
Gene Annotation and Transcriptome Delineation on a De Novo Genome Assembly for the Reference Leishmania major Friedlin Strain

 ,
, 
Abstract
1. Introduction
2. Materials and Methods
2.1. Leishmania Parasites and DNA Isolation
2.2. Illumina Sequencing
2.3. PacBio Sequencing and De Novo Assembly
2.4. Assembly Refinements
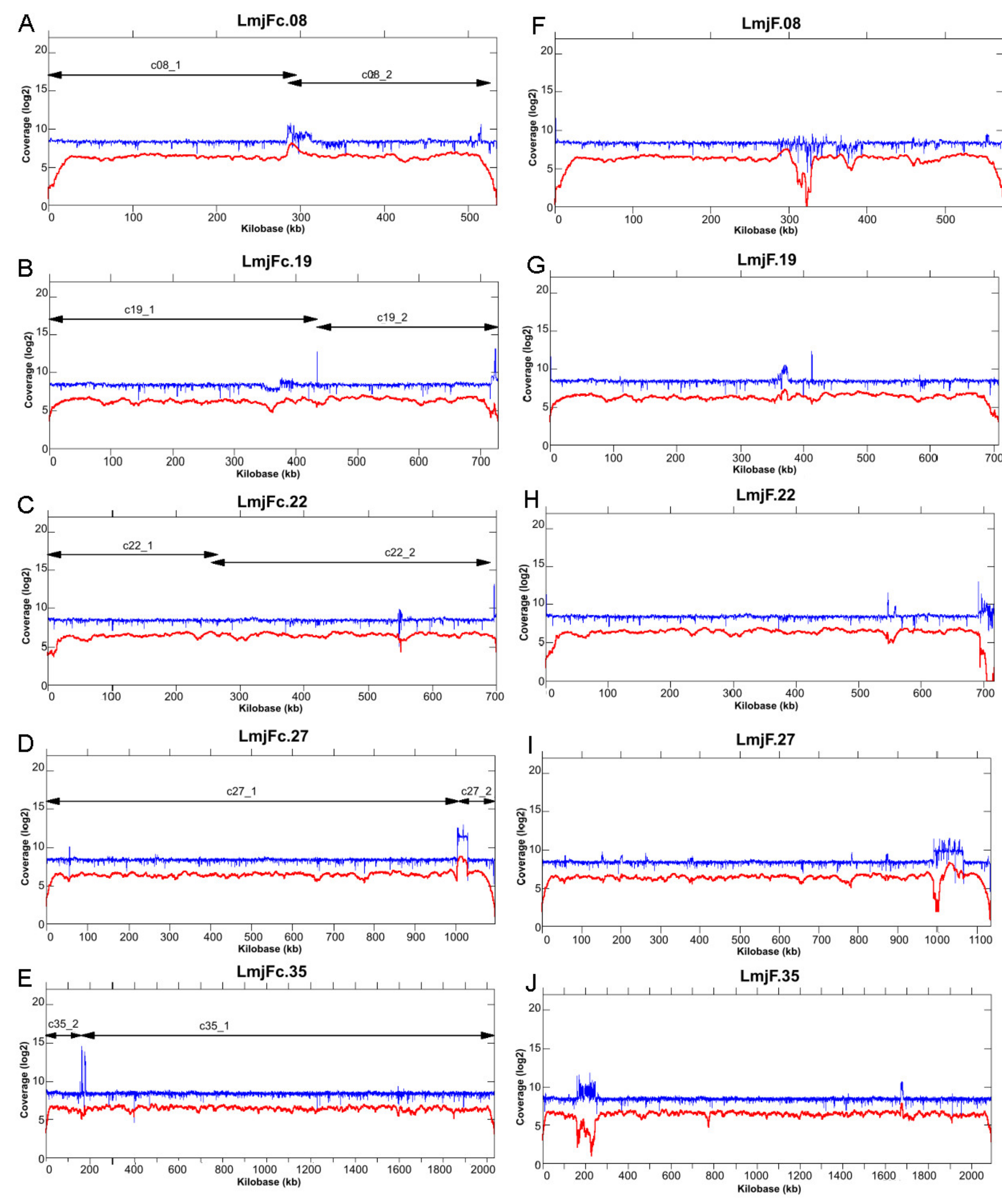
2.5. Coverage and Alignment
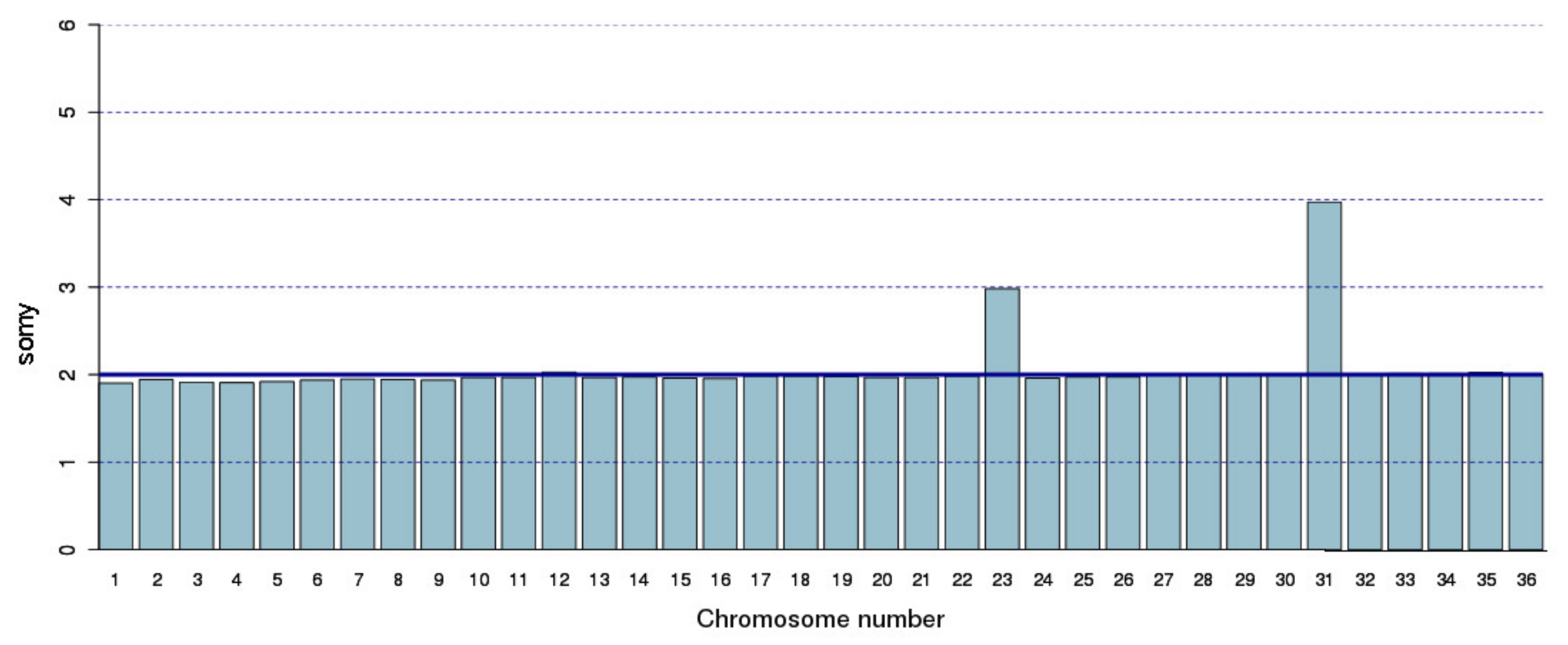
2.6. Somy Analysis
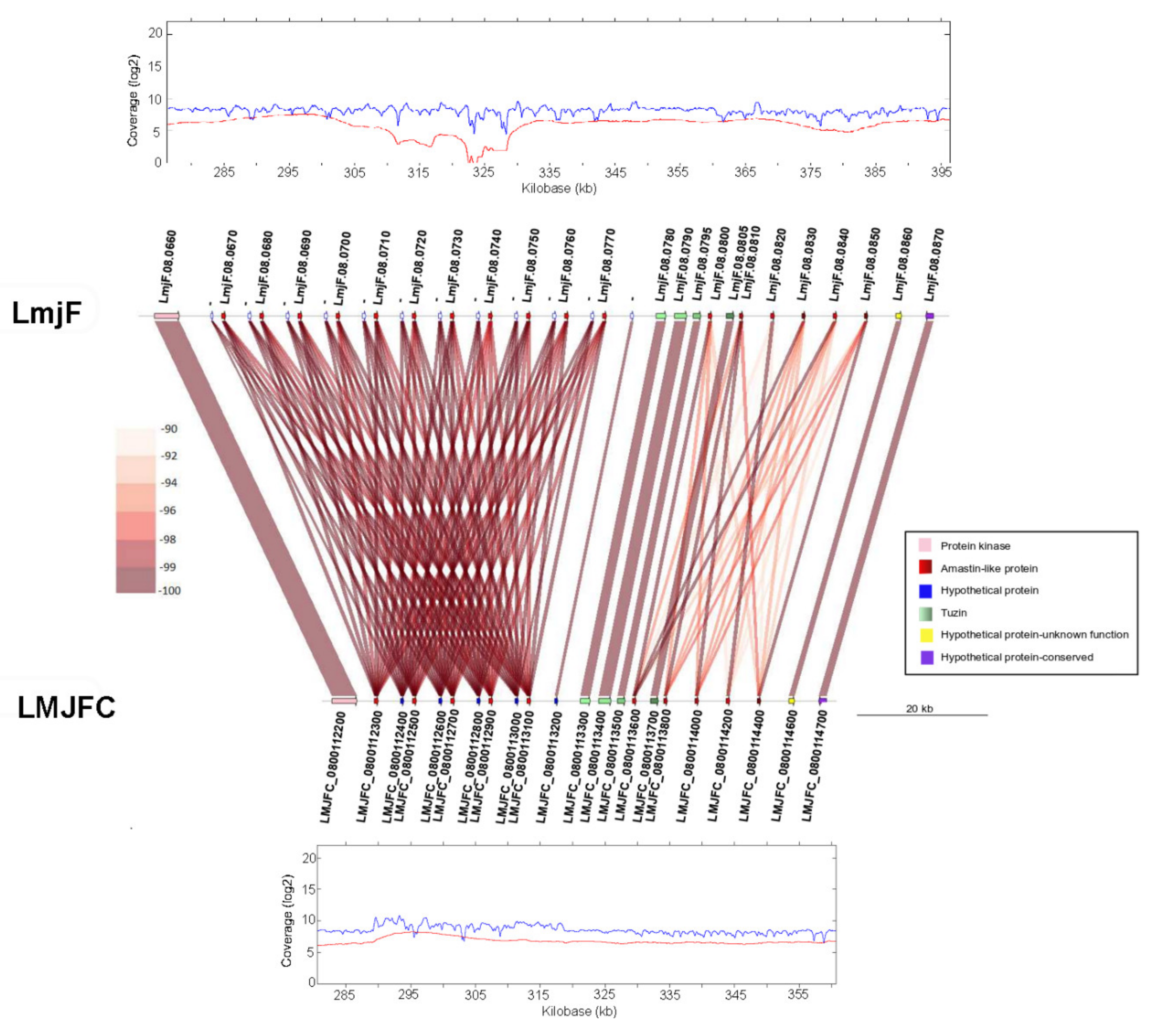
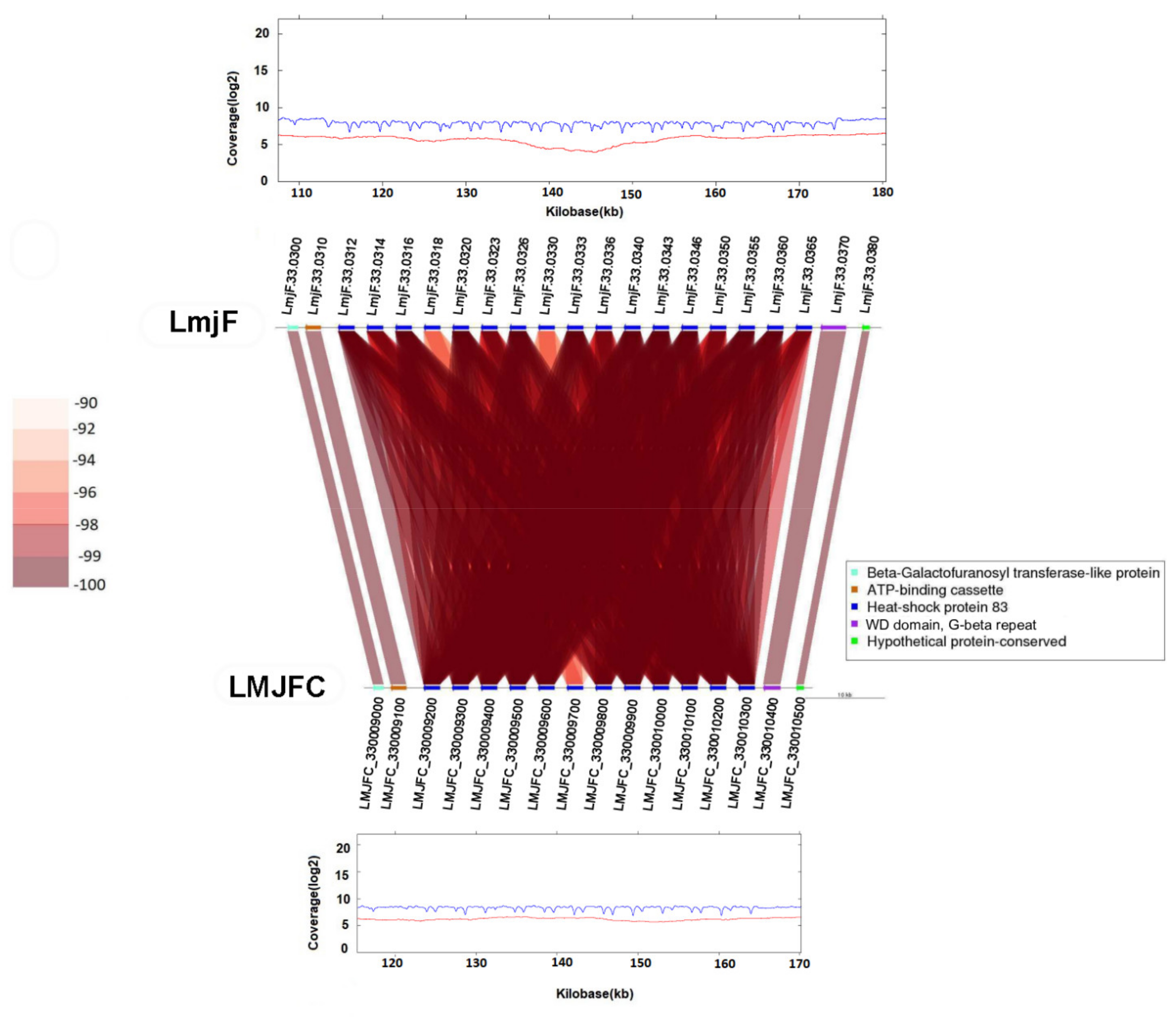
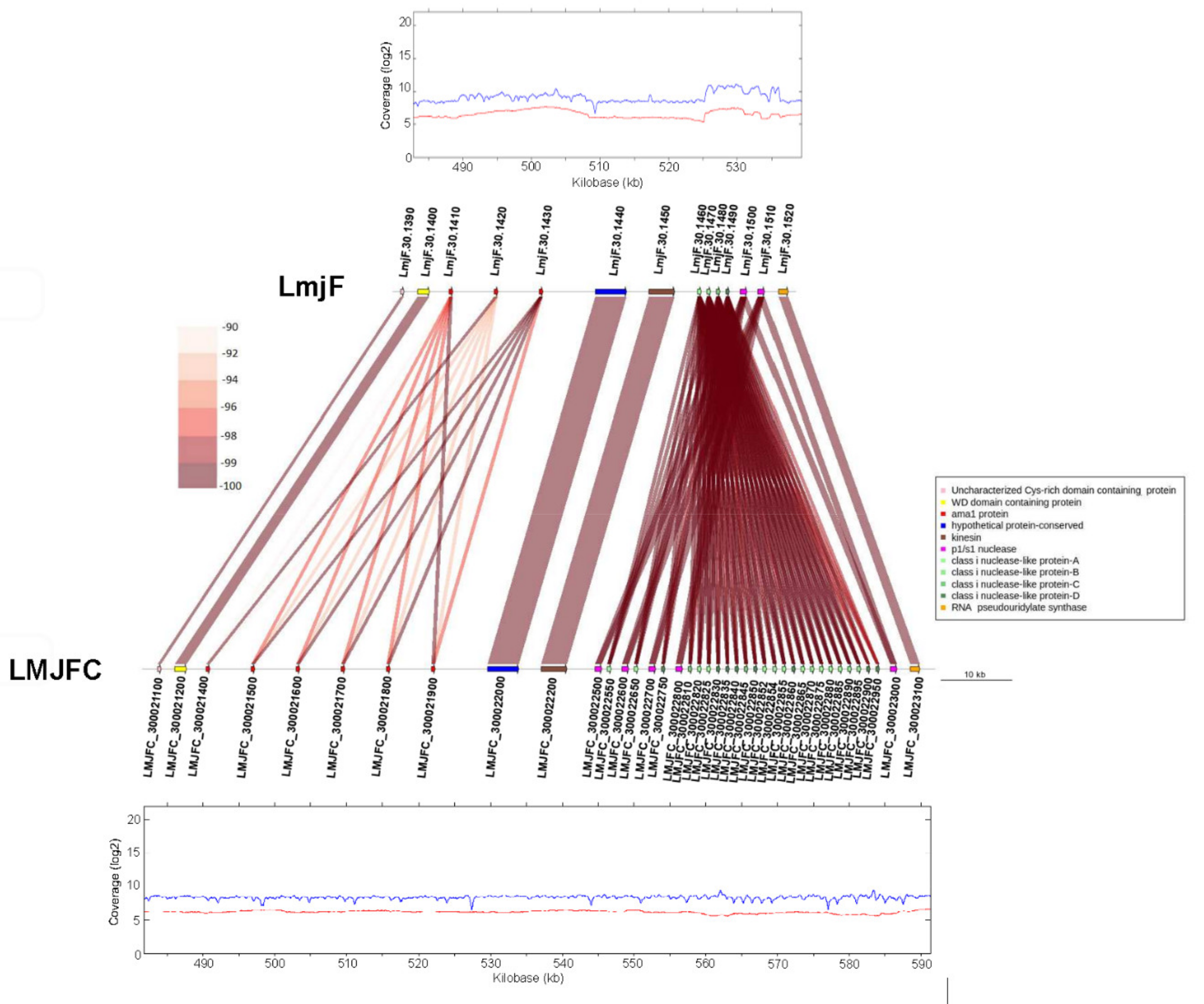
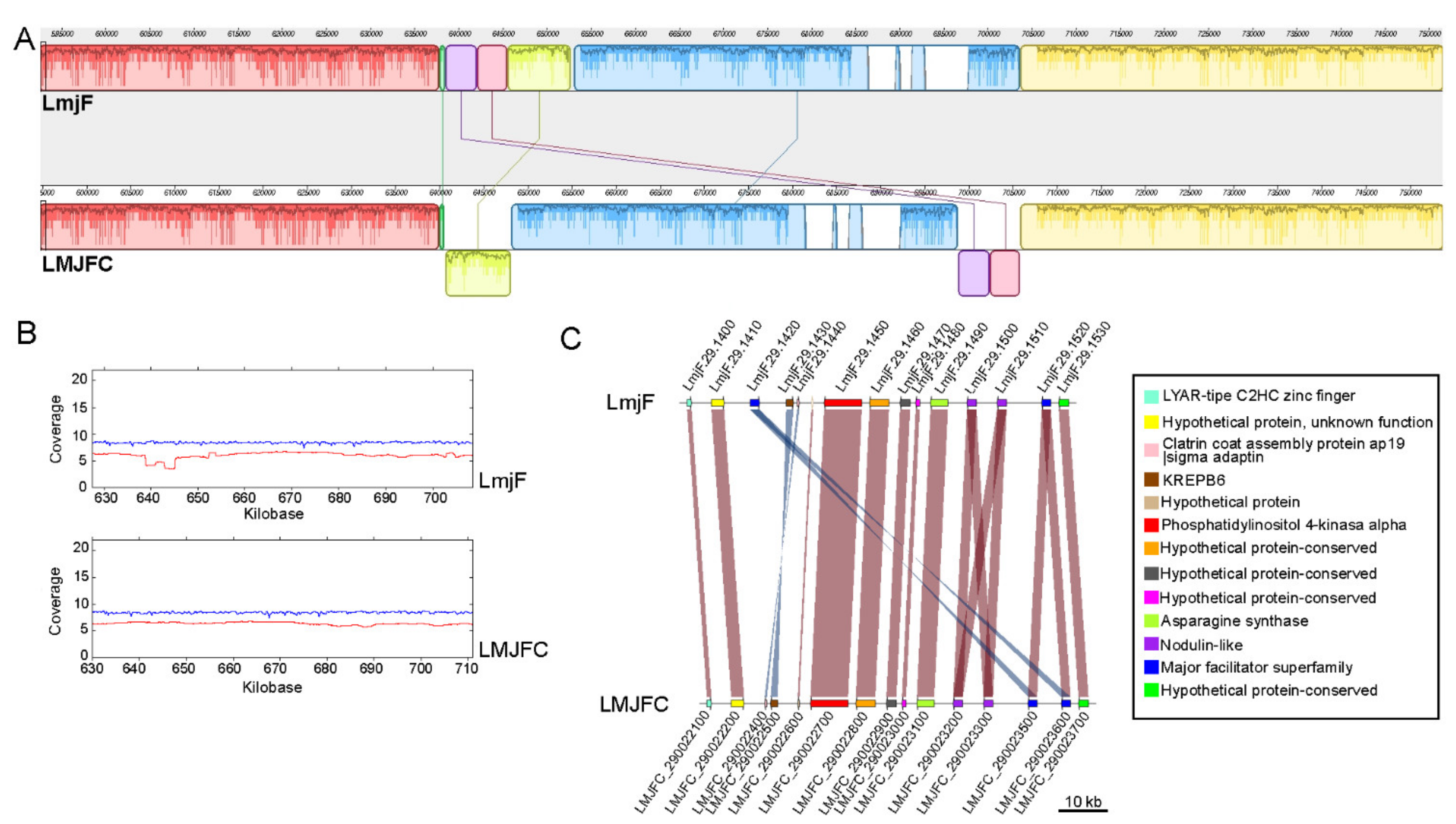
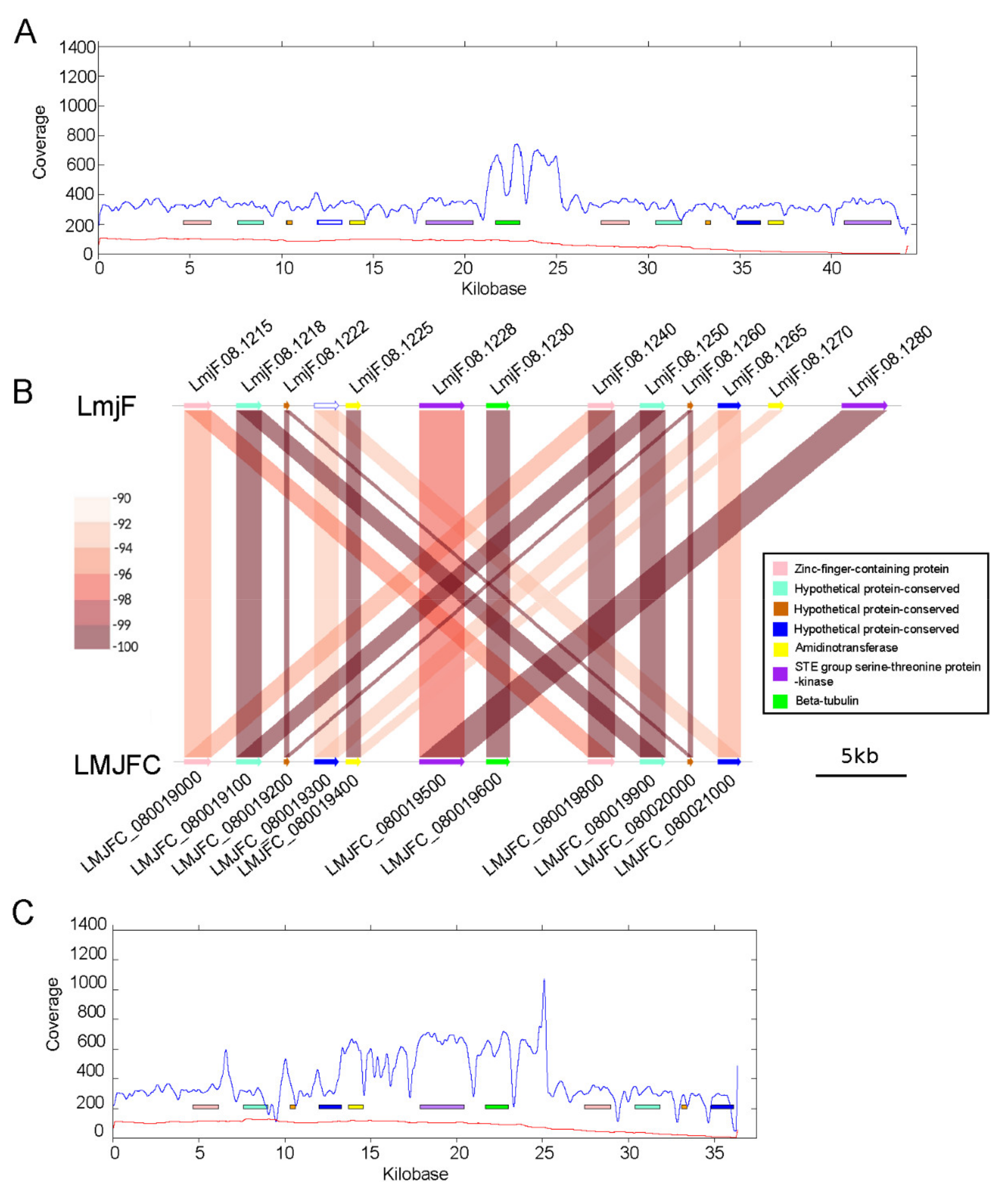
2.7. Synteny Analysis
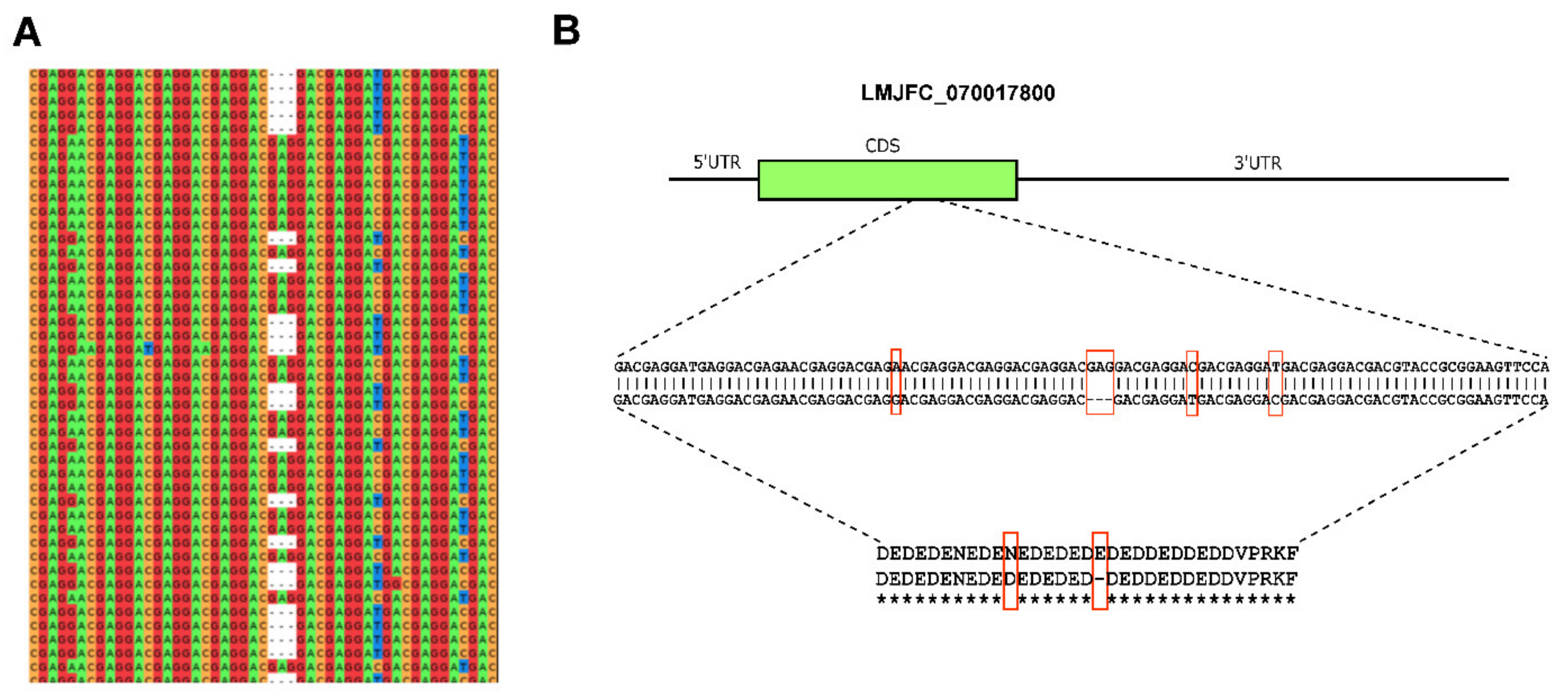
2.8. Haplotype Detection
2.9. Annotation of Protein-Coding Sequences, Known Non-Coding RNAs and Structural RNAs
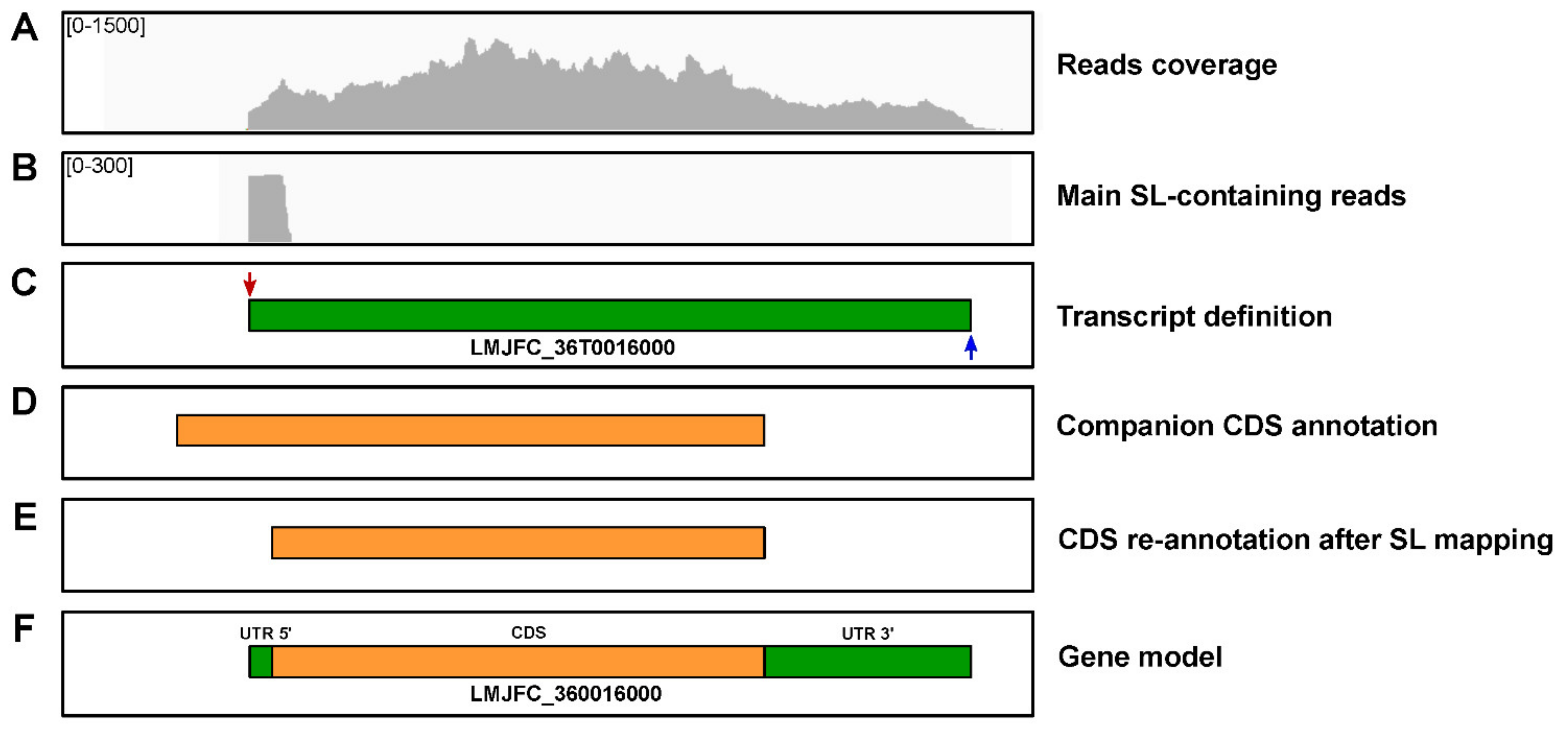
2.10. Transcriptome Definition and Annotation
2.11. Generation and Annotation of Gene Models
2.12. Data Availability
3. Results and Discussion
3.1. Re-Sequencing and De Novo Assembly of the L. major (Friedlin Strain) Genome
3.2. Transcriptome of L. major Friedlin Strain Based on the New Assembly (LMJFC)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Burza, S.; Croft, S.L.; Boelaert, M. Leishmaniasis. Lancet 2018, 392, 951–970. [Google Scholar] [CrossRef]
- Iborra, S.; Solana, J.C.; Requena, J.M.; Soto, M. Vaccine candidates against leishmania under current research. Expert Rev. Vaccines 2018, 17, 323–334. [Google Scholar] [CrossRef] [PubMed]
- Hefnawy, A.; Berg, M.; Dujardin, J.C.; De Muylder, G. Exploiting Knowledge on Leishmania Drug Resistance to Support the Quest for New Drugs. Trends Parasitol. 2017, 33, 162–174. [Google Scholar] [CrossRef] [PubMed]
- Ivens, A.C.; Blackwell, J.M. Unravelling the Leishmania genome. Curr. Opin. Genet. Dev. 1996, 6, 704–710. [Google Scholar] [CrossRef]
- Ivens, A.C.; Lewis, S.M.; Bagherzadeh, A.; Zhang, L.; Chan, H.M.; Smith, D.F. A physical map of the Leishmania major Friedlin genome. Genome Res. 1998, 8, 135–145. [Google Scholar] [CrossRef]
- Zhou, S.; Kile, A.; Kvikstad, E.; Bechner, M.; Severin, J.; Forrest, D.; Runnheim, R.; Churas, C.; Anantharaman, T.S.; Myler, P.; et al. Shotgun optical mapping of the entire Leishmania major Friedlin genome. Mol. Biochem. Parasitol. 2004, 138, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Ivens, A.C.; Peacock, C.S.; Worthey, E.A.; Murphy, L.; Aggarwal, G.; Berriman, M.; Sisk, E.; Rajandream, M.A.; Adlem, E.; Aert, R.; et al. The Genome of the Kinetoplastid Parasite, Leishmania major. Science 2005, 309, 436–442. [Google Scholar] [CrossRef]
- Cohen-Freue, G.; Holzer, T.R.; Forney, J.D.; McMaster, W.R. Global gene expression in Leishmania. Int. J. Parasitol. 2007, 37, 1077–1086. [Google Scholar] [CrossRef]
- Peacock, C.S.; Seeger, K.; Harris, D.; Murphy, L.; Ruiz, J.C.; Quail, M.A.; Peters, N.; Adlem, E.; Tivey, A.; Aslett, M.; et al. Comparative genomic analysis of three Leishmania species that cause diverse human disease. Nat. Genet. 2007, 39, 839–847. [Google Scholar] [CrossRef] [PubMed]
- Butenko, A.; Kostygov, A.Y.; Sadlova, J.; Kleschenko, Y.; Becvar, T.; Podesvova, L.; Macedo, D.H.; Zihala, D.; Lukes, J.; Bates, P.A.; et al. Comparative genomics of Leishmania (Mundinia). BMC Genom. 2019, 20, 726. [Google Scholar] [CrossRef]
- Real, F.; Vidal, R.O.; Carazzolle, M.F.; Mondego, J.M.; Costa, G.G.; Herai, R.H.; Wurtele, M.; de Carvalho, L.M.; Carmona e Ferreira, R.; Mortara, R.A.; et al. The genome sequence of Leishmania (Leishmania) amazonensis: Functional annotation and extended analysis of gene models. DNA Res. 2013, 20, 567–581. [Google Scholar] [CrossRef] [PubMed]
- Llanes, A.; Restrepo, C.M.; Del Vecchio, G.; Anguizola, F.J.; Lleonart, R. The genome of Leishmania panamensis: Insights into genomics of the L. (Viannia) subgenus. Sci. Rep. 2015, 5, 8550. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.K.; Srivastava, S.; Singh, A.; Singh, S. De Novo Whole-Genome Sequence and Annotation of a Leishmania Strain Isolated from a Case of Post-Kala-Azar Dermal Leishmaniasis. Genome Announc. 2015, 3, e00809-15. [Google Scholar] [CrossRef]
- Downing, T.; Imamura, H.; Decuypere, S.; Clark, T.G.; Coombs, G.H.; Cotton, J.A.; Hilley, J.D.; de Doncker, S.; Maes, I.; Mottram, J.C.; et al. Whole genome sequencing of multiple Leishmania donovani clinical isolates provides insights into population structure and mechanisms of drug resistance. Genome Res. 2011, 21, 2143–2156. [Google Scholar] [CrossRef] [PubMed]
- Raymond, F.; Boisvert, S.; Roy, G.; Ritt, J.F.; Legare, D.; Isnard, A.; Stanke, M.; Olivier, M.; Tremblay, M.J.; Papadopoulou, B.; et al. Genome sequencing of the lizard parasite Leishmania tarentolae reveals loss of genes associated to the intracellular stage of human pathogenic species. Nucleic Acids Res. 2012, 40, 1131–1147. [Google Scholar] [CrossRef]
- Valdivia, H.O.; Almeida, L.V.; Roatt, B.M.; Reis-Cunha, J.L.; Pereira, A.A.S.; Gontijo, C.; Fujiwara, R.T.; Reis, A.B.; Sanders, M.J.; Cotton, J.A.; et al. Comparative genomics of canine-isolated Leishmania (Leishmania) amazonensis from an endemic focus of visceral leishmaniasis in Governador Valadares, southeastern Brazil. Sci. Rep. 2017, 7, 40804. [Google Scholar] [CrossRef]
- Urrea, D.A.; Duitama, J.; Imamura, H.; Alzate, J.F.; Gil, J.; Munoz, N.; Villa, J.A.; Dujardin, J.C.; Ramirez-Pineda, J.R.; Triana-Chavez, O. Genomic Analysis of Colombian Leishmania panamensis strains with different level of virulence. Sci. Rep. 2018, 8, 17336. [Google Scholar] [CrossRef] [PubMed]
- Rogers, M.B.; Hilley, J.D.; Dickens, N.J.; Wilkes, J.; Bates, P.A.; Depledge, D.P.; Harris, D.; Her, Y.; Herzyk, P.; Imamura, H.; et al. Chromosome and gene copy number variation allow major structural change between species and strains of Leishmania. Genome Res. 2011, 21, 2129–2142. [Google Scholar] [CrossRef] [PubMed]
- Johner, A.; Kunz, S.; Linder, M.; Shakur, Y.; Seebeck, T. Cyclic nucleotide specific phosphodiesterases of Leishmania major. BMC Microbiol. 2006, 6, 25. [Google Scholar] [CrossRef]
- Alonso, G.; Rastrojo, A.; López-Pérez, S.; Requena, J.M.; Aguado, B. Resequencing and assembly of seven complex loci to improve the Leishmania major (Friedlin strain) reference genome. Parasites Vectors 2016, 9, 74. [Google Scholar] [CrossRef]
- Pita, S.; Diaz-Viraque, F.; Iraola, G.; Robello, C. The Tritryps Comparative Repeatome: Insights on Repetitive Element Evolution in Trypanosomatid Pathogens. Genome Biol. Evol. 2019, 11, 546–551. [Google Scholar] [CrossRef]
- Ubeda, J.M.; Raymond, F.; Mukherjee, A.; Plourde, M.; Gingras, H.; Roy, G.; Lapointe, A.; Leprohon, P.; Papadopoulou, B.; Corbeil, J.; et al. Genome-wide stochastic adaptive DNA amplification at direct and inverted DNA repeats in the parasite Leishmania. PLoS Biol. 2014, 12, e1001868. [Google Scholar] [CrossRef]
- Requena, J.M.; Rastrojo, A.; Garde, E.; López, M.C.; Thomas, M.C.; Aguado, B.; Lopez, M.C.; Thomas, M.C.; Aguado, B. Genomic cartography and proposal of nomenclature for the repeated, interspersed elements of the Leishmania major SIDER2 family and identification of SIDER2-containing transcripts. Mol. Biochem. Parasitol. 2017, 212, 9–15. [Google Scholar] [CrossRef]
- Requena, J.M. Lights and shadows on gene organization and regulation of gene expression in Leishmania. Front. Biosci. 2011, 16, 2069–2085. [Google Scholar] [CrossRef]
- van Dijk, E.L.; Jaszczyszyn, Y.; Naquin, D.; Thermes, C. The Third Revolution in Sequencing Technology. Trends Genet. 2018, 34, 666–681. [Google Scholar] [CrossRef] [PubMed]
- Lypaczewski, P.; Hoshizaki, J.; Zhang, W.W.; McCall, L.I.; Torcivia-Rodriguez, J.; Simonyan, V.; Kaur, A.; Dewar, K.; Matlashewski, G. A complete Leishmania donovani reference genome identifies novel genetic variations associated with virulence. Sci. Rep. 2018, 8, 16549. [Google Scholar] [CrossRef]
- Gonzalez-de la Fuente, S.; Camacho, E.; Peiro-Pastor, R.; Rastrojo, A.; Carrasco-Ramiro, F.; Aguado, B.; Requena, J.M. Complete and de novo assembly of the Leishmania braziliensis (M2904) genome. Mem. Inst. Oswaldo Cruz. 2018, 114, e180438. [Google Scholar] [CrossRef]
- Lin, W.; Batra, D.; Narayanan, V.; Rowe, L.A.; Sheth, M.; Zheng, Y.; Juieng, P.; Loparev, V.; de Almeida, M. First Draft Genome Sequence of Leishmania (Viannia) lainsoni Strain 216-34, Isolated from a Peruvian Clinical Case. Microbiol. Resour Announc. 2019, 8, e01524. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-de la Fuente, S.; Peiro-Pastor, R.; Rastrojo, A.; Moreno, J.; Carrasco-Ramiro, F.; Requena, J.M.; Aguado, B. Resequencing of the Leishmania infantum (strain JPCM5) genome and de novo assembly into 36 contigs. Sci. Rep. 2017, 7, 18050. [Google Scholar] [CrossRef]
- Camacho, E.; Gonzalez-de la Fuente, S.; Rastrojo, A.; Peiro-Pastor, R.; Solana, J.C.; Tabera, L.; Gamarro, F.; Carrasco-Ramiro, F.; Requena, J.M.; Aguado, B. Complete assembly of the Leishmania donovani (HU3 strain) genome and transcriptome annotation. Sci. Rep. 2019, 9, 6127. [Google Scholar] [CrossRef] [PubMed]
- Batra, D.; Lin, W.; Narayanan, V.; Rowe, L.A.; Sheth, M.; Zheng, Y.; Loparev, V.; de Almeida, M. Draft Genome Sequences of Leishmania (Leishmania) amazonensis, Leishmania (Leishmania) mexicana, and Leishmania (Leishmania) aethiopica, Potential Etiological Agents of Diffuse Cutaneous Leishmaniasis. Microbiol. Resour Announc. 2019, 8, e00269. [Google Scholar] [CrossRef]
- Steinbiss, S.; Silva-Franco, F.; Brunk, B.; Foth, B.; Hertz-Fowler, C.; Berriman, M.; Otto, T.D. Companion: A web server for annotation and analysis of parasite genomes. Nucleic Acids Res. 2016, 44, W29–W34. [Google Scholar] [CrossRef]
- Smith, M.; Blanchette, M.; Papadopoulou, B. Improving the prediction of mRNA extremities in the parasitic protozoan Leishmania. BMC Bioinform. 2008, 9, 158. [Google Scholar] [CrossRef]
- Rastrojo, A.; Carrasco-Ramiro, F.; Martín, D.; Crespillo, A.; Reguera, R.M.; Aguado, B.; Requena, J.M. The transcriptome of Leishmania major in the axenic promastigote stage: Transcript annotation and relative expression levels by RNA-seq. BMC Genom. 2013, 14, 223. [Google Scholar] [CrossRef] [PubMed]
- Fiebig, M.; Kelly, S.; Gluenz, E. Comparative Life Cycle Transcriptomics Revises Leishmania mexicana Genome Annotation and Links a Chromosome Duplication with Parasitism of Vertebrates. PLoS Pathog. 2015, 11, e1005186. [Google Scholar] [CrossRef] [PubMed]
- Soto, M.; Requena, J.M.; Alonso, C. Isolation, characterization and analysis of the expression of the Leishmania ribosomal PO protein genes. Mol. Biochem. Parasitol. 1993, 61, 265–274. [Google Scholar] [CrossRef]
- Soto, M.; Requena, J.M.; Garcia, M.; Gómez, L.C.; Navarrete, I.; Alonso, C. Genomic organization and expression of two independent gene arrays coding for two antigenic acidic ribosomal proteins of Leishmania. J. Biol. Chem. 1993, 268, 21835–21843. [Google Scholar] [CrossRef]
- Requena, J.M.; Soto, M.; Quijada, L.; Alonso, C. Genes and Chromosomes of Leishmania infantum. Mem. Inst. Oswaldo Cruz 1997, 92, 853–858. [Google Scholar] [CrossRef][Green Version]
- Zilka, A.; Garlapati, S.; Dahan, E.; Yaolsky, V.; Shapira, M. Developmental Regulation of Heat Shock Protein 83 in Leishmania. 3′ Processing and mRNA stability control transcript abundance, and translation is directed by a determinant in the 3′-untranslated region. J. Biol. Chem. 2001, 276, 47922–47929. [Google Scholar] [CrossRef]
- Soto, M.; Quijada, L.; Larreta, R.; Iborra, S.; Alonso, C.; Requena, J.M. Leishmania infantum possesses a complex family of histone H2A genes: Structural characterization and analysis of expression. Parasitology 2003, 127, 95–105. [Google Scholar] [CrossRef] [PubMed]
- Larreta, R.; Soto, M.; Quijada, L.; Folgueira, C.; Abanades, D.R.; Alonso, C.; Requena, J.M. The expression of HSP83 genes in Leishmania infantum is affected by temperature and by stage-differentiation and is regulated at the levels of mRNA stability and translation. BMC Mol. Biol. 2004, 5, 3. [Google Scholar] [CrossRef][Green Version]
- Folgueira, C.; Quijada, L.; Soto, M.; Abanades, D.R.; Alonso, C.; Requena, J.M. The translational efficiencies of the two Leishmania infantum HSP70 mRNAs, differing in their 3′-untranslated regions, are affected by shifts in the temperature of growth through different mechanisms. J. Biol. Chem. 2005, 280, 35172–35183. [Google Scholar] [CrossRef] [PubMed]
- Requena, J.M.; López, M.C.; Jimnez-Ruiz, A.; de la Torre, J.C.; Alonso, C. A head-to-tail tandem organization of hsp70 genes in Trypanosoma cruzi. Nucleic Acids Res. 1988, 16, 1393–1406. [Google Scholar] [CrossRef] [PubMed]
- Eid, J.; Fehr, A.; Gray, J.; Luong, K.; Lyle, J.; Otto, G.; Peluso, P.; Rank, D.; Baybayan, P.; Bettman, B.; et al. Real-time DNA sequencing from single polymerase molecules. Science 2009, 323, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Chin, C.S.; Alexander, D.H.; Marks, P.; Klammer, A.A.; Drake, J.; Heiner, C.; Clum, A.; Copeland, A.; Huddleston, J.; Eichler, E.E.; et al. Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat. Methods 2013, 10, 563–569. [Google Scholar] [CrossRef]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef]
- Sommer, D.D.; Delcher, A.L.; Salzberg, S.L.; Pop, M. Minimus: A fast, lightweight genome assembler. BMC Bioinform. 2007, 8, 64. [Google Scholar] [CrossRef]
- Katoh, K.; Kuma, K.; Toh, H.; Miyata, T. MAFFT version 5: Improvement in accuracy of multiple sequence alignment. Nucleic Acids Res. 2005, 33, 511–518. [Google Scholar] [CrossRef] [PubMed]
- Boetzer, M.; Henkel, C.V.; Jansen, H.J.; Butler, D.; Pirovano, W. Scaffolding pre-assembled contigs using SSPACE. Bioinformatics 2011, 27, 578–579. [Google Scholar] [CrossRef]
- Nadalin, F.; Vezzi, F.; Policriti, A. GapFiller: A de novo assembly approach to fill the gap within paired reads. BMC Bioinform. 2012, 13 (Suppl. 1), S8. [Google Scholar] [CrossRef]
- Sacristán-Horcajada, E.; González-de la Fuente, S.; Peiró-Pastor, R.; Carrasco-Ramiro, F.; Amils, R.; Requena, J.; Berenguer, J.; Aguado, B. ARAMIS: From systematic errors of NGS long reads to accurate assemblies. Brief. Bioinform. 2021. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Koren, S.; Walenz, B.P.; Berlin, K.; Miller, J.R.; Bergman, N.H.; Phillippy, A.M. Canu: Scalable and accurate long-read assembly via adaptive κ-mer weighting and repeat separation. Genome Res. 2017, 27, 722–736. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Chaisson, M.J.; Tesler, G. Mapping single molecule sequencing reads using basic local alignment with successive refinement (BLASR): Application and theory. BMC Bioinform. 2012, 13, 238. [Google Scholar] [CrossRef] [PubMed]
- Mondelaers, A.; Sanchez-Cañete, M.P.; Hendrickx, S.; Eberhardt, E.; Garcia-Hernandez, R.; Lachaud, L.; Cotton, J.; Sanders, M.; Cuypers, B.; Imamura, H.; et al. Genomic and Molecular Characterization of Miltefosine Resistance in Leishmania infantum Strains with Either Natural or Acquired Resistance through Experimental Selection of Intracellular Amastigotes. PLoS ONE 2016, 11, e0154101. [Google Scholar] [CrossRef]
- Darling, A.E.; Mau, B.; Perna, N.T. progressiveMauve: Multiple genome alignment with gene gain, loss and rearrangement. PLoS ONE 2010, 5, e11147. [Google Scholar] [CrossRef]
- Guy, L.; Kultima, J.R.; Andersson, S.G. genoPlotR: Comparative gene and genome visualization in R. Bioinformatics 2010, 26, 2334–2335. [Google Scholar] [CrossRef]
- Van der Auwera, G.A.; Carneiro, M.O.; Hartl, C.; Poplin, R.; Del Angel, G.; Levy-Moonshine, A.; Jordan, T.; Shakir, K.; Roazen, D.; Thibault, J.; et al. From FastQ data to high confidence variant calls: The Genome Analysis Toolkit best practices pipeline. Curr. Protoc Bioinforma 2013, 43. [Google Scholar] [CrossRef]
- Edge, P.; Bafna, V.; Bansal, V. HapCUT2: Robust and accurate haplotype assembly for diverse sequencing technologies. Genome Res. 2017, 27, 801–812. [Google Scholar] [CrossRef]
- Thorvaldsdottir, H.; Robinson, J.T.; Mesirov, J.P. Integrative Genomics Viewer (IGV): High-performance genomics data visualization and exploration. Br. Bioinform 2013, 14, 178–192. [Google Scholar] [CrossRef]
- Li, L.; Stoeckert, C.J., Jr.; Roos, D.S. OrthoMCL: Identification of ortholog groups for eukaryotic genomes. Genome Res. 2003, 13, 2178–2189. [Google Scholar] [CrossRef]
- Rastrojo, A.; Corvo, L.; Lombraña, R.; Solana, J.C.; Aguado, B.; Requena, J.M. Analysis by RNA-seq of transcriptomic changes elicited by heat shock in Leishmania major. Sci. Rep. 2019, 9, 6919. [Google Scholar] [CrossRef]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef]
- Dillon, L.A.L.; Okrah, K.; Hughitt, V.K.; Suresh, R.; Li, Y.; Fernandes, M.C.; Belew, A.T.; Corrada Bravo, H.; Mosser, D.M.; El-Sayed, N.M. Transcriptomic profiling of gene expression and RNA processing during Leishmania major differentiation. Nucleic Acids Res. 2015, 43, 6799–6813. [Google Scholar] [CrossRef]
- Martinez-Calvillo, S.; Sunkin, S.M.; Yan, S.; Fox, M.; Stuart, K.; Myler, P.J. Genomic organization and functional characterization of the Leishmania major Friedlin ribosomal RNA gene locus. Mol. Biochem. Parasitol. 2001, 116, 147–157. [Google Scholar] [CrossRef]
- Lypaczewski, P.; Zhang, W.W.; Matlashewski, G. Evidence that a naturally occurring single nucleotide polymorphism in the RagC gene of Leishmania donovani contributes to reduced virulence. PLoS Negl. Trop. Dis. 2021, 15, e0009079. [Google Scholar] [CrossRef] [PubMed]
- Wagner, G.P.; Kin, K.; Lynch, V.J. Measurement of mRNA abundance using RNA-seq data: RPKM measure is inconsistent among samples. Theory Biosci. 2012, 131, 281–285. [Google Scholar] [CrossRef]
- Carter, N.S.; Drew, M.E.; Sanchez, M.; Vasudevan, G.; Landfear, S.M.; Ullman, B. Cloning of a novel inosine-guanosine transporter gene from Leishmania donovani by functional rescue of a transport-deficient mutant. J. Biol. Chem. 2000, 275, 20935–20941. [Google Scholar] [CrossRef] [PubMed]
- Da Costa, K.S.; Galucio, J.M.P.; Leonardo, E.S.; Cardoso, G.; Leal, E.; Conde, G.; Lameira, J. Structural and evolutionary analysis of Leishmania Alba proteins. Mol. Biochem. Parasitol. 2017, 217, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Dupe, A.; Dumas, C.; Papadopoulou, B. An Alba-domain protein contributes to the stage-regulated stability of amastin transcripts in Leishmania. Mol. Microbiol. 2014, 91, 548–561. [Google Scholar] [CrossRef] [PubMed]
- Mishra, A.K.; Singh, N.; Agnihotri, P.; Mishra, S.; Singh, S.P.; Kolli, B.K.; Chang, K.P.; Sahasrabuddhe, A.A.; Siddiqi, M.I.; Pratap, J. V Discovery of novel inhibitors for Leishmania nucleoside diphosphatase kinase (NDK) based on its structural and functional characterization. J. Comput Aided Mol. Des. 2017, 31, 547–562. [Google Scholar] [CrossRef] [PubMed]
- Rascher, C.; Pahl, A.; Pecht, A.; Brune, K.; Solbach, W.; Bang, H. Leishmania major parasites express cyclophilin isoforms with an unusual interaction with calcineurin. Biochem. J. 1998, 334, 659–667. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, C.; Carvalho, P.C.; Alves, L.R.; Goldenberg, S. The role of the trypanosoma cruzi TcNRBD1 protein in translation. PLoS ONE 2016, 11, e0164650. [Google Scholar] [CrossRef] [PubMed]
- Bates, P.A. The lipophosphoglycan-associated molecules of Leishmania. Parasitol. Today 1995, 11, 317–318. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters/Genome | LMJFC [This Work] | LmjF [v44-TritrypDB] |
|---|---|---|
| Number of chromosomes | 36 | 36 |
| Protein-coding genes | 8596 | 8400 |
| Pseudogenes | 88 | 88 |
| rRNAs | 30 | 63 |
| tRNAs | 93 | 83 |
| snoRNA+snRNA+slRNA | 1128 | 747 |
| Number of gaps | 0 | 9 |
| Number of Ns | 0 | 13 |
| Genome size (bp) | 32,792,963 | 32,855,082 |
| Annotated Transcripts | 9828 |
|---|---|
| Protein-coding transcripts | 8517 |
| Transcripts with mapped SL addition (SLA) site | 9745 (99.1 %) |
| Transcripts with alternative SLA sites | 9341 (95 %) |
| Transcripts with mapped poly-A addition site (PAS) | 8677/9336 1 |
| Transcripts with alternative PASs | 6668 |
| Annotated CDS lacking a defined transcript | 10 |
| Transcripts with two or more CDS | 47 (43 are bicistronic) |
| Transcript ID | TPM ± SD 1 | Name of the Encoded Protein |
|---|---|---|
| LMJFC_27T0019600 | 3660.01 ± 344.91 | histone H1 |
| LMJFC_27T0019100 | 3629.05 ± 386.87 | histone H1 |
| LMJFC_35T0007800 | 3477.13 ± 722.67 | ribosomal protein L30 |
| LMJFC_06T0005100 | 3034.31 ± 646.98 | histone H4 |
| LMJFC_29T0026200 | 3027.80 ± 656.33 | histone H2A |
| LMJFC_36T0028500 | 2876.01 ± 261.79 | inosine-guanosine transporter (NT2) |
| LMJFC_19T0005400 | 2862.36 ± 308.82 | histone H2B |
| LMJFC_19T0005500 | 2684.51 ± 221.06 | histone H2B |
| LMJFC_13T0009700 | 2578.58 ± 230.03 | ALBA-domain protein 1 (ALBA1) |
| LMJFC_19T0005600 | 2555.44 ± 211.16 | histone H2B |
| LMJFC_30T0045400 | 2430.21 ± 261.54 | ribosomal protein L9 |
| LMJFC_15T0005100 | 2294.60 ± 265.01 | histone H4 |
| LMJFC_16T0012400 | 2285.04 ± 250.45 | histone H3 |
| LMJFC_35T0048600 | 2246.30 ± 380.22 | ribosomal protein L23 |
| LMJFC_31T0048800 | 2078.23 ± 130.10 | histone H4 |
| LMJFC_29T0026000 | 2068.24 ± 167.92 | histone H2A |
| LMJFC_24T0032000 | 2060.45 ± 60.25 | ribosomal protein L12 |
| LMJFC_19T0005700 | 2057.42 ± 51.88 | ribosomal protein S2 |
| LMJFC_13T0011100 | 2052.88 ± 103.60 | ribosomal protein S12 |
| LMJFC_09T0020600 | 1994.99 ± 193.32 | histone H2B |
| LMJFC_32T0040100 | 1964.77 ± 53.15 | nucleoside diphosphate kinase b |
| LMJFC_35T0011400 | 1939.67 ± 357.53 | ribosomal protein L18a |
| LMJFC_28T0032200 | 1935.03 ± 70.94 | ribosomal protein S29 |
| LMJFC_35T0026300 | 1899.65 ± 350.17 | ribosomal protein L15 |
| LMJFC_25T0016400 | 1898.52 ± 326.43 | cyclophilin A | CyP1 |
| LMJFC_29T0039100 | 1898.13 ± 80.33 | ribosomal protein S19-like protein |
| LMJFC_14T0020500 | 1887.76 ± 63.85 | ubiquitin/ribosomal protein S27a |
| LMJFC_32T0010100 | 1857.84 ± 87.17 | ribosomal protein L17 |
| LMJFC_32T0014200 | 1836.37 ± 60.13 | RNA binding protein |
| LMJFC_32T0010300 | 1833.43 ± 53.95 | ribosomal protein S2 |
| LMJFC_35T0027400 | 1814.80 ± 300.02 | ribosomal protein S6 |
| LMJFC_36T0051800 | 1798.18 ± 96.35 | ribosomal protein L34 |
| LMJFC_35T0042900 | 1794.95 ± 276.06 | ribosomal subunit protein L31 |
| LMJFC_32T0016000 | 1782.48 ± 98.87 | ribosomal protein L18a |
| LMJFC_31T0016500 | 1780.58 ± 476.99 | hypothetical protein-conserved |
| LMJFC_35T0048200 | 1779.35 ± 288.58 | ribosomal protein L27A/L29 |
| LMJFC_29T0034600 | 1779.35 ± 55.64 | ribosomal protein L13 |
| LMJFC_35T0048400 | 1770.18 ± 217.83 | ribosomal protein L27A/L29 |
| LMJFC_35T0029900 | 1754.88 ± 188.03 | kinetoplastid membrane protein 11 (KMP11) |
| LMJFC_25T0035900 | 1745.73 ± 104.64 | histone H4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Camacho, E.; González-de la Fuente, S.; Solana, J.C.; Rastrojo, A.; Carrasco-Ramiro, F.; Requena, J.M.; Aguado, B. Gene Annotation and Transcriptome Delineation on a De Novo Genome Assembly for the Reference Leishmania major Friedlin Strain. Genes 2021, 12, 1359. https://doi.org/10.3390/genes12091359
Camacho E, González-de la Fuente S, Solana JC, Rastrojo A, Carrasco-Ramiro F, Requena JM, Aguado B. Gene Annotation and Transcriptome Delineation on a De Novo Genome Assembly for the Reference Leishmania major Friedlin Strain. Genes. 2021; 12(9):1359. https://doi.org/10.3390/genes12091359
Chicago/Turabian StyleCamacho, Esther, Sandra González-de la Fuente, Jose C. Solana, Alberto Rastrojo, Fernando Carrasco-Ramiro, Jose M. Requena, and Begoña Aguado. 2021. "Gene Annotation and Transcriptome Delineation on a De Novo Genome Assembly for the Reference Leishmania major Friedlin Strain" Genes 12, no. 9: 1359. https://doi.org/10.3390/genes12091359
APA StyleCamacho, E., González-de la Fuente, S., Solana, J. C., Rastrojo, A., Carrasco-Ramiro, F., Requena, J. M., & Aguado, B. (2021). Gene Annotation and Transcriptome Delineation on a De Novo Genome Assembly for the Reference Leishmania major Friedlin Strain. Genes, 12(9), 1359. https://doi.org/10.3390/genes12091359