
Mining Candidate Genes Related to Heavy Metals in Mature Melon (Cucumis melo L.) Peel and Pulp Using WGCNA

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials
2.2. Heavy Metal Content Detection
2.3. RNA Sequencing
2.4. RNA-Seq Data Analysis
2.5. Identification, Annotation and Cluster Analysis of DEGs
2.6. Hormone Biosynthesis, Signal Transduction and Transcription Factor Analysis
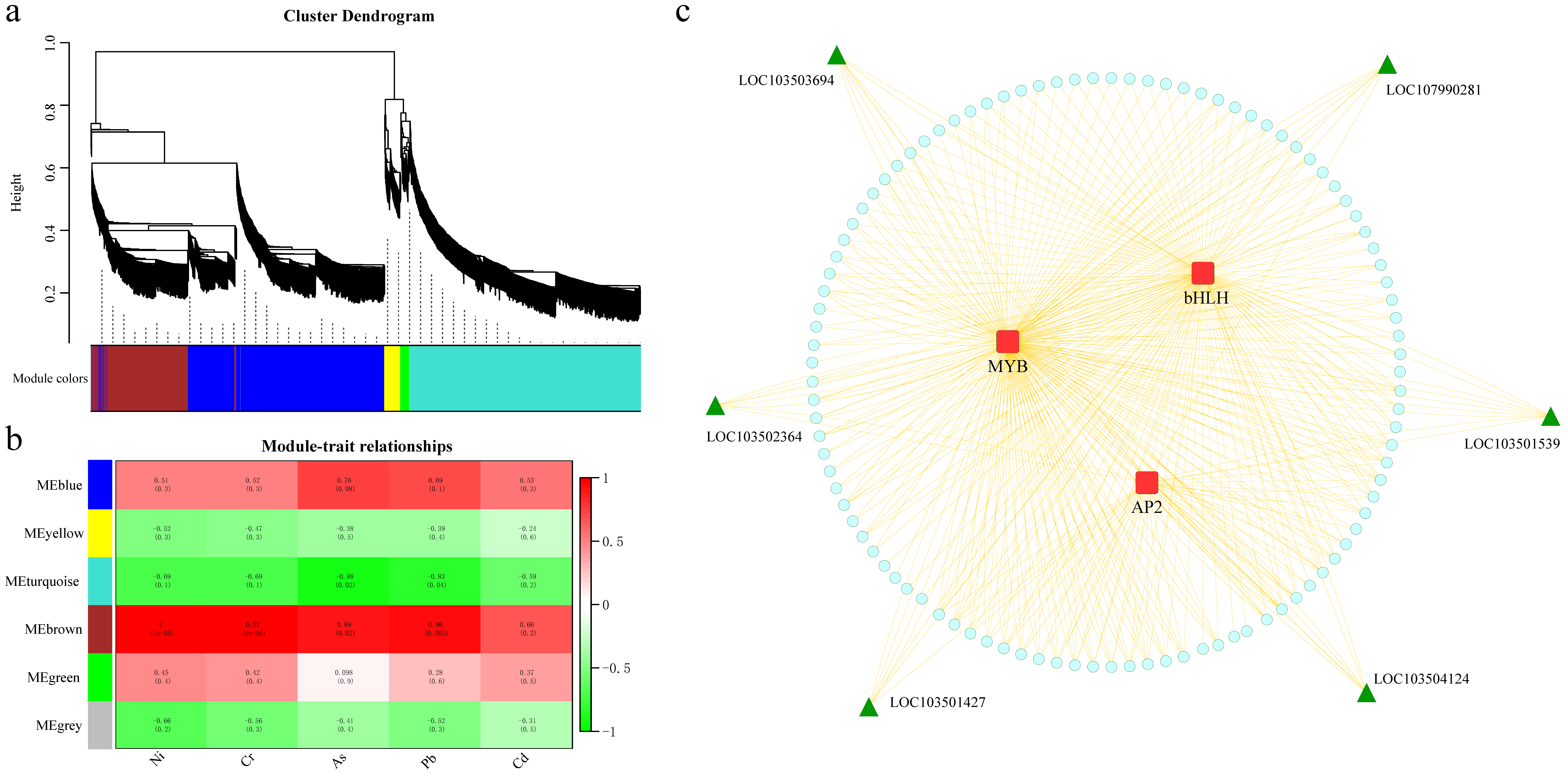
2.7. WGCNA
2.8. qRT–PCR
3. Results
3.1. Determination of Metal Ion Content in Melon Peel and Fruit Tissue
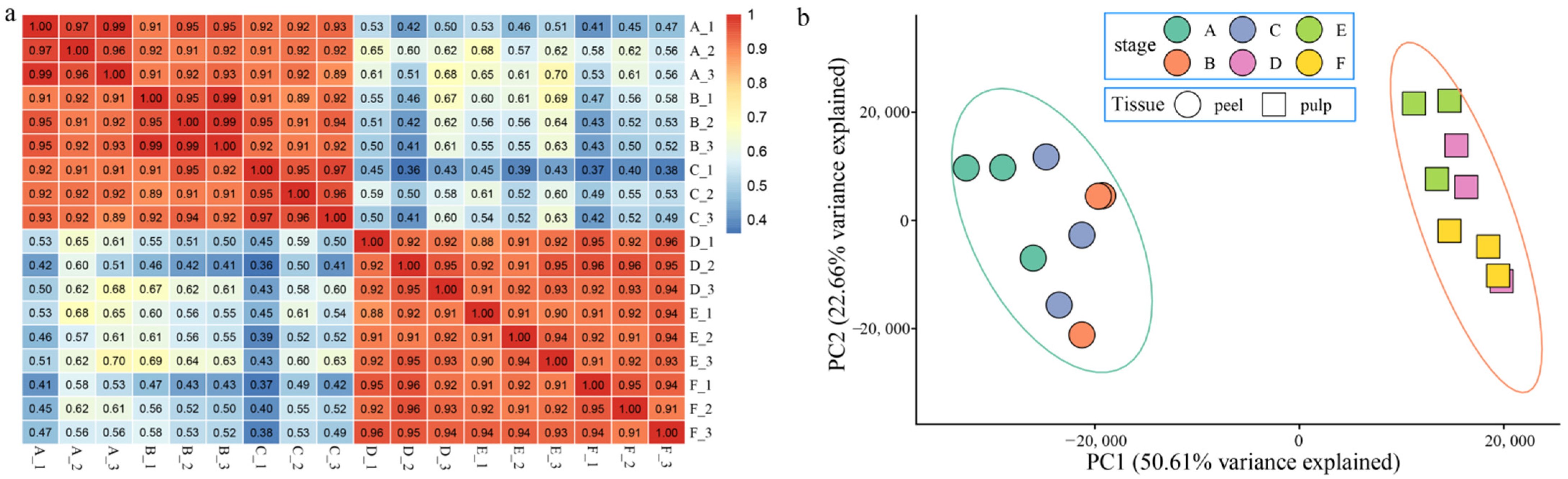
3.2. Global Transcriptome Changes in Melon Peel and Fruit Tissue
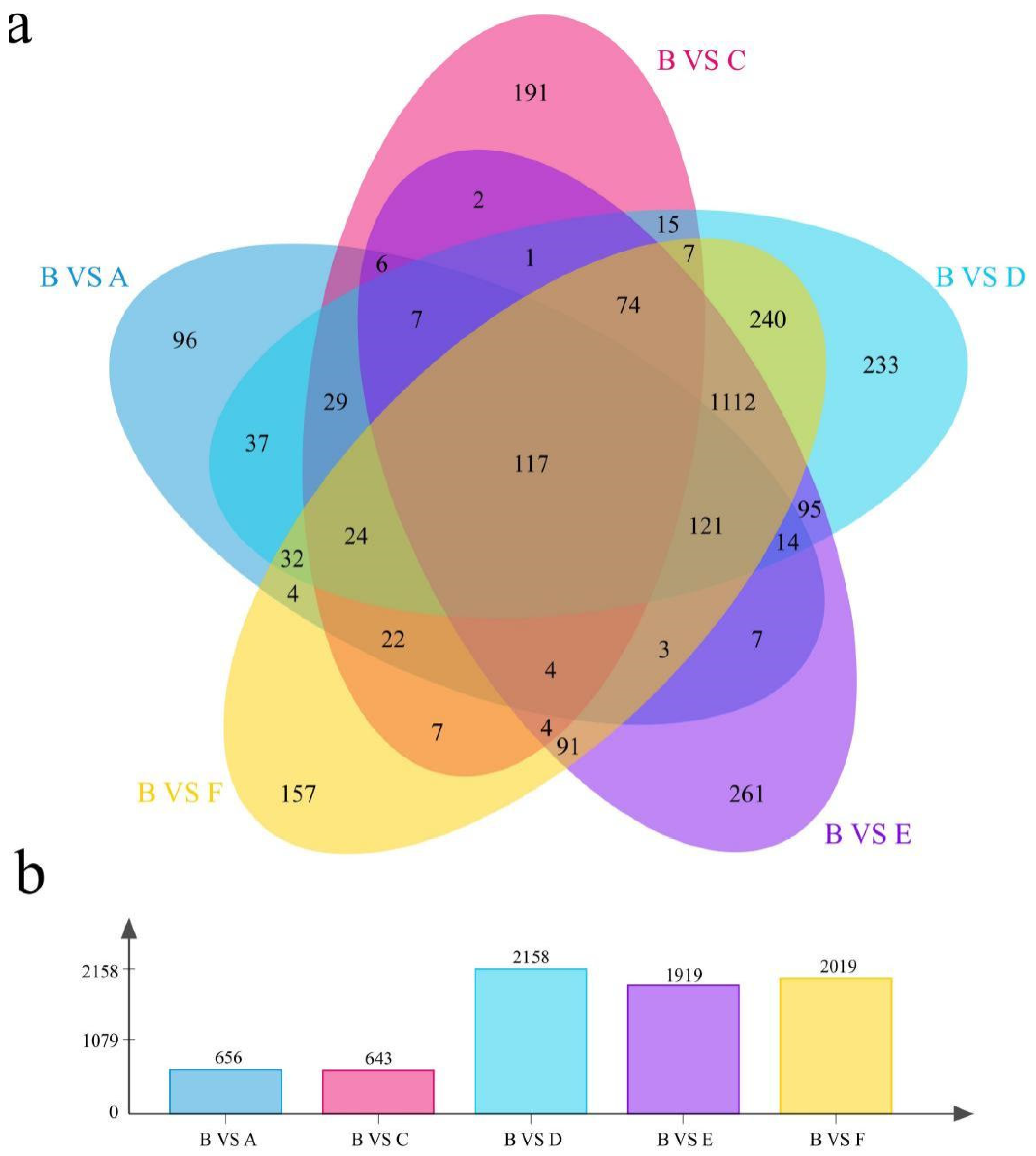
3.3. Difference Analysis
3.4. GO and KEGG Enrichment Analysis of DEGs
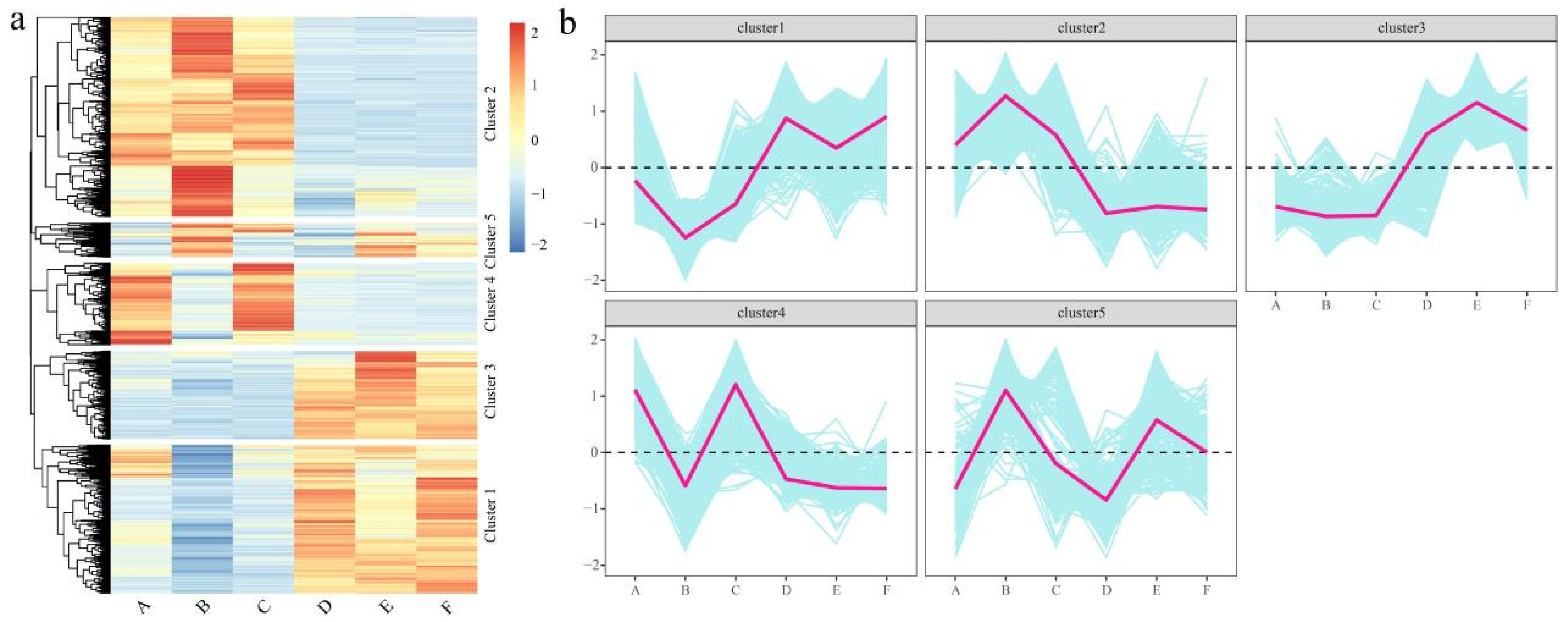
3.5. Expression Pattern Analysis of DEGs
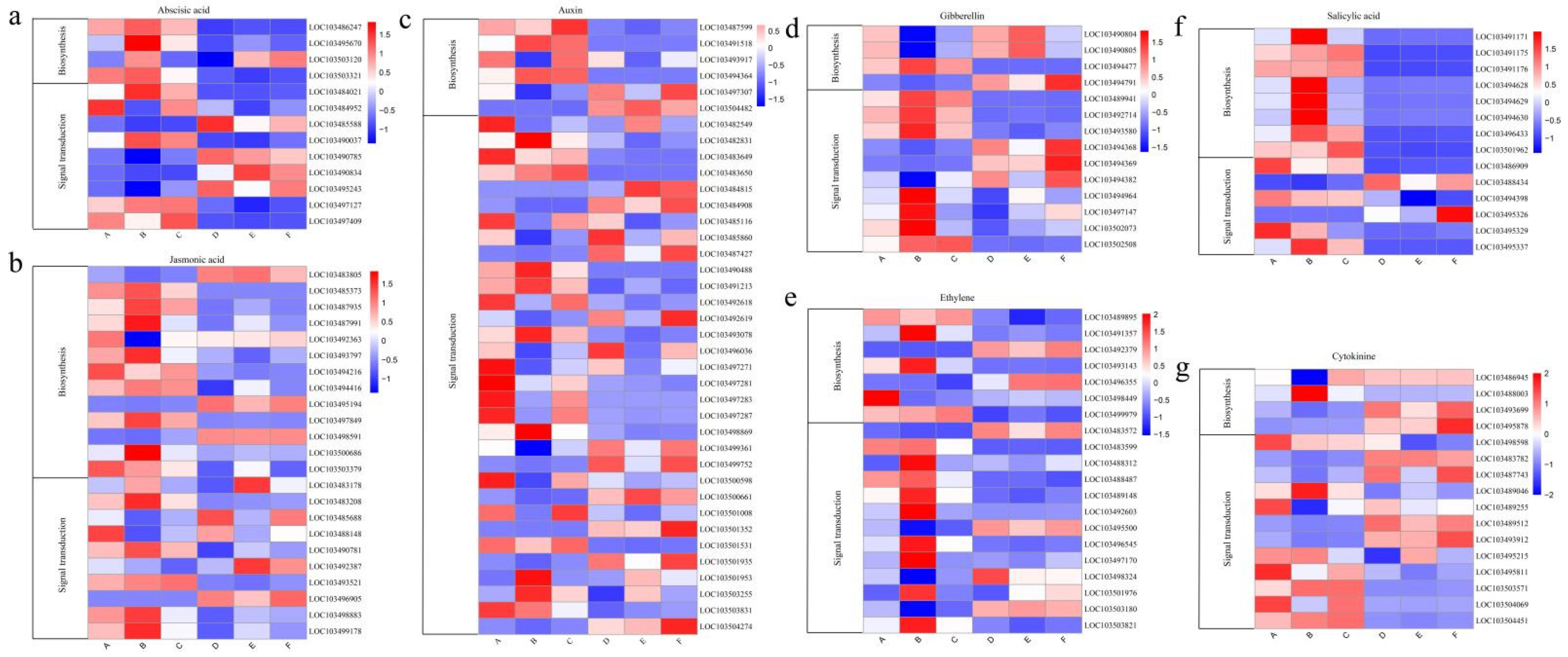
3.6. Expression Analysis of Hormone-Related Genes
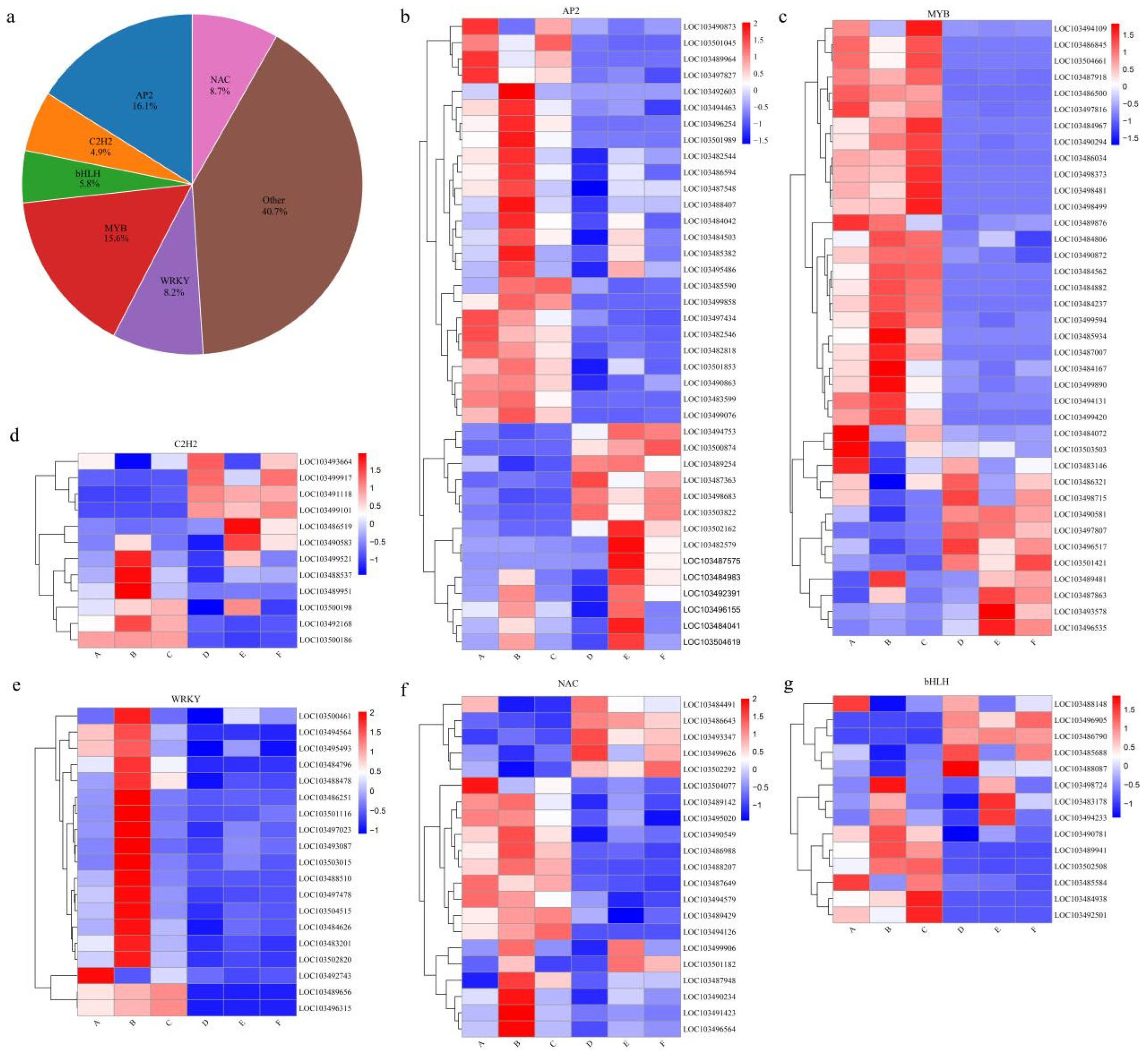
3.7. Expression Analysis of Specific TFs
3.8. Mining of Candidate Genes Related to WGCNA and Metal Ion Content
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wu, Z.; Shi, Z.; Yang, X.; Xie, C.; Xu, J.; Yu, Z. Comparative metabolomics profiling reveals the molecular information of whole and fresh-cut melon fruit (cv. Xizhoumi-17) during storage. Sci. Hortic. 2022, 296, 110914. [Google Scholar] [CrossRef]
- Akash, M.; Awad, N.; Kasrawi, M. Genetic diversity among snake melon landraces (Cucumis melo Var. Flexuosus) using molecular descriptors. Plant Biosyst. 2020, 154, 206–212. [Google Scholar] [CrossRef]
- Falade, O.S.; Otemuyiwa, I.O.; Adekunle, A.S.; Adewusi, S.A.; Oluwasefunmi, O. Nutrient composition of watermelon (Citrullis lanatus (Thunb.) Matsum. & Nakai) and egusi melon (Citrullus colocynthis (L.) Schrad.) seeds. Agric. Conspec. Sci. 2020, 85, 43–49. [Google Scholar]
- Gómez-García, R.; Campos, D.A.; Oliveira, A.; Aguilar, C.N.; Madureira, A.R.; Pintado, M. A chemical valorisation of melon peels towards functional food ingredients: Bioactives profile and antioxidant properties. Food Chem. 2021, 335, 127579. [Google Scholar] [CrossRef] [PubMed]
- Lopes, A.P.; Galuch, M.B.; Petenuci, M.E.; Oliveira, J.H.; Canesin, E.A.; Schneider, V.V.A.; Visentainer, J.V. Quantification of phenolic compounds in ripe and unripe bitter melons (Momordica charantia) and evaluation of the distribution of phenolic compounds in different parts of the fruit by UPLC–MS/MS. Chem. Pap. 2020, 74, 2613–2625. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhang, Z.; Han, X.; Wu, J.; Zhang, L.; Wang, J.; Wang-Pruski, G. Specific response mechanism to autotoxicity in melon (Cucumis melo L.) root revealed by physiological analyses combined with transcriptome profiling. Ecotoxicol. Environ. Saf. 2020, 200, 110779. [Google Scholar] [CrossRef] [PubMed]
- Azeez, N.A.; Dash, S.S.; Gummadi, S.N.; Deepa, V.S. Nsano-remediation of toxic heavy metal contamination: Hexavalent chromium [Cr (VI)]. Chemosphere 2021, 266, 129204. [Google Scholar] [CrossRef]
- Gasparatos, D. Soil Contamination by Heavy Metals and Metalloids. Environments 2022, 9, 32. [Google Scholar] [CrossRef]
- Kolawole, T.O.; Ajibade, O.M.; Olajide-Kayode, J.O.; Fomba, K.W. Level, distribution, ecological, and human health risk assessment of heavy metals in soils and stream sediments around a used-automobile spare part market in Nigeria. Environ. Geochem. Health 2022, 1–26. [Google Scholar] [CrossRef]
- Wang, X.W.; Liu, H.Y.; Gu, X.F.; Tu, Y.; Yu, E.J.; Wu, P. Distribution Characteristics of Heavy Metals in Soils Affected by Different Land Use Types in a Superimposed Pollution Area with High Geological Background. Huan Jing Ke Xue 2022, 43, 2094–2103. [Google Scholar]
- Widowati, H.; Sutanto, A.; Thresia, F.; Hendri, N. Potential of heavy metal cd, pb in affecting non-organic agricultural land levels. J. Phys. Conf. Ser. 2020, 1594, 012029. [Google Scholar] [CrossRef]
- Zhang, H.; Xu, Z.; Guo, K.; Huo, Y.; He, G.; Sun, H.; Guan, Y.; Xu, N.; Yang, W.; Sun, G. Toxic effects of heavy metal Cd and Zn on chlorophyll, carotenoid metabolism and photosynthetic function in tobacco leaves revealed by physiological and proteomics analysis. Ecotoxicol. Environ. Saf. 2020, 202, 110856. [Google Scholar] [CrossRef] [PubMed]
- Gou, L. Research on Remediation Methods of Soil Arsenic Pollution. Sci. J. Intell. Syst. Res. 2021, 3, 34–37. [Google Scholar]
- Matzen, S.L.; Lobo, G.P.; Fakra, S.C.; Kakouridis, A.; Nico, P.S.; Pallud, C.E. Arsenic hyperaccumulator Pteris vittata shows reduced biomass in soils with high arsenic and low nutrient availability, leading to increased arsenic leaching from soil. Sci. Total Environ. 2022, 818, 151803. [Google Scholar] [CrossRef] [PubMed]
- Boquete, M.T.; Lang, I.; Weidinger, M.; Richards, C.L.; Alonso, C. Patterns and mechanisms of heavy metal accumulation and tolerance in two terrestrial moss species with contrasting habitat specialization. Environ. Exp. Bot. 2021, 182, 104336. [Google Scholar] [CrossRef]
- Patra, D.K.; Acharya, S.; Pradhan, C.; Patra, H.K. Poaceae plants as potential phytoremediators of heavy metals and eco-restoration in contaminated mining sites. Environ. Technol. Innov. 2021, 21, 101293. [Google Scholar] [CrossRef]
- Ishak, A.R.; Mohamad, S.; Soo, T.K.; Hamid, F.S. Leachate and surface water characterization and heavy metal health risk on cockles in Kuala Selangor. Procedia Soc. Behav. Sci. 2016, 222, 263–271. [Google Scholar] [CrossRef]
- Uluisik, S.; Oney-Birol, S. Uncovering candidate genes involved in postharvest ripening of tomato using the Solanum pennellii introgression line population by integrating phenotypic data, RNA-Seq, and SNP analyses. Sci. Hortic. 2021, 288, 1103. [Google Scholar] [CrossRef]
- Fan, Y.; Zheng, Y.; Teixeira da Silva, J.A.; Yu, X. Comparative transcriptomics and WGCNA reveal candidate genes involved in petaloid stamens in Paeonia lactiflora. J. Hortic. Sci. Biotechnol. 2021, 96, 588–603. [Google Scholar] [CrossRef]
- Tan, M.; Cheng, D.; Yang, Y.; Zhang, G.; Qin, M.; Chen, J.; Chen, Y.; Jiang, M. Co-expression network analysis of the transcriptomes of rice roots exposed to various cadmium stresses reveals universal cadmium-responsive genes. BMC Plant Biol. 2017, 17, 194. [Google Scholar] [CrossRef]
- Zou, X.; Liu, A.; Zhang, Z.; Ge, Q.; Fan, S.; Gong, W.; Li, J.; Gong, J.; Shi, Y.; Tian, B.; et al. Co-Expression Network Analysis and Hub Gene Selection for High-Quality Fiber in Upland Cotton (Gossypium hirsutum) Using RNA Sequencing Analysis. Genes 2019, 10, 119. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Wang, Z.; Ge, H.; Liu, Y.; Chen, H. Weighted gene co-expression network analysis identifies genes related to anthocyanin biosynthesis and functional verification of hub gene SmWRKY44. Plant Sci. 2021, 309, 110935. [Google Scholar] [CrossRef] [PubMed]
- Shilev, S.; Babrikova, I.; Babrikov, T. Consortium of plant growth-promoting bacteria improves spinach (Spinacea oleracea L.) growth under heavy metal stress conditions. J. Chem. Technol. Biotechnol. 2020, 95, 932–939. [Google Scholar] [CrossRef]
- Giordano, A.; Domingo, M.S.; Quadrana, L.; Pujol, M.; Martín-Hernández, A.M.; Garcia-Mas, J. CRISPR/Cas9 gene editing uncovers the role of CTR1 and ROS1 in melon fruit ripening and epigenetic regulation. J. Exp. Bot. 2022, 73, 4022–4033. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Martínez, C.; Gonzalo, M.J.; Sipowicz, P.; Campos, M.; Martínez-Fernández, I.; Leida, C.; Zouine, M.; Alexiou, K.G.; Garcia-Mas, J.; Gómez, M.D.; et al. A cryptic variation in a member of the Ovate Family Proteins is underlying the melon fruit shape QTL fsqs8.1. Theor. Appl. Genet. 2022, 135, 785–801. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Yang, K.; Xia, L. Application of ICP-MS Method in Environmental Field. IOP Conf. Ser. Earth Environ. Sci. 2021, 769, 022028. [Google Scholar] [CrossRef]
- Lee, C.; Harris, R.A.; Wall, J.K.; Mayfield, R.D.; Wilke, C.O. RNaseIII and T4 polynucleotide Kinase sequence biases and solutions during RNA-seq library construction. Biol. Direct 2013, 8, 16. [Google Scholar] [CrossRef]
- Wang, D.; Karamyshev, A.L. Next Generation Sequencing (NGS) Application in Multiparameter Gene Expression Analysis. Methods Mol. Biol. 2020, 2102, 17–34. [Google Scholar]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef]
- Zhang, X.; Wen, B.; Zhang, Y.; Li, Y.; Yu, C.; Peng, Z.; Wang, K.; Liu, Z.; Huang, J.; Xiong, L.; et al. Transcriptomic and biochemical analysis reveal differential regulatory mechanisms of photosynthetic pigment and characteristic secondary metabolites between high amino acids green-leaf and albino tea cultivars. Sci. Hortic. 2022, 295, 110823. [Google Scholar] [CrossRef]
- Liu, S.; Wang, Z.; Zhu, R.; Wang, F.; Cheng, Y.; Liu, Y. Three Differential Expression Analysis Methods for RNA Sequencing: Limma, EdgeR, DESeq2. J. Vis. Exp. 2021, 175, e62528. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef] [PubMed]
- Hua, J.; Liu, H.; Zhang, B.; Jin, S. LAK: Lasso and K-means based single-cell RNA-Seq data clustering analysis. IEEE Access 2020, 8, 129679–129688. [Google Scholar] [CrossRef]
- Jin, J.; Tian, F.; Yang, D.C.; Meng, Y.Q.; Kong, L.; Luo, J.; Gao, G. PlantTFDB 4.0: Toward a central hub for transcription factors and regulatory interactions in plants. Nucleic Acids Res. 2017, 45, D1040–D1045. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef]
- Mousavian, Z.; Khodabandeh, M.; Sharifi-Zarchi, A.; Nadafian, A.; Mahmoudi, A. StrongestPath: A Cytoscape application for protein–protein interaction analysis. BMC Bioinform. 2021, 22, 352. [Google Scholar] [CrossRef]
- Li, C.; Wang, X.; Huang, H.; Wang, L.; Wei, F.; Zhang, C.; Rong, Q. Effect of multiple heavy metals pollution to bacterial diversity and community structure in farmland soils. Hum. Ecol. Risk Assess. 2021, 27, 724–741. [Google Scholar] [CrossRef]
- Singh, S.; Parihar, P.; Singh, R.; Singh, V.P.; Prasad, S.M. Heavy Metal Tolerance in Plants: Role of Transcriptomics, Proteomics, Metabolomics, and Ionomics. Front. Plant Sci. 2016, 6, 1143. [Google Scholar] [CrossRef]
- Hu, H.Q.; Xu, X.H.; Huang, L.; Yao, M.Y.; Chen, T.B.; Liu, M.H.; Wang, C.H. Study on the enhancement intensity of Cd in rice with microwave-assisted laser-induced breakdown spectroscopy. Guang Pu Xue Yu Guang Pu Fen Xi 2016, 36, 1180–1185. [Google Scholar]
- Wu, T.; Li, C.; Xing, X.; Pan, X.; Liu, C.; Tian, Y.; Wang, Z.; Zhao, J.; Wang, J.; He, B. Straw return and organic fertilizers instead of chemical fertilizers on growth, yield and quality of rice. Earth Sci. Inform. 2022, 15, 1363–1369. [Google Scholar] [CrossRef]
- Kuang, J.F.; Wu, C.J.; Guo, Y.F.; Walther, D.; Shan, W.; Chen, J.Y.; Chen, L.; Lu, W.J. Deciphering transcriptional regulators of banana fruit ripening by regulatory network analysis. Plant Biotechnol. J. 2021, 19, 477–489. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Gu, L.; Zhang, X.; Liu, M.; Jiang, H.; Cai, R.; Zhao, Y.; Cheng, B. Global transcriptome and weighted gene co-expression network analyses reveal hybrid-specific modules and candidate genes related to plant height development in maize. Plant Mol. Biol. 2018, 98, 187–203. [Google Scholar] [CrossRef] [PubMed]
- Urano, K.; Maruyama, K.; Jikumaru, Y.; Kamiya, Y.; Yamaguchi-Shinozaki, K.; Shinozaki, K. Analysis of plant hormone profiles in response to moderate dehydration stress. Plant J. 2017, 90, 17–36. [Google Scholar] [CrossRef]
- Nguyen, T.Q.; Sesin, V.; Kisiala, A.; Emery, R. Phytohormonal Roles in Plant Responses to Heavy Metal Stress: Implications for Using Macrophytes in Phytoremediation of Aquatic Ecosystems. Environ. Toxicol. Chem. 2021, 40, 7–22. [Google Scholar] [CrossRef]
- Chen, H.; Zhang, Q.; Lv, W.; Yu, X.; Zhang, Z. Ethylene positively regulates Cd tolerance via reactive oxygen species scavenging and apoplastic transport barrier formation in rice. Environ. Pollut. 2022, 302, 119063. [Google Scholar] [CrossRef]
- Jia, H.; Wang, X.; Wei, T.; Wang, M.; Liu, X.; Hua, L.; Ren, X.; Guo, J.; Li, J. Exogenous salicylic acid regulates cell wall polysaccharides synthesis and pectin methylation to reduce Cd accumulation of tomato. Ecotoxicol. Environ. Saf. 2021, 207, 111550. [Google Scholar] [CrossRef]
- Xu, B.; Chen, J.; Yu, J.; Guo, Y.; Cai, Q.; Wu, Y.; Li, Y.; Xie, T.; Chen, Y.; Wang, G. Effects of 24-epibrassinolide and 28-homobrassinolide on iron plaque formation and the uptake of As and Cd by rice seedlings (Oryza sativa L.) in solution culture. Environ. Technol. Innov. 2020, 19, 100802. [Google Scholar] [CrossRef]
- Zhang, Z.W.; Deng, Z.L.; Tao, Q.; Peng, H.Q.; Wu, F.; Fu, Y.F.; Yang, X.Y.; Xu, P.Z.; Li, Y.; Wang, C.Q.; et al. Salicylate and glutamate mediate different Cd accumulation and tolerance between Brassica napus and B. juncea. Chemosphere 2022, 292, 133466. [Google Scholar] [CrossRef]
- Rolón-Cárdenas, G.A.; Arvizu-Gómez, J.L.; Soria-Guerra, R.E.; Pacheco-Aguilar, J.R.; Alatorre-Cobos, F.; Hernández-Morales, A. The role of auxins and auxin-producing bacteria in the tolerance and accumulation of cadmium by plants. Environ. Geochem. Health 2022. advance online publication. [Google Scholar] [CrossRef]
- Gong, J.M.; Lee, D.A.; Schroeder, J.I. Long-distance root-to-shoot transport of phytochelatins and cadmium in Arabidopsis. Proc. Natl. Acad. Sci. USA 2003, 100, 10118–10123. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.F.; Zhu, H.H.; He, Q.Y.; Li, P.F.; Fan, W.; Xu, J.M.; Yang, J.L.; Chen, W.W. The Tomato Transcription Factor SlNAC063 Is Required for Aluminum Tolerance by Regulating SlAAE3-1 Expression. Front. Plant Sci. 2022, 13, 826954. [Google Scholar] [CrossRef] [PubMed]
- Romero-Puertas, M.C.; Corpas, F.J.; Rodríguez-Serrano, M.; Gómez, M.; Del Río, L.A.; Sandalio, L.M. Differential expression and regulation of antioxidative enzymes by cadmium in pea plants. J. Plant Physiol. 2007, 164, 1346–1357. [Google Scholar] [CrossRef]
- Ren, Y.B. The Function of Arabidopsis thaliana AtMYB50 and AtMYB61 Transcription Factors in Response to Heavy Metal, Low Phosphorus and Osmotic Stress. Master’s Thesis, Hefei University of Technology, Hefei, China, 2010. [Google Scholar]
- Shen, N.; Hou, S.; Tu, G.; Lan, W.; Jing, Y. Transcription Factor WRKY33 Mediates the Phosphate Deficiency-Induced Remodeling of Root Architecture by Modulating Iron Homeostasis in Arabidopsis Roots. Int. J. Mol. Sci. 2021, 22, 9275. [Google Scholar] [CrossRef] [PubMed]
- Lei, R.; Li, Y.; Cai, Y.; Li, C.; Pu, M.; Lu, C.; Yang, Y.; Liang, G. bHLH121 Functions as a Direct Link that Facilitates the Activation of FIT by bHLH IVc Transcription Factors for Maintaining Fe Homeostasis in Arabidopsis. Mol. Plant 2020, 13, 634–649. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shen, Q.; Wu, X.; Tao, Y.; Yan, G.; Wang, X.; Cao, S.; Wang, C.; He, W. Mining Candidate Genes Related to Heavy Metals in Mature Melon (Cucumis melo L.) Peel and Pulp Using WGCNA. Genes 2022, 13, 1767. https://doi.org/10.3390/genes13101767
Shen Q, Wu X, Tao Y, Yan G, Wang X, Cao S, Wang C, He W. Mining Candidate Genes Related to Heavy Metals in Mature Melon (Cucumis melo L.) Peel and Pulp Using WGCNA. Genes. 2022; 13(10):1767. https://doi.org/10.3390/genes13101767
Chicago/Turabian StyleShen, Qi, Xiaonan Wu, Yongxia Tao, Guorong Yan, Xian Wang, Shuangyu Cao, Cheng Wang, and Weizhong He. 2022. "Mining Candidate Genes Related to Heavy Metals in Mature Melon (Cucumis melo L.) Peel and Pulp Using WGCNA" Genes 13, no. 10: 1767. https://doi.org/10.3390/genes13101767
APA StyleShen, Q., Wu, X., Tao, Y., Yan, G., Wang, X., Cao, S., Wang, C., & He, W. (2022). Mining Candidate Genes Related to Heavy Metals in Mature Melon (Cucumis melo L.) Peel and Pulp Using WGCNA. Genes, 13(10), 1767. https://doi.org/10.3390/genes13101767