
Allele-Specific Disruption of a Common STAT3 Autosomal Dominant Allele Is Not Sufficient to Restore Downstream Signaling in Patient-Derived T Cells
 , ,
, ,  and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Plasmids
2.2. Cell Culture
2.3. STAT3 Reporter Assay
2.4. Next Generation Sequencing
2.5. Functional Evaluation of STAT3 Signaling
3. Results
3.1. Selection of Designer Nucleases Specific for the Dominant Negative STAT3 Alleles
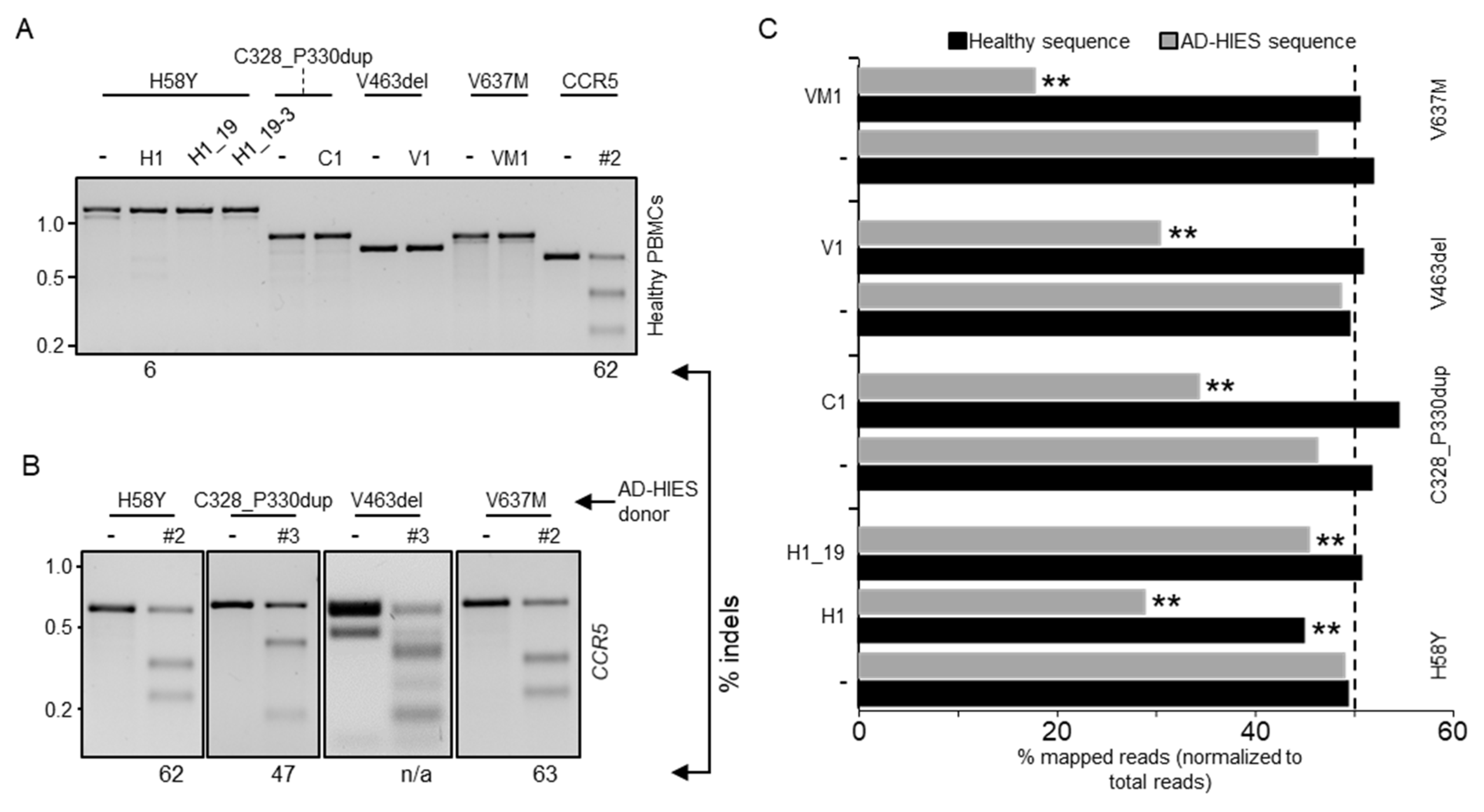
3.2. Validation of Allele-Specificity at Genomic Sites
3.3. Allele-Specific Disruption of Autosomal Dominant STAT3 in Patient-Derived Human Cells
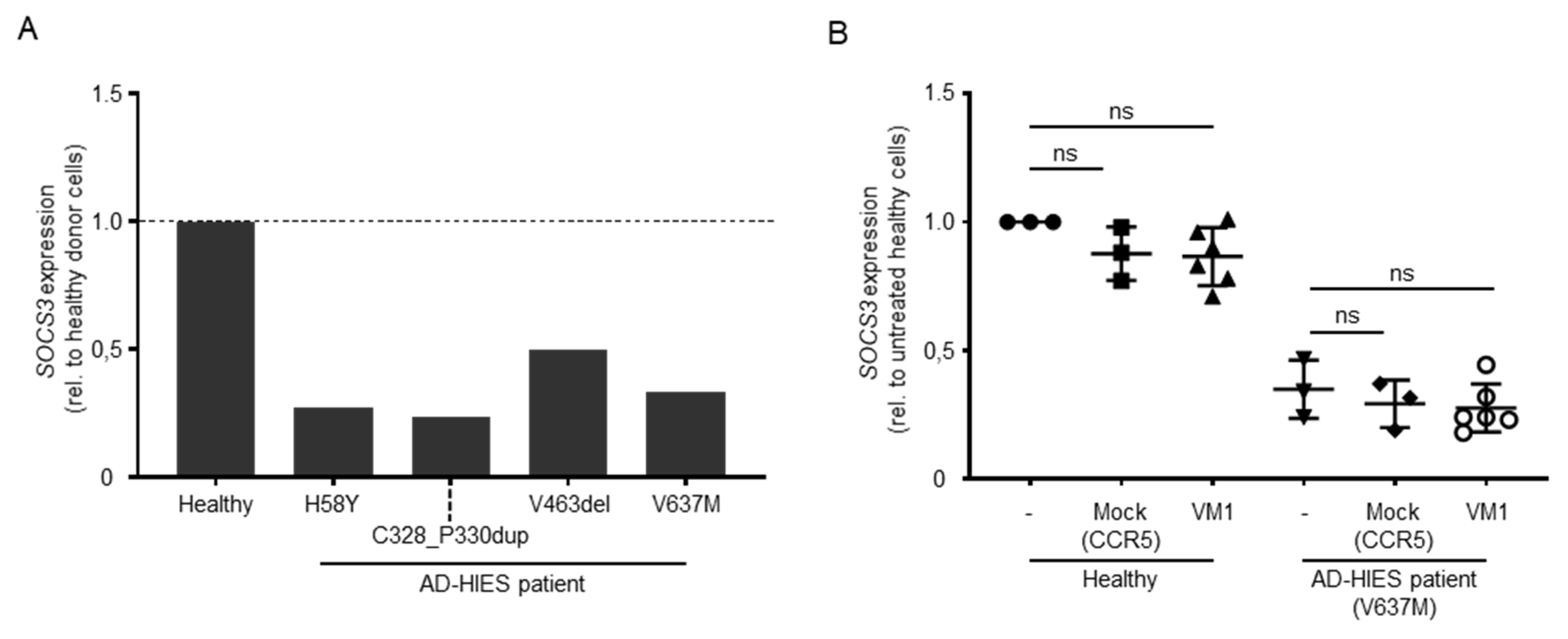
3.4. Disruption of the Mutated DN-STAT3 Alleles Does Not Rescue STAT3 Signaling
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Morris, R.; Kershaw, N.J.; Babon, J.J. The molecular details of cytokine signaling via the JAK/STAT pathway. Protein Sci. 2018, 27, 1984–2009. [Google Scholar] [CrossRef] [PubMed]
- O’Shea, J.J. Jaks, STATs, cytokine signal transduction, and immunoregulation: Are we there yet? Immunity 1997, 7, 1–11. [Google Scholar] [CrossRef]
- Hillmer, E.J.; Zhang, H.; Li, H.S.; Watowich, S.S. STAT3 signaling in immunity. Cytokine Growth Factor Rev. 2016, 31, 1–15. [Google Scholar] [CrossRef]
- Faletti, L.; Ehl, S.; Heeg, M. Germline STAT3 gain-of-function mutations in primary immunodeficiency: Impact on the cellular and clinical phenotype. Biomed. J. 2021, 44, 412–421. [Google Scholar] [CrossRef] [PubMed]
- Holland, S.M.; DeLeo, F.R.; Elloumi, H.Z.; Hsu, A.P.; Uzel, G.; Brodsky, N.; Freeman, A.F.; Demidowich, A.; Davis, J.; Turner, M.L.; et al. STAT3 mutations in the hyper-IgE syndrome. N. Engl. J. Med. 2007, 357, 1608–1619. [Google Scholar] [CrossRef] [PubMed]
- Grimbacher, B.; Holland, S.M.; Gallin, J.I.; Greenberg, F.; Hill, S.C.; Malech, H.L.; Miller, J.A.; O’Connell, A.C.; Puck, J.M. Hyper-IgE syndrome with recurrent infections—An autosomal dominant multisystem disorder. N. Engl. J. Med. 1999, 340, 692–702. [Google Scholar] [CrossRef]
- Asano, T.; Khourieh, J.; Zhang, P.; Rapaport, F.; Spaan, A.N.; Li, J.; Lei, W.T.; Pelham, S.J.; Hum, D.; Chrabieh, M.; et al. Human STAT3 variants underlie autosomal dominant hyper-IgE syndrome by negative dominance. J. Exp. Med. 2021, 218, e20202592. [Google Scholar] [CrossRef]
- Tsilifis, C.; Freeman, A.F.; Gennery, A.R. STAT3 Hyper-IgE Syndrome-an Update and Unanswered Questions. J. Clin. Immunol. 2021, 41, 864–880. [Google Scholar] [CrossRef]
- Freeman, A.F.; Kleiner, D.E.; Nadiminti, H.; Davis, J.; Quezado, M.; Anderson, V.; Puck, J.M.; Holland, S.M. Causes of death in hyper-IgE syndrome. J. Allergy Clin. Immunol. 2007, 119, 1234–1240. [Google Scholar] [CrossRef]
- Gennery, A.R.; Flood, T.J.; Abinun, M.; Cant, A.J. Bone marrow transplantation does not correct the hyper IgE syndrome. Bone Marrow Transplant. 2000, 25, 1303–1305. [Google Scholar] [CrossRef]
- Harrison, S.C.; Tsilifis, C.; Slatter, M.A.; Nademi, Z.; Worth, A.; Veys, P.; Ponsford, M.J.; Jolles, S.; Al-Herz, W.; Flood, T.; et al. Hematopoietic Stem Cell Transplantation Resolves the Immune Deficit Associated with STAT3-Dominant-Negative Hyper-IgE Syndrome. J. Clin. Immunol. 2021, 41, 934–943. [Google Scholar] [CrossRef]
- Oikonomopoulou, C.; Goussetis, E. Autosomal dominant hyper-IgE syndrome: When hematopoietic stem cell transplantation should be considered? Pediatr. Transpl. 2020, 24, e13699. [Google Scholar] [CrossRef]
- Hierlmeier, S.; Eyrich, M.; Wolfl, M.; Schlegel, P.G.; Wiegering, V. Early and late complications following hematopoietic stem cell transplantation in pediatric patients—A retrospective analysis over 11 years. PLoS ONE 2018, 13, e0204914. [Google Scholar] [CrossRef]
- Takeda, K.; Noguchi, K.; Shi, W.; Tanaka, T.; Matsumoto, M.; Yoshida, N.; Kishimoto, T.; Akira, S. Targeted disruption of the mouse Stat3 gene leads to early embryonic lethality. Proc. Natl. Acad. Sci. USA 1997, 94, 3801–3804. [Google Scholar] [CrossRef]
- Carroll, D. Genome engineering with targetable nucleases. Annu. Rev. Biochem. 2014, 83, 409–439. [Google Scholar] [CrossRef] [PubMed]
- Carusillo, A.; Mussolino, C. DNA Damage: From Threat to Treatment. Cells 2020, 9, 1665. [Google Scholar] [CrossRef]
- Frangoul, H.; Altshuler, D.; Cappellini, M.D.; Chen, Y.S.; Domm, J.; Eustace, B.K.; Foell, J.; de la Fuente, J.; Grupp, S.; Handgretinger, R.; et al. CRISPR-Cas9 Gene Editing for Sickle Cell Disease and beta-Thalassemia. N. Engl. J. Med. 2021, 384, 252–260. [Google Scholar] [CrossRef]
- Gillmore, J.D.; Gane, E.; Taubel, J.; Kao, J.; Fontana, M.; Maitland, M.L.; Seitzer, J.; O’Connell, D.; Walsh, K.R.; Wood, K.; et al. CRISPR-Cas9 In Vivo Gene Editing for Transthyretin Amyloidosis. N. Engl. J. Med. 2021, 385, 493–502. [Google Scholar] [CrossRef] [PubMed]
- Clement, K.; Rees, H.; Canver, M.C.; Gehrke, J.M.; Farouni, R.; Hsu, J.Y.; Cole, M.A.; Liu, D.R.; Joung, J.K.; Bauer, D.E.; et al. CRISPResso2 provides accurate and rapid genome editing sequence analysis. Nat. Biotechnol. 2019, 37, 224–226. [Google Scholar] [CrossRef]
- Woellner, C.; Gertz, E.M.; Schaffer, A.A.; Lagos, M.; Perro, M.; Glocker, E.O.; Pietrogrande, M.C.; Cossu, F.; Franco, J.L.; Matamoros, N.; et al. Mutations in STAT3 and diagnostic guidelines for hyper-IgE syndrome. J. Allergy Clin. Immunol. 2010, 125, 424–432.e8. [Google Scholar] [CrossRef] [PubMed]
- Farasat, I.; Salis, H.M. A Biophysical Model of CRISPR/Cas9 Activity for Rational Design of Genome Editing and Gene Regulation. PLoS Comput. Biol. 2016, 12, e1004724. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Sander, J.D.; Reyon, D.; Cascio, V.M.; Joung, J.K. Improving CRISPR-Cas nuclease specificity using truncated guide RNAs. Nat. Biotechnol. 2014, 32, 279–284. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Stroukov, W.; Cathomen, T.; Mussolino, C. Chimerization Enables Gene Synthesis and Lentiviral Delivery of Customizable TALE-Based Effectors. Int. J. Mol. Sci. 2020, 21, 795. [Google Scholar] [CrossRef] [PubMed]
- Mussolino, C.; Alzubi, J.; Fine, E.J.; Morbitzer, R.; Cradick, T.J.; Lahaye, T.; Bao, G.; Cathomen, T. TALENs facilitate targeted genome editing in human cells with high specificity and low cytotoxicity. Nucleic Acids Res. 2014, 42, 6762–6773. [Google Scholar] [CrossRef]
- Turchiano, G.; Andrieux, G.; Klermund, J.; Blattner, G.; Pennucci, V.; El Gaz, M.; Monaco, G.; Poddar, S.; Mussolino, C.; Cornu, T.I.; et al. Quantitative evaluation of chromosomal rearrangements in gene-edited human stem cells by CAST-Seq. Cell Stem Cell 2021, 28, 1136–1147.e5. [Google Scholar] [CrossRef]
- Tripathi, S.K.; Chen, Z.; Larjo, A.; Kanduri, K.; Nousiainen, K.; Aijo, T.; Ricano-Ponce, I.; Hrdlickova, B.; Tuomela, S.; Laajala, E.; et al. Genome-wide Analysis of STAT3-Mediated Transcription during Early Human Th17 Cell Differentiation. Cell Rep. 2017, 19, 1888–1901. [Google Scholar] [CrossRef]
- Boyman, O.; Sprent, J. The role of interleukin-2 during homeostasis and activation of the immune system. Nat. Rev. Immunol. 2012, 12, 180–190. [Google Scholar] [CrossRef]
- Bocchini, C.E.; Nahmod, K.; Katsonis, P.; Kim, S.; Kasembeli, M.M.; Freeman, A.; Lichtarge, O.; Makedonas, G.; Tweardy, D.J. Protein stabilization improves STAT3 function in autosomal dominant hyper-IgE syndrome. Blood 2016, 128, 3061–3072. [Google Scholar] [CrossRef]
- Ledford, H. CRISPR treatment inserted directly into the body for first time. Nature 2020, 579, 185. [Google Scholar] [CrossRef]
- Ernst, M.P.T.; Broeders, M.; Herrero-Hernandez, P.; Oussoren, E.; van der Ploeg, A.T.; Pijnappel, W. Ready for Repair? Gene Editing Enters the Clinic for the Treatment of Human Disease. Mol. Therapy. Methods Clin. Dev. 2020, 18, 532–557. [Google Scholar] [CrossRef]
- Natarajan, M.; Hsu, A.P.; Weinreich, M.A.; Zhang, Y.; Niemela, J.E.; Butman, J.A.; Pittaluga, S.; Sugui, J.; Collar, A.L.; Lim, J.K.; et al. Aspergillosis, eosinophilic esophagitis, and allergic rhinitis in signal transducer and activator of transcription 3 haploinsufficiency. J. Allergy Clin. Immunol. 2018, 142, 993–997.e3. [Google Scholar] [CrossRef] [PubMed]
- Bchetnia, M.; Dionne Gagne, R.; Powell, J.; Morin, C.; McCuaig, C.; Duperee, A.; Germain, L.; Tremblay, J.P.; Laprise, C. Allele-Specific Inactivation of an Autosomal Dominant Epidermolysis Bullosa Simplex Mutation Using CRISPR-Cas9. CRISPR J. 2022, 5, 586–597. [Google Scholar] [CrossRef] [PubMed]
- Porto, E.M.; Komor, A.C.; Slaymaker, I.M.; Yeo, G.W. Base editing: Advances and therapeutic opportunities. Nat. Rev. Drug Discov. 2020, 19, 839–859. [Google Scholar] [CrossRef] [PubMed]
- Mussolino, C. Precise Epigenome Editing on the Stage: A Novel Approach to Modulate Gene Expression. Epigenet. Insights 2018, 11, 2516865718818838. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target Allele | ID | Protospacer+PAM (5’-->3’) a | Notes |
|---|---|---|---|
| CCR5 | mock/#2 | CAATGTGTCAACTCTTGACAGGG | Full-length |
| #3 | ATTTCCAAAGTCCCACTGGGCGG | ||
| STAT3 | S_KO | TAAGACCCAGATCCAGTCCGTGG | |
| H58Y | H1 | GAGATTATAAAACACCAAAGNGG | |
| H2 | CAGGAGATTATAAAACACCANAG | ||
| C328_P330dup | C1 | ATGGGCATGCAGGGCATGCANGG | |
| C2 | CATGGGCATGCAGGGCATGCNGG | ||
| R382W | R2 | ATCCTGGAAATTTAACATTCNGG | |
| R3 | TTAAATTTCCAGGATCCTCTNAG | ||
| V463del | V1 | CAGATGTTGGAGATCACAACNGG | |
| V637M | VM1 | TAAGACCCAGATCCAGTCCANGG | |
| VM3 | TTGTGTATGGTTCCATGGACNGG | ||
| H58Y | H1_19 | GATTATAAAACACCAAAGNGG | Truncated |
| H2_19 | GGAGATTATAAAACACCANAG | ||
| V463del | V1_19 | GATGTTGGAGATCACAACNGG | |
| V637M | VM1_17 | GACCCAGATCCAGTCCANGG | |
| H58Y | H1_19+1 | GATTAAAAAACACCAAAGNGG | truncated and mismatched |
| H1_19+2 | GATTTTAAAACACCAAAGNGG | ||
| H1_19+3 | GATAATAAAACACCAAAGNGG | ||
| H1_19-1 | GATTATATAACACCAAAGNGG | ||
| H1_19-2 | GATTATAATACACCAAAGNGG | ||
| H1_19-3 | GATTATAAATCACCAAAGNGG | ||
| R382W | R1_19 | GCCCAGAATGTTAAATTTCNAG | |
| R2_19+4 | GTCCTGGAAATTTAACATTCNGG | ||
| R2_18+3 | GCCTGGAAATTTAACATTCNGG | ||
| R2_17+2 | GCTGGAAATTTAACATTCNGG | ||
| R3_19+10 | GTAAATTTCCAGGATCCTCTNAG | ||
| R3_18+9 | GAAATTTCCAGGATCCTCTNAG | ||
| R3_17+8 | GAATTTCCAGGATCCTCTNAG |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
König, S.; Fliegauf, M.; Rhiel, M.; Grimbacher, B.; Cornu, T.I.; Cathomen, T.; Mussolino, C. Allele-Specific Disruption of a Common STAT3 Autosomal Dominant Allele Is Not Sufficient to Restore Downstream Signaling in Patient-Derived T Cells. Genes 2022, 13, 1912. https://doi.org/10.3390/genes13101912
König S, Fliegauf M, Rhiel M, Grimbacher B, Cornu TI, Cathomen T, Mussolino C. Allele-Specific Disruption of a Common STAT3 Autosomal Dominant Allele Is Not Sufficient to Restore Downstream Signaling in Patient-Derived T Cells. Genes. 2022; 13(10):1912. https://doi.org/10.3390/genes13101912
Chicago/Turabian StyleKönig, Saskia, Manfred Fliegauf, Manuel Rhiel, Bodo Grimbacher, Tatjana I. Cornu, Toni Cathomen, and Claudio Mussolino. 2022. "Allele-Specific Disruption of a Common STAT3 Autosomal Dominant Allele Is Not Sufficient to Restore Downstream Signaling in Patient-Derived T Cells" Genes 13, no. 10: 1912. https://doi.org/10.3390/genes13101912
APA StyleKönig, S., Fliegauf, M., Rhiel, M., Grimbacher, B., Cornu, T. I., Cathomen, T., & Mussolino, C. (2022). Allele-Specific Disruption of a Common STAT3 Autosomal Dominant Allele Is Not Sufficient to Restore Downstream Signaling in Patient-Derived T Cells. Genes, 13(10), 1912. https://doi.org/10.3390/genes13101912