman, B.; Richard, G.; Juusola, J. Whole-exome sequencing on deceased fetuses with ultrasound anomalies: Expanding our knowledge of genetic disease during fetal development. Genet. Med. 2017, 19, 1171–1178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiorentino, F.; Bono, S.; Biricik, A.; Nuccitelli, A.; Cotroneo, E.; Cottone, G.; Kokocinski, F.; Michel, C.E.; Minasi, M.G.; Greco, E. Application of next-generation sequencing technology for comprehensive aneuploidy screening of blastocysts in clinical preimplantation genetic screening cycles. Hum Reprod. 2014, 29, 2802–2813. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Emms, A.; Castleman, J.; Allen, S.; Williams, D.; Kinning, E.; Kilby, M. Next Generation Sequencing after Invasive Prenatal Testing in Fetuses with Congenital Malformations: Prenatal or Neonatal Investigation. Genes 2022, 13, 1517. https://doi.org/10.3390/genes13091517
Emms A, Castleman J, Allen S, Williams D, Kinning E, Kilby M. Next Generation Sequencing after Invasive Prenatal Testing in Fetuses with Congenital Malformations: Prenatal or Neonatal Investigation. Genes. 2022; 13(9):1517. https://doi.org/10.3390/genes13091517
Chicago/Turabian StyleEmms, Alexandra, James Castleman, Stephanie Allen, Denise Williams, Esther Kinning, and Mark Kilby. 2022. "Next Generation Sequencing after Invasive Prenatal Testing in Fetuses with Congenital Malformations: Prenatal or Neonatal Investigation" Genes 13, no. 9: 1517. https://doi.org/10.3390/genes13091517
APA StyleEmms, A., Castleman, J., Allen, S., Williams, D., Kinning, E., & Kilby, M. (2022). Next Generation Sequencing after Invasive Prenatal Testing in Fetuses with Congenital Malformations: Prenatal or Neonatal Investigation. Genes, 13(9), 1517. https://doi.org/10.3390/genes13091517