
A Splice Variant of the MYH7 Gene Is Causative in a Family with Isolated Left Ventricular Noncompaction Cardiomyopathy

 , ,
, ,  , , , , , , , ,
, , , , , , , ,  ,
,  , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Clinical Investigation of the Patients
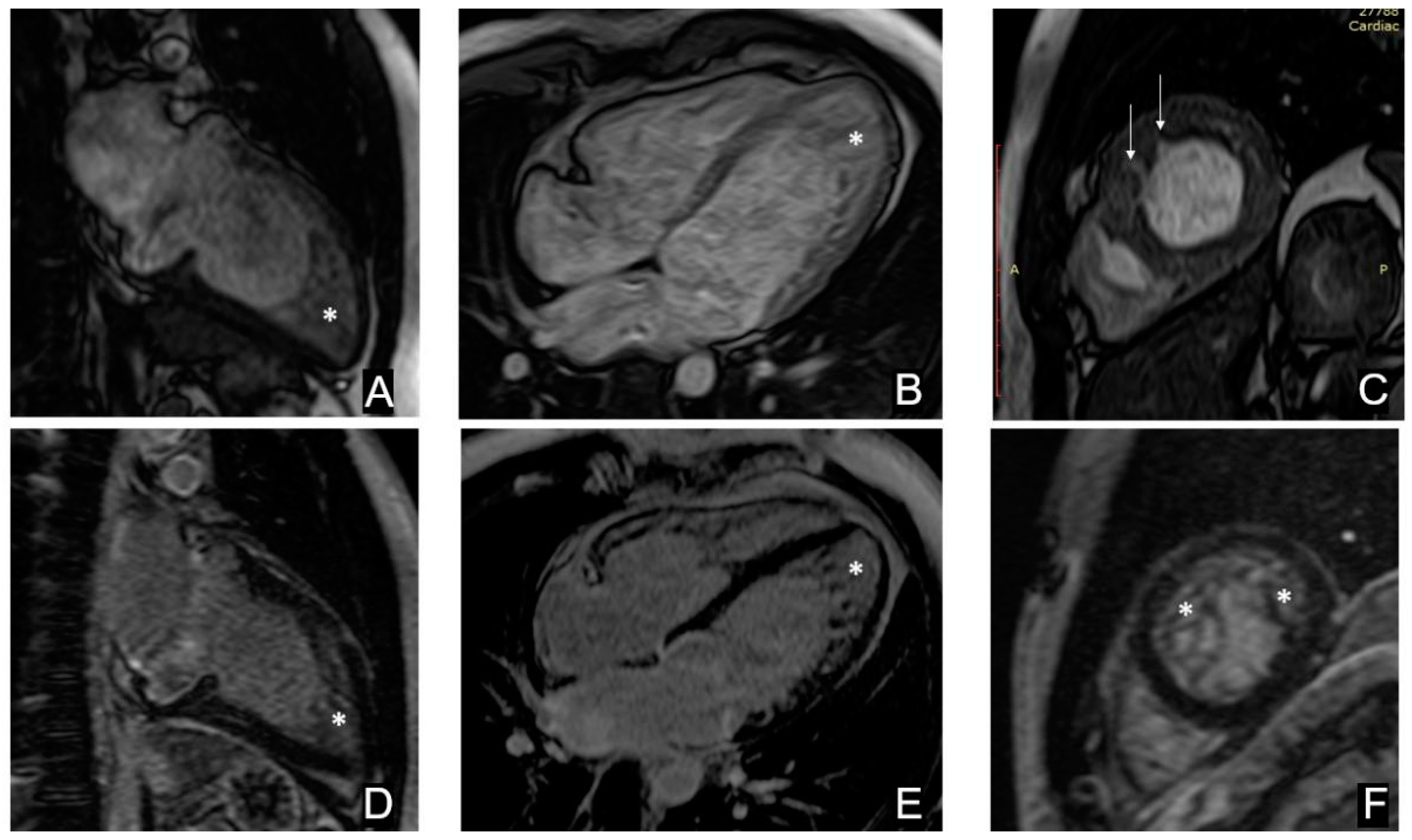
2.2. Cardiac Magnetic Resonance Imaging
2.3. Whole Genome Sequencing and Bioinformatic Analysis
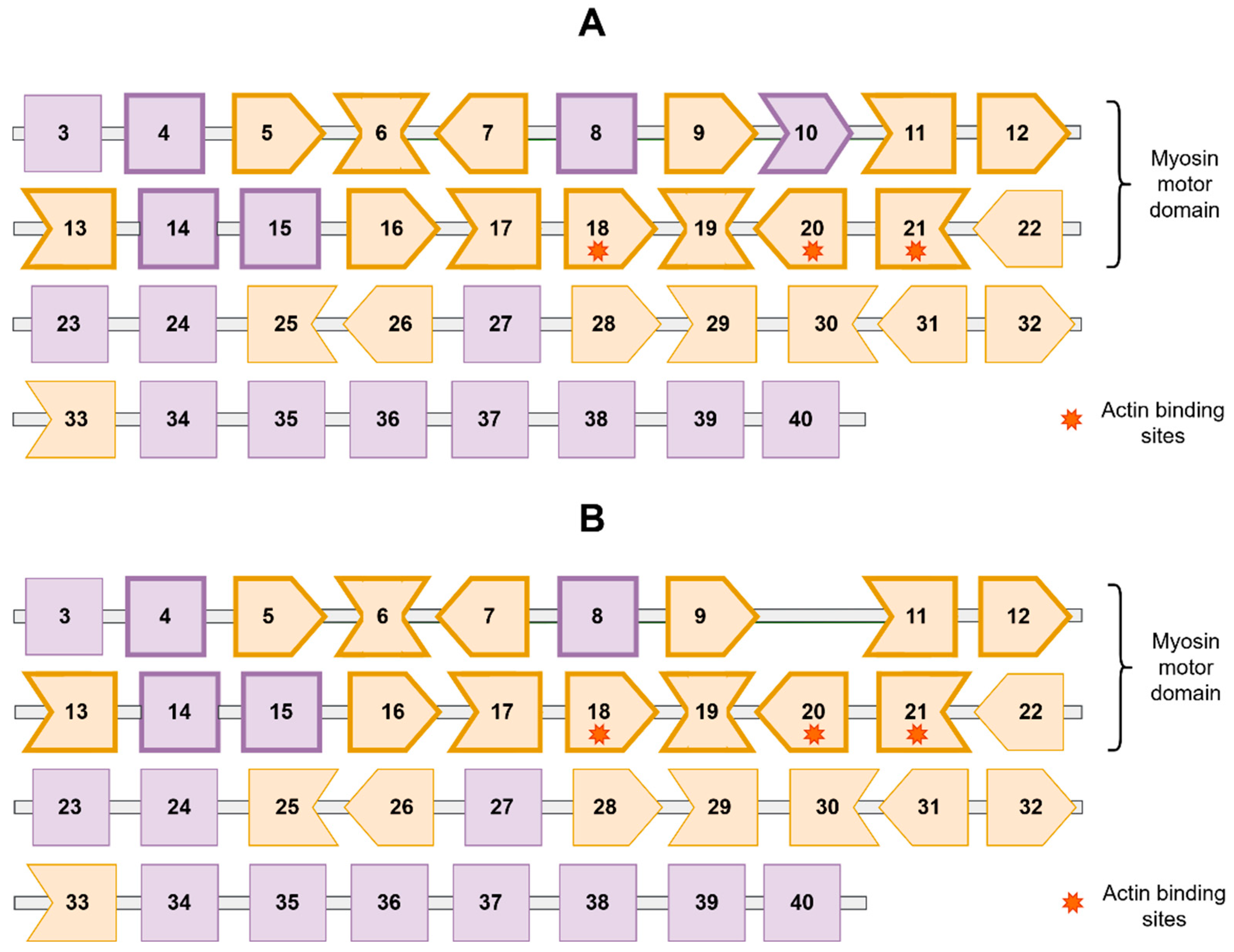
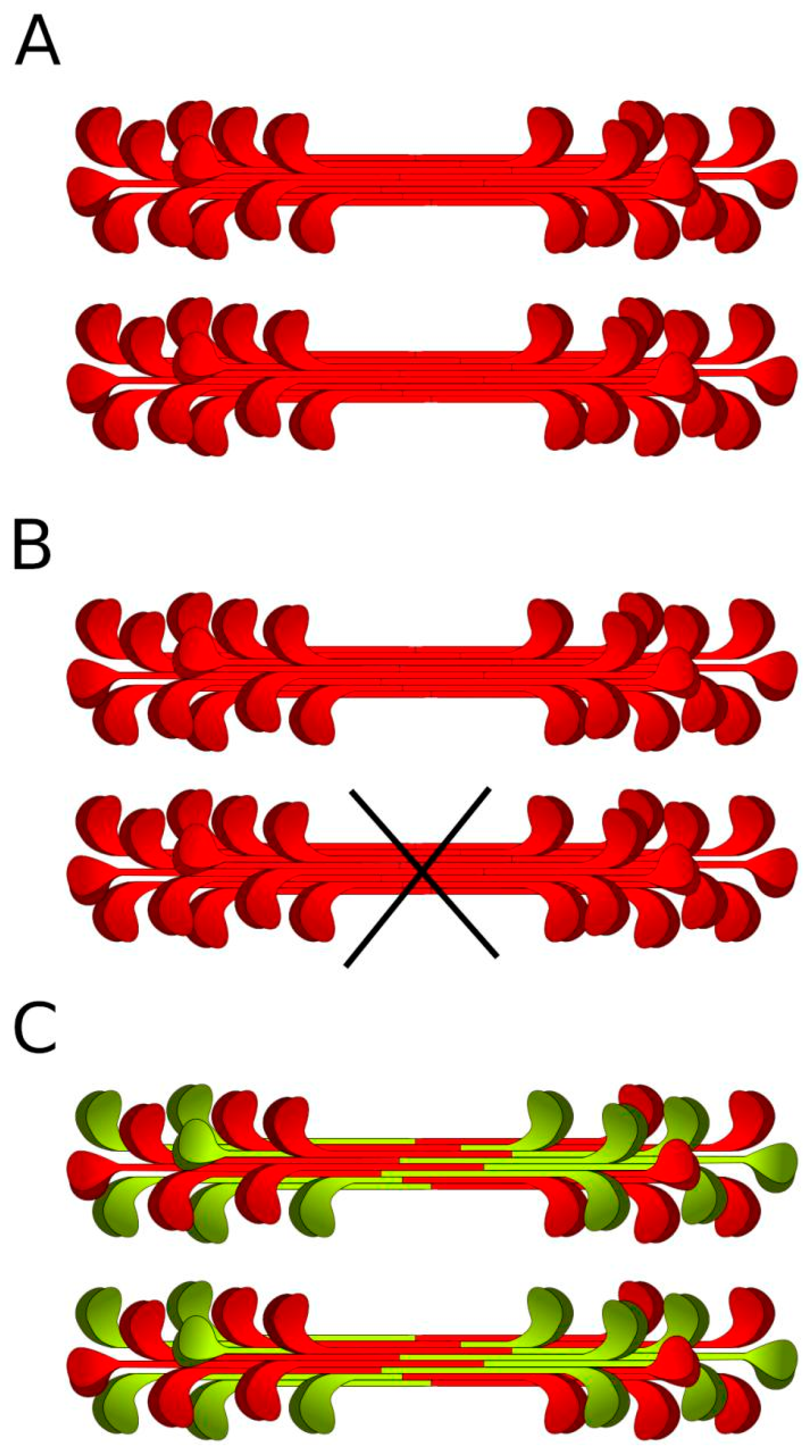
3. Results
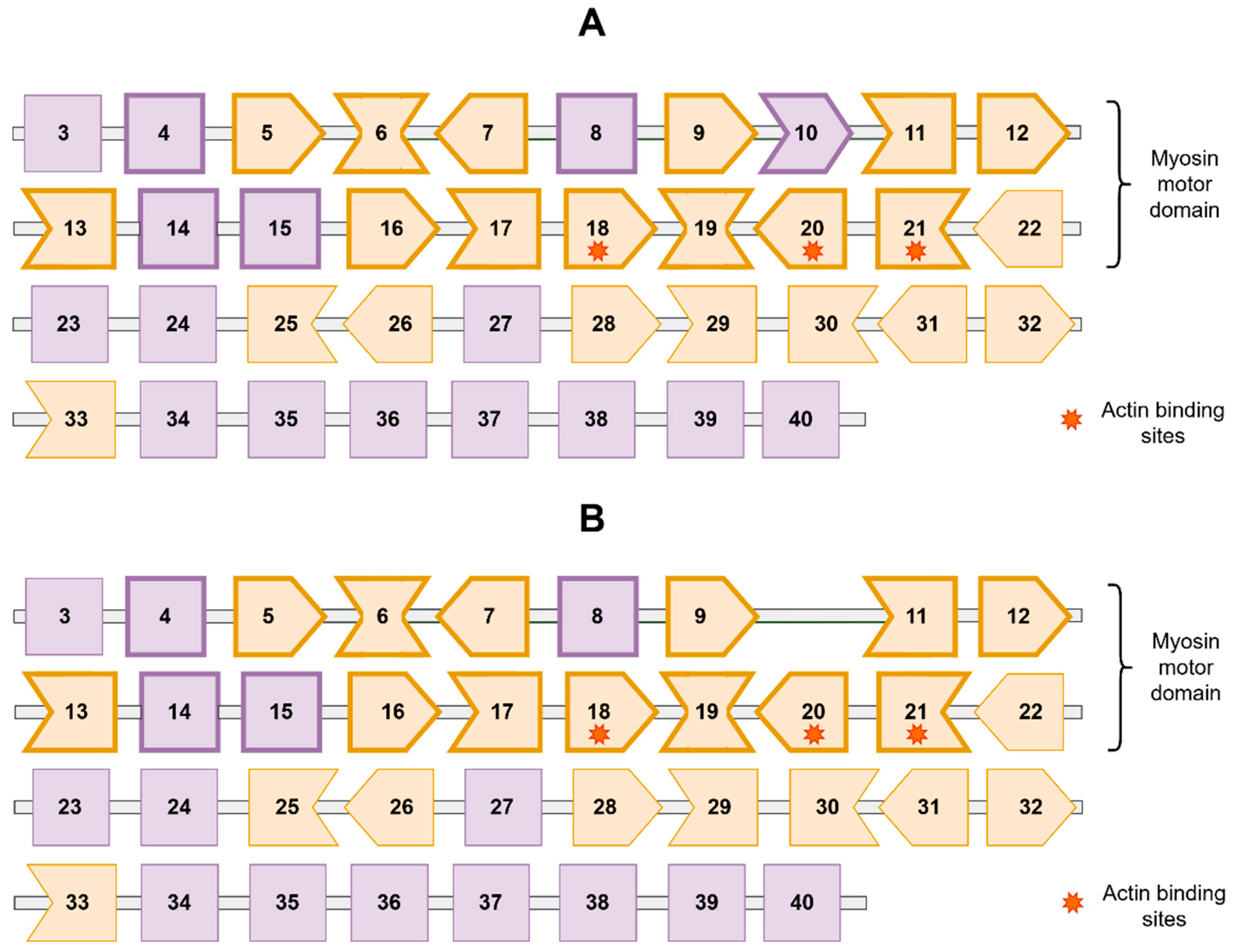
4. Discussion
5. Conclusions
6. Limitations of the Study
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Vergani, V.; Lazzeroni, D.; Peretto, G. Bridging the Gap between Hypertrabeculation Phenotype, Noncompaction Phenotype and Left Ventricular Noncompaction Cardiomyopathy. J. Cardiovasc. Med. 2020, 21, 192–199. [Google Scholar] [CrossRef] [PubMed]
- Arbustini, E.; Favalli, V.; Narula, N.; Serio, A.; Grasso, M. Left Ventricular Noncompaction: A Distinct Genetic Cardiomyopathy? J. Am. Coll. Cardiol. 2016, 68, 949–966. [Google Scholar] [CrossRef] [PubMed]
- Yotti, R.; Seidman, C.E.; Seidman, J.G. Advances in the Genetic Basis and Pathogenesis of Sarcomere Cardiomyopathies. Annu. Rev. Genom. Hum. Genet. 2019, 20, 129–153. [Google Scholar] [CrossRef] [PubMed]
- Savarese, M.; Sarparanta, J.; Vihola, A.; Jonson, P.H.; Johari, M.; Rusanen, S.; Hackman, P.; Udd, B. Panorama of the Distal Myopathies. Acta Myol. 2020, 39, 245–265. [Google Scholar] [CrossRef]
- Claeys, K.G. Congenital Myopathies: An Update. Dev. Med. Child Neurol. 2020, 62, 297–302. [Google Scholar] [CrossRef] [PubMed]
- Mazzarotto, F.; Hawley, M.H.; Beltrami, M.; Beekman, L.; de Marvao, A.; McGurk, K.A.; Statton, B.; Boschi, B.; Girolami, F.; Roberts, A.M.; et al. Systematic Large-Scale Assessment of the Genetic Architecture of Left Ventricular Noncompaction Reveals Diverse Etiologies. Genet. Med. 2021, 23, 856–864. [Google Scholar] [CrossRef] [PubMed]
- Modrek, B.; Resch, A.; Grasso, C.; Lee, C. Genome-Wide Detection of Alternative Splicing in Expressed Sequences of Human Genes. Nucleic Acids Res. 2001, 29, 2850–2859. [Google Scholar] [CrossRef]
- Kayvanpour, E.; Sedaghat-Hamedani, F.; Amr, A.; Lai, A.; Haas, J.; Holzer, D.B.; Frese, K.S.; Keller, A.; Jensen, K.; Katus, H.A.; et al. Genotype-Phenotype Associations in Dilated Cardiomyopathy: Meta-Analysis on More than 8000 Individuals. Clin. Res. Cardiol. 2017, 106, 127–139. [Google Scholar] [CrossRef]
- Han, L.; Li, Y.; Tchao, J.; Kaplan, A.D.; Lin, B.; Li, Y.; Mich-Basso, J.; Lis, A.; Hassan, N.; London, B.; et al. Study Familial Hypertrophic Cardiomyopathy Using Patient-Specific Induced Pluripotent Stem Cells. Cardiovasc. Res. 2014, 104, 258–269. [Google Scholar] [CrossRef]
- Surikova, Y.; Filatova, A.; Polyak, M.; Skoblov, M.; Zaklyazminskaya, E. Common Pathogenic Mechanism in Patients with Dropped Head Syndrome Caused by Different Mutations in the MYH7 Gene. Gene 2019, 697, 159–164. [Google Scholar] [CrossRef]
- Jenni, R.; Oechslin, E.; Schneider, J.; Attenhofer Jost, C.; Kaufmann, P.A. Echocardiographic and Pathoanatomical Characteristics of Isolated Left Ventricular Non-Compaction: A Step towards Classification as a Distinct Cardiomyopathy. Heart 2001, 86, 666–671. [Google Scholar] [CrossRef] [PubMed]
- Petersen, S.E.; Selvanayagam, J.B.; Wiesmann, F.; Robson, M.D.; Francis, J.M.; Anderson, R.H.; Watkins, H.; Neubauer, S. Left Ventricular Non-Compaction: Insights from Cardiovascular Magnetic Resonance Imaging. J. Am. Coll. Cardiol. 2005, 46, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Meshkov, A.; Ershova, A.; Kiseleva, A.; Zotova, E.; Sotnikova, E.; Petukhova, A.; Zharikova, A.; Malyshev, P.; Rozhkova, T.; Blokhina, A.; et al. The LDLR, APOB, and PCSK9 Variants of Probands with Familial Hypercholesterolemia in Russia. Genes 2021, 12, 66. [Google Scholar] [CrossRef] [PubMed]
- Kelly, M.A.; Caleshu, C.; Morales, A.; Buchan, J.; Wolf, Z.; Harrison, S.M.; Cook, S.; Dillon, M.W.; Garcia, J.; Haverfield, E.; et al. Adaptation and Validation of the ACMG/AMP Variant Classification Framework for MYH7-Associated Inherited Cardiomyopathies: Recommendations by ClinGen’s Inherited Cardiomyopathy Expert Panel. Genet. Med. 2018, 20, 351–359. [Google Scholar] [CrossRef] [PubMed]
- Abou Tayoun, A.N.; Pesaran, T.; DiStefano, M.T.; Oza, A.; Rehm, H.L.; Biesecker, L.G.; Harrison, S.M. ClinGen Sequence Variant Interpretation Working Group (ClinGen SVI) Recommendations for Interpreting the Loss of Function PVS1 ACMG/AMP Variant Criterion. Hum. Mutat. 2018, 39, 1517–1524. [Google Scholar] [CrossRef] [PubMed]
- CeGaT Pedigree Chart Designer Software. Available online: https://www.cegat.com/for-physicians/pedigree-chart-designer/ (accessed on 28 July 2022).
- dbSNP. Available online: https://www.ncbi.nlm.nih.gov/snp/rs111547156 (accessed on 28 July 2022).
- ClinVar. Available online: https://www.ncbi.nlm.nih.gov/clinvar/variation/432302/ (accessed on 28 July 2022).
- Yeo, G.; Burge, C.B. Maximum Entropy Modeling of Short Sequence Motifs with Applications to RNA Splicing Signals. J. Comput. Biol. 2004, 11, 377–394. [Google Scholar] [CrossRef]
- Jaganathan, K.; Kyriazopoulou Panagiotopoulou, S.; McRae, J.F.; Darbandi, S.F.; Knowles, D.; Li, Y.I.; Kosmicki, J.A.; Arbelaez, J.; Cui, W.; Schwartz, G.B.; et al. Predicting Splicing from Primary Sequence with Deep Learning. Cell 2019, 176, 535–548.e24. [Google Scholar] [CrossRef]
- Kayvanpour, E.; Sedaghat-Hamedani, F.; Gi, W.-T.; Tugrul, O.F.; Amr, A.; Haas, J.; Zhu, F.; Ehlermann, P.; Uhlmann, L.; Katus, H.A.; et al. Clinical and Genetic Insights into Non-Compaction: A Meta-Analysis and Systematic Review on 7598 Individuals. Clin. Res. Cardiol. 2019, 108, 1297–1308. [Google Scholar] [CrossRef]
- Kolokotronis, K.; Kühnisch, J.; Klopocki, E.; Dartsch, J.; Rost, S.; Huculak, C.; Mearini, G.; Störk, S.; Carrier, L.; Klaassen, S.; et al. Biallelic Mutation in MYH7 and MYBPC3 Leads to Severe Cardiomyopathy with Left Ventricular Noncompaction Phenotype. Hum. Mutat. 2019, 40, 1101–1114. [Google Scholar] [CrossRef]
- Lefter, M.; Vis, J.K.; Vermaat, M.; den Dunnen, J.T.; Taschner, P.E.M.; Laros, J.F.J. Next Generation HGVS Nomenclature Checker. Bioinformatics 2021, 37, 2811–2817. [Google Scholar] [CrossRef]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The Mutational Constraint Spectrum Quantified from Variation in 141,456 Humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef] [PubMed]
- van Waning, J.I.; Caliskan, K.; Hoedemaekers, Y.M.; van Spaendonck-Zwarts, K.Y.; Baas, A.F.; Boekholdt, S.M.; van Melle, J.P.; Teske, A.J.; Asselbergs, F.W.; Backx, A.P.C.M.; et al. Genetics, Clinical Features, and Long-Term Outcome of Noncompaction Cardiomyopathy. J. Am. Coll. Cardiol. 2018, 71, 711–722. [Google Scholar] [CrossRef] [PubMed]
- Sedaghat-Hamedani, F.; Haas, J.; Zhu, F.; Geier, C.; Kayvanpour, E.; Liss, M.; Lai, A.; Frese, K.; Pribe-Wolferts, R.; Amr, A.; et al. Clinical Genetics and Outcome of Left Ventricular Non-Compaction Cardiomyopathy. Eur. Heart J. 2017, 38, 3449–3460. [Google Scholar] [CrossRef] [PubMed]
- Myasnikov, R.P.; Kulikova, O.V.; Meshkov, A.N.; Kiseleva, A.V.; Shumarina, A.O.; Koretskiy, S.N.; Zharikova, A.A.; Divashuk, M.G.; Kharlap, M.S.; Serduk, S.E.; et al. New Variant of MYH7 Gene Nucleotide Sequence in Familial Non-Compaction Cardiomyopathy with Benign Course. Ration. Pharmacother. Cardiol. 2020, 16, 383–391. [Google Scholar] [CrossRef]
- Tu, P.; Sun, H.; Zhang, X.; Ran, Q.; He, Y.; Ran, S. Diverse Cardiac Phenotypes among Different Carriers of the Same MYH7 Splicing Variant Allele (c.732+1G>A) from a Family. BMC Med. Genom. 2022, 15, 36. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
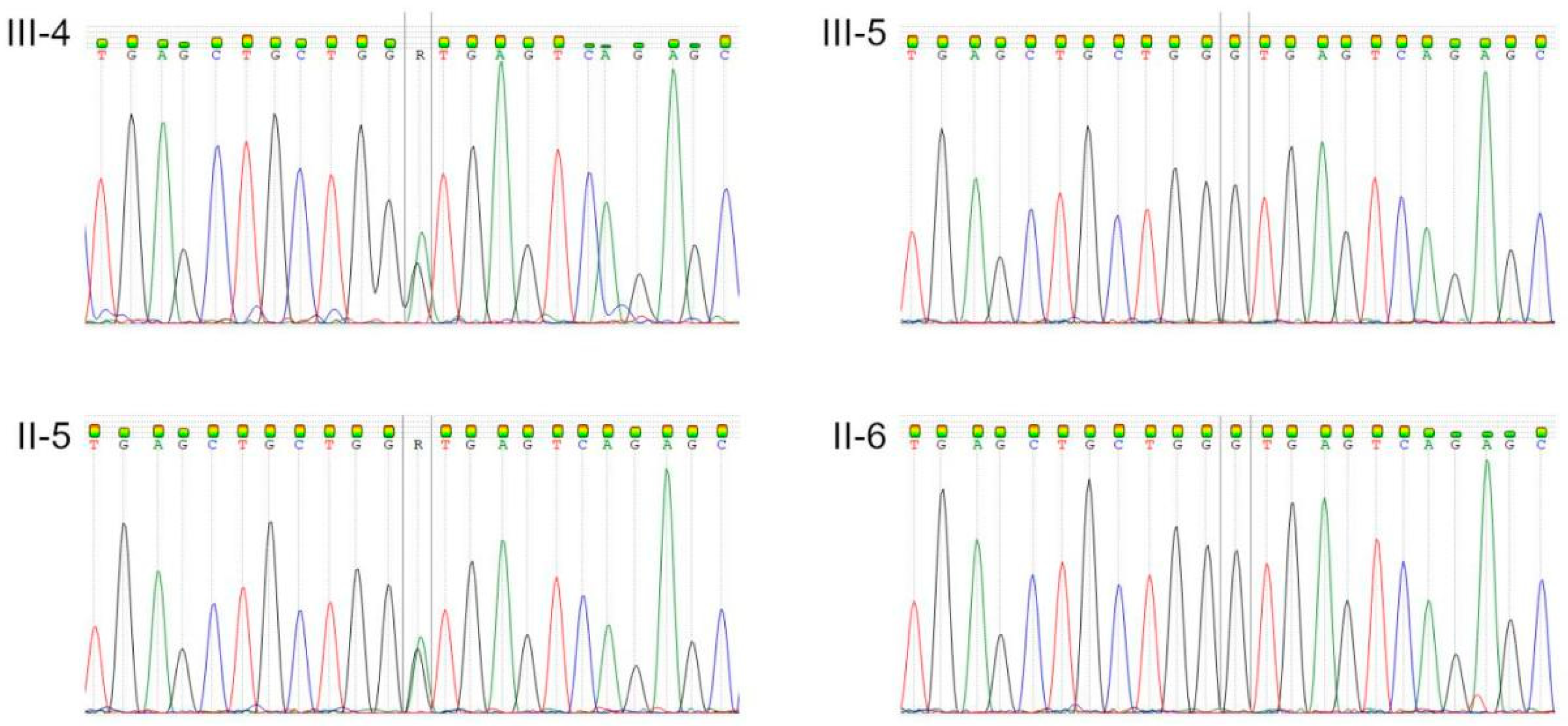
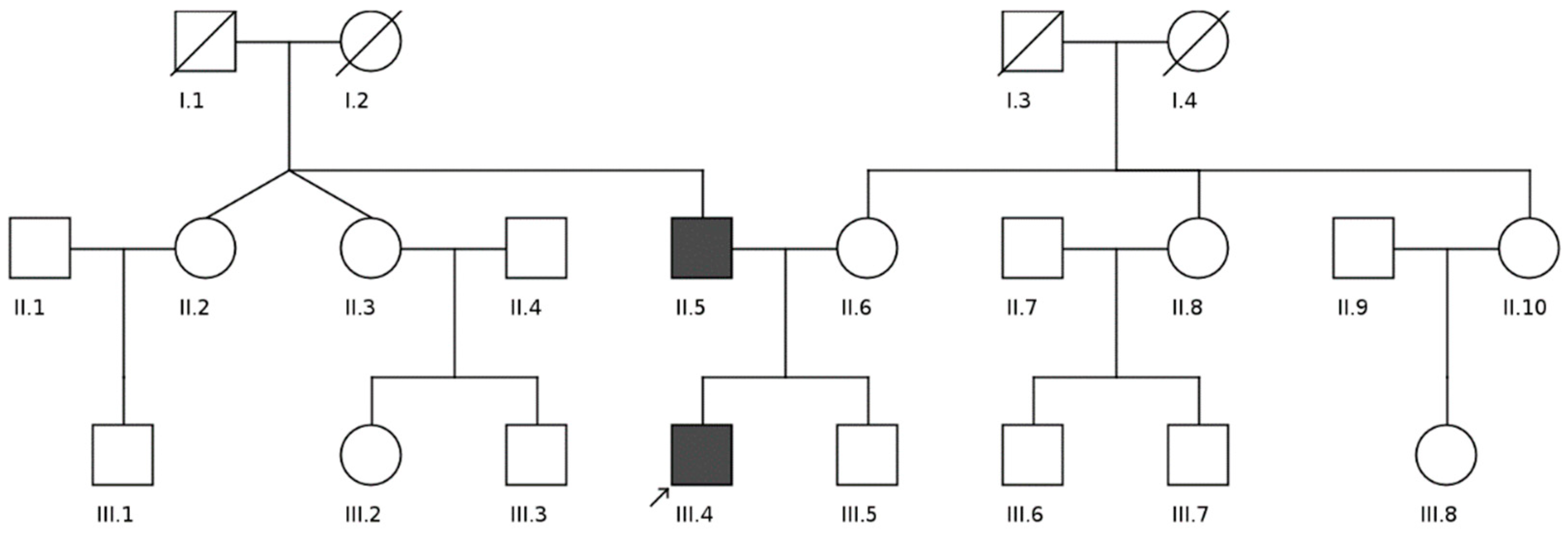
| Family Member | WGS | Sanger Sequencing | Phenotype |
|---|---|---|---|
| I-1 | – | – | Died at 70 |
| I-2 | – | – | Died at 90 |
| I-3 | – | – | Died at 54, congenital heart disease, pulmonary embolism |
| I-4 | – | – | Died at 77, enterocolitis |
| II-1 | – | – | 60 y.o, not examined |
| II-2 | – | – | 60 y.o., hypertension |
| II-3 | – | – | 60 y.o., hypertension |
| II-4 | – | – | 61 y.o., not examined |
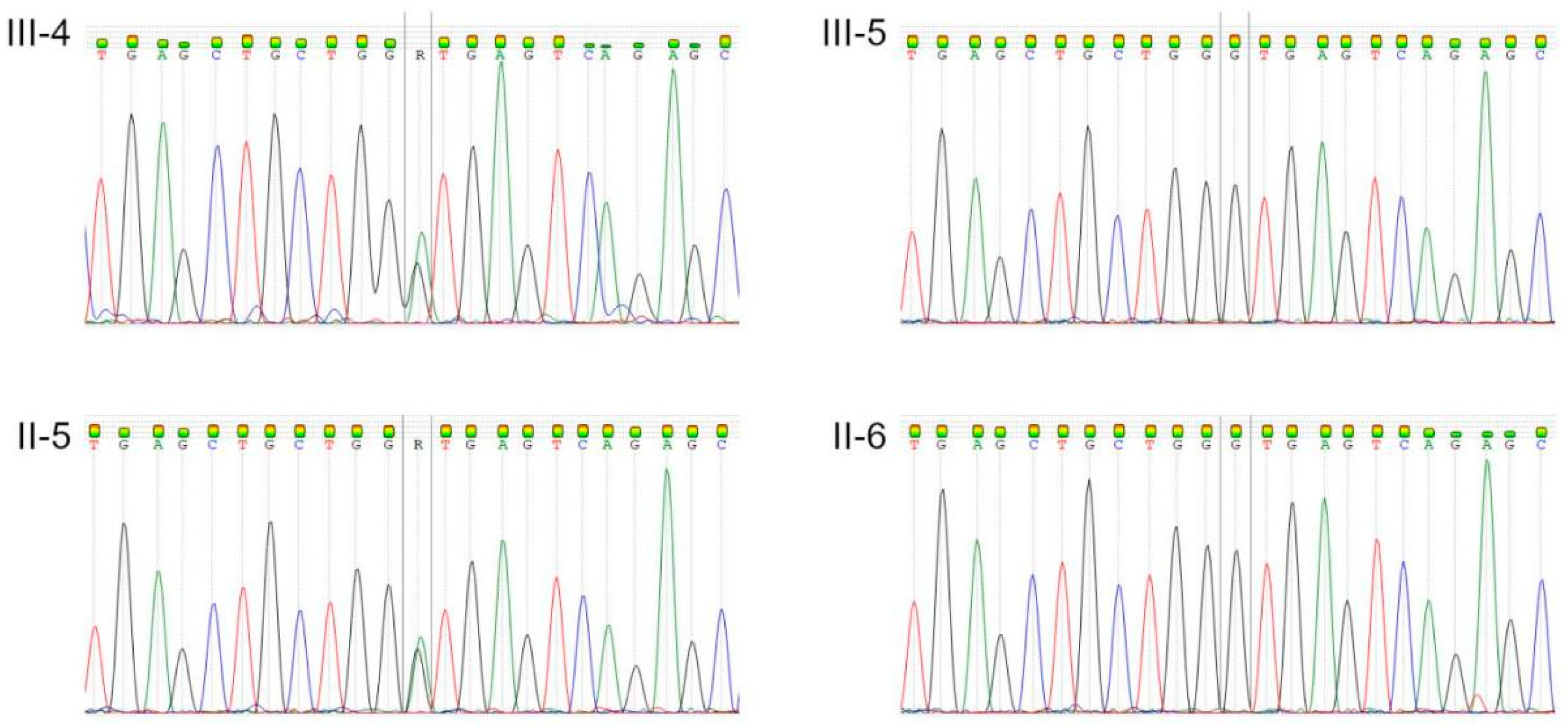
| II-5 | + | + | 53 y.o., LVNC |
| II-6 | + | + | 52 y.o., healthy |
| II-7–II-10; II-8–III-3 | – | – | Not examined |
| III-4 | + | + | 22 y.o., LVNC, heart failure |
| III-5 | + | + | 17 y.o., healthy |
| III-6–III-8 | – | – | Not examined |
| Splicing Prediction Parameter | MaxEntScan_ref | MaxEntScan_alt | MaxEntScan_diff | SpliceAI_pred_DS_DL | SpliceAI_pred_DP_DL |
|---|---|---|---|---|---|
| Value | 8.731 | 0.549 | 8.182 | 0.99 | 1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Myasnikov, R.P.; Kulikova, O.V.; Meshkov, A.N.; Bukaeva, A.A.; Kiseleva, A.V.; Ershova, A.I.; Petukhova, A.V.; Divashuk, M.G.; Zotova, E.D.; Sotnikova, E.A.; et al. A Splice Variant of the MYH7 Gene Is Causative in a Family with Isolated Left Ventricular Noncompaction Cardiomyopathy. Genes 2022, 13, 1750. https://doi.org/10.3390/genes13101750
Myasnikov RP, Kulikova OV, Meshkov AN, Bukaeva AA, Kiseleva AV, Ershova AI, Petukhova AV, Divashuk MG, Zotova ED, Sotnikova EA, et al. A Splice Variant of the MYH7 Gene Is Causative in a Family with Isolated Left Ventricular Noncompaction Cardiomyopathy. Genes. 2022; 13(10):1750. https://doi.org/10.3390/genes13101750
Chicago/Turabian StyleMyasnikov, Roman P., Olga V. Kulikova, Alexey N. Meshkov, Anna A. Bukaeva, Anna V. Kiseleva, Alexandra I. Ershova, Anna V. Petukhova, Mikhail G. Divashuk, Evgenia D. Zotova, Evgeniia A. Sotnikova, and et al. 2022. "A Splice Variant of the MYH7 Gene Is Causative in a Family with Isolated Left Ventricular Noncompaction Cardiomyopathy" Genes 13, no. 10: 1750. https://doi.org/10.3390/genes13101750
APA StyleMyasnikov, R. P., Kulikova, O. V., Meshkov, A. N., Bukaeva, A. A., Kiseleva, A. V., Ershova, A. I., Petukhova, A. V., Divashuk, M. G., Zotova, E. D., Sotnikova, E. A., Abisheva, A. A., Muraveva, A. V., Koretskiy, S. N., Popov, S. V., Utkina, M. V., Snigir, E. A., Mitrofanov, S. I., Konureeva, K. D., Mershina, E. A., ... Drapkina, O. M. (2022). A Splice Variant of the MYH7 Gene Is Causative in a Family with Isolated Left Ventricular Noncompaction Cardiomyopathy. Genes, 13(10), 1750. https://doi.org/10.3390/genes13101750