
Genome Analysis of Endotrypanum and Porcisia spp., Closest Phylogenetic Relatives of Leishmania, Highlights the Role of Amastins in Shaping Pathogenicity

 ,
,  , , , ,
, , , , 
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cultivation, DNA Isolation and Species Verification
2.2. Whole-Genome and Transcriptome Sequencing and Annotation
2.3. Repeats Identification and Synteny Analysis
2.4. Genome Coverage Analysis and Ploidy Estimation
2.5. Variant Calling
2.6. Orthology and Phylogenomic Analyses
2.7. Gene Ontology Analysis and Functional Annotation
2.8. Analyses of Amastin Surface Proteins and Biopterin Transporters
2.9. Selection Analysis
3. Results
3.1. Endotrypanum Sp. ATCC 30507 (MCHO/PA/72/3130) Is E. monterogeii
3.2. General Features of Endotrypanum and Porcisia Genomes
3.3. Genome Coverage Analysis, Ploidy Estimation and Synteny Analysis
3.4. Analysis of Repetitive Sequences
3.5. Gene Family Sharing Analysis
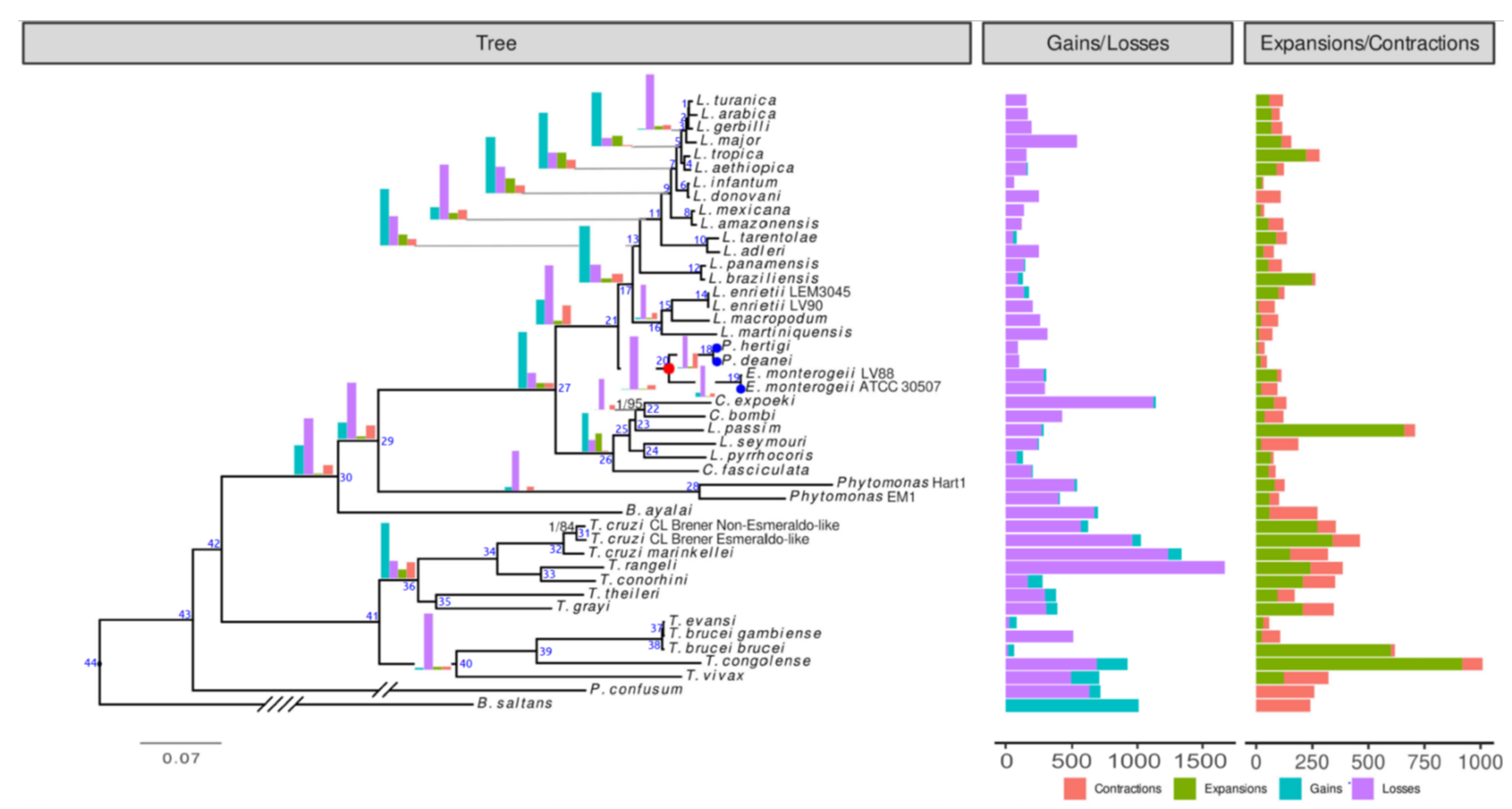
3.6. Phylogenomic Analysis
3.7. Evolution of Gene Families
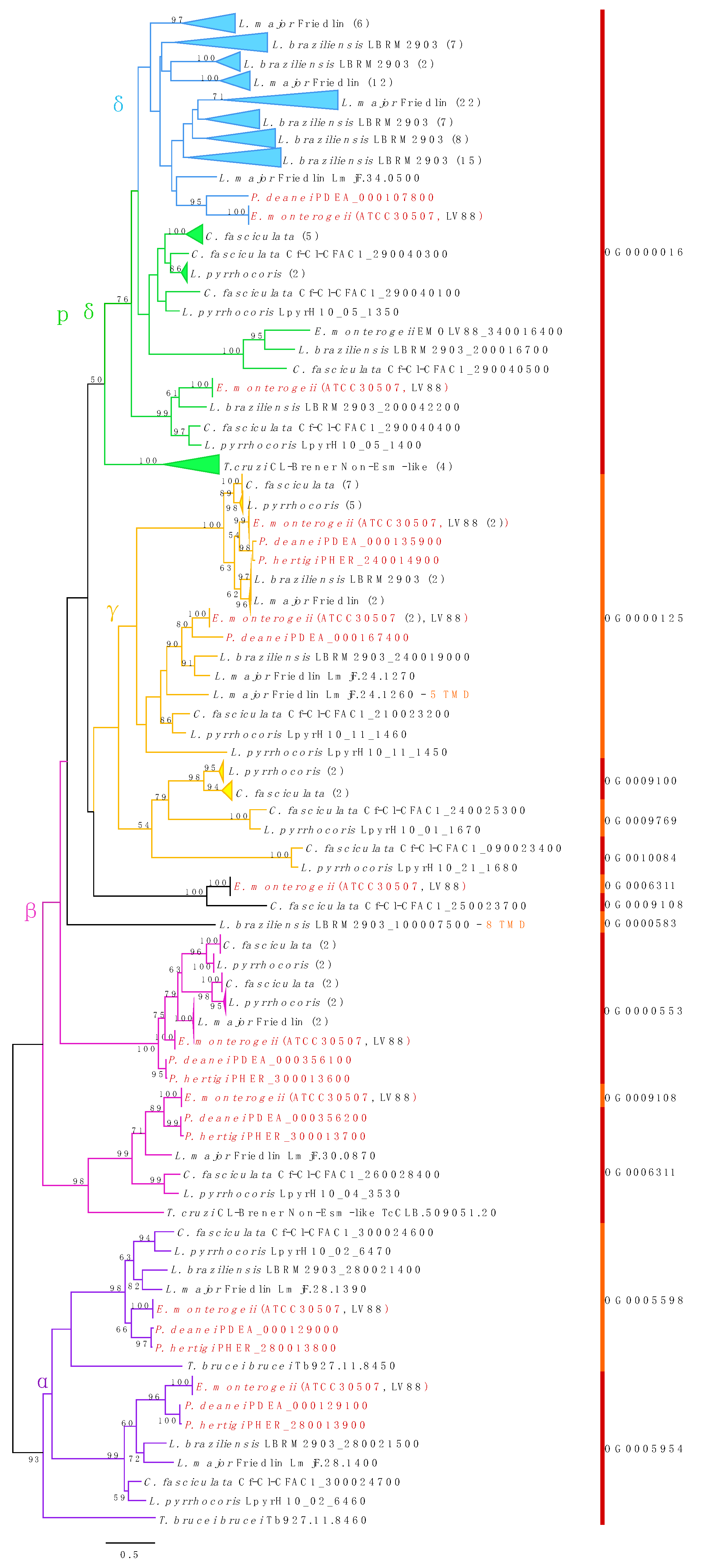
3.8. Amastins
3.9. Biopterin Transporter BT1
3.10. Notes on Metabolism of Endotrypanum and Porcisia
3.11. Selection Analysis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Maslov, D.A.; Opperdoes, F.R.; Kostygov, A.Y.; Hashimi, H.; Lukeš, J.; Yurchenko, V. Recent advances in trypanosomatid research: Genome organization, expression, metabolism, taxonomy and evolution. Parasitology 2019, 146, 1–27. [Google Scholar] [CrossRef] [PubMed]
- Lukeš, J.; Butenko, A.; Hashimi, H.; Maslov, D.A.; Votýpka, J.; Yurchenko, V. Trypanosomatids are much more than just trypanosomes: Clues from the expanded family tree. Trends Parasitol. 2018, 34, 466–480. [Google Scholar] [CrossRef] [PubMed]
- Lukeš, J.; Skalický, T.; Týč, J.; Votýpka, J.; Yurchenko, V. Evolution of parasitism in kinetoplastid flagellates. Mol. Biochem. Parasitol. 2014, 195, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Kostygov, A.Y.; Yurchenko, V. Revised classification of the subfamily Leishmaniinae (Trypanosomatidae). Folia Parasitol. 2017, 64, 020. [Google Scholar] [CrossRef] [PubMed]
- Jirků, M.; Yurchenko, V.; Lukeš, J.; Maslov, D.A. New species of insect trypanosomatids from Costa Rica and the proposal for a new subfamily within the Trypanosomatidae. J. Eukaryot. Microbiol. 2012, 59, 537–547. [Google Scholar] [CrossRef] [PubMed]
- Bruschi, F.; Gradoni, L. The Leishmaniases: Old Neglected Tropical Diseases; Springer: Cham, Switzerland, 2018; p. 245. [Google Scholar] [CrossRef]
- Akhoundi, M.; Downing, T.; Votýpka, J.; Kuhls, K.; Lukeš, J.; Cannet, A.; Ravel, C.; Marty, P.; Delaunay, P.; Kasbari, M.; et al. Leishmania infections: Molecular targets and diagnosis. Mol. Asp. Med. 2017, 57, 1–29. [Google Scholar] [CrossRef]
- Espinosa, O.A.; Serrano, M.G.; Camargo, E.P.; Teixeira, M.M.; Shaw, J.J. An appraisal of the taxonomy and nomenclature of trypanosomatids presently classified as Leishmania and Endotrypanum. Parasitology 2018, 145, 430–442. [Google Scholar] [CrossRef]
- Butenko, A.; Kostygov, A.Y.; Sádlová, J.; Kleschenko, Y.; Bečvář, T.; Podešvová, L.; Macedo, D.H.; Žihala, D.; Lukeš, J.; Bates, P.A.; et al. Comparative genomics of Leishmania (Mundinia). BMC Genom. 2019, 20, 726. [Google Scholar] [CrossRef]
- Coughlan, S.; Taylor, A.S.; Feane, E.; Sanders, M.; Schonian, G.; Cotton, J.A.; Downing, T. Leishmania naiffi and Leishmania guyanensis reference genomes highlight genome structure and gene evolution in the Viannia subgenus. R. Soc. Open Sci. 2018, 5, 172212. [Google Scholar] [CrossRef]
- Coughlan, S.; Mulhair, P.; Sanders, M.; Schonian, G.; Cotton, J.A.; Downing, T. The genome of Leishmania adleri from a mammalian host highlights chromosome fission in Sauroleishmania. Sci. Rep. 2017, 7, 43747. [Google Scholar] [CrossRef] [PubMed]
- Valdivia, H.O.; Reis-Cunha, J.L.; Rodrigues-Luiz, G.F.; Baptista, R.P.; Baldeviano, G.C.; Gerbasi, R.V.; Dobson, D.E.; Pratlong, F.; Bastien, P.; Lescano, A.G.; et al. Comparative genomic analysis of Leishmania (Viannia) peruviana and Leishmania (Viannia) braziliensis. BMC Genom. 2015, 16, 715. [Google Scholar] [CrossRef]
- Peacock, C.S.; Seeger, K.; Harris, D.; Murphy, L.; Ruiz, J.C.; Quail, M.A.; Peters, N.; Adlem, E.; Tivey, A.; Aslett, M.; et al. Comparative genomic analysis of three Leishmania species that cause diverse human disease. Nat. Genet. 2007, 39, 839–847. [Google Scholar] [CrossRef]
- Mesnil, F.; Brimont, E. Sur un hématozoaire nouveau (Endotrypanum n. gen.) d’un édenté de la Guyane. C.R. Séances Soc. Biol. Ses. Fil. 1908, 65, 581–583. [Google Scholar]
- Shaw, J.J.; Bird, R.G. The endoerythrocytic habitat of a member of the Trypanosomatidae, Endotrypanum schaudinni, Mesnil and Brimont, 1908. Z. Trop. Parasitol. 1969, 20, 144–150. [Google Scholar]
- Cunha, A.M.; Muniz, J. Pesquisas sôbre o Endotrypanum schaudinni Mesnil e Brimont, 1908, parasita do Choloepus didactylus (L.). Mem. Do Inst. Oswaldo Cruz 1944, 41, 179–193. [Google Scholar] [CrossRef]
- Cupolillo, E.; Medina-Acosta, E.; Noyes, H.; Momen, H.; Grimaldi, G., Jr. A revised classification for Leishmania and Endotrypanum. Parasitol. Today 2000, 16, 142–144. [Google Scholar] [CrossRef]
- Shaw, J.J. The Haemoflagellates of Sloths; H. K. Lewis: London, UK, 1969; p. 132. [Google Scholar]
- Kreutzer, R.D.; Corredor, A.; Grimaldi, G., Jr.; Grogl, M.; Rowton, E.D.; Young, D.G.; Morales, A.; McMahon-Pratt, D.; Guzman, H.; Tesh, R.B. Characterization of Leishmania colombiensis sp. n (Kinetoplastida: Trypanosomatidae), a new parasite infecting humans, animals, and phlebotomine sand flies in Colombia and Panama. Am. J. Trop. Med. Hyg. 1991, 44, 662–675. [Google Scholar] [CrossRef]
- Zeledón, R.; Ponce, C.; Murillo, J. Leishmania herreri sp. n. from sloths and sandflies of Costa Rica. J. Parasitol. 1979, 65, 275–279. [Google Scholar] [CrossRef] [PubMed]
- Delgado, O.; Castes, M.; White, A.C., Jr.; Kreutzer, R.D. Leishmania colombiensis in Venezuela. Am. J. Trop. Med. Hyg. 1993, 48, 145–147. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Bonfante, C.; Bonfante-Garrido, R.; Grimaldi, G., Jr.; Momen, H.; Cupolillo, E. Genotypically distinct Leishmania colombiensis isolates from Venezuela cause both cutaneous and visceral leishmaniasis in humans. Infect. Genet. Evol. J. Mol. Epidemiol. Evol. Genet. Infect. Dis. 2003, 3, 119–124. [Google Scholar] [CrossRef]
- Herrer, A. Leishmania hertigi sp. n., from the tropical porcupine, Coendou rothschildi Thomas. J. Parasitol. 1971, 57, 626–629. [Google Scholar] [CrossRef] [PubMed]
- Lainson, R.; Shaw, J.J. Leishmanias of neotropical porcupines: Leishmania hertigi deanei nov. subsp. Acta Amaz. 1977, 7, 51–57. [Google Scholar] [CrossRef]
- Gardener, P.J.; Chance, M.L.; Peters, W. Biochemical taxonomy of Leishmania. II: Electrophoretic variation of malate dehydrogenase. Ann. Trop. Med. Parasitol. 1974, 68, 317–325. [Google Scholar] [CrossRef] [PubMed]
- da Silva, D.A.; Madeira Mde, F.; Barbosa Filho, C.J.; Schubach, E.Y.; Barros, J.H.; Figueiredo, F.B. Leishmania (Leishmania) hertigi in a porcupine (Coendou sp.) found in Brasilia, Federal District, Brazil. Rev. Bras. Parasitol. Vet. 2013, 22, 297–299. [Google Scholar] [CrossRef]
- Deane, L.M.; da Silva, J.E.; de Figueiredo, P.Z. Leishmaniae in the viscera of porcupines from the state of Piaui, Brazil. Rev. Do Inst. De Med. Trop. De Sao Paulo 1974, 16, 68–69. [Google Scholar]
- Pothirat, T.; Tantiworawit, A.; Chaiwarith, R.; Jariyapan, N.; Wannasan, A.; Siriyasatien, P.; Supparatpinyo, K.; Bates, M.D.; Kwakye-Nuako, G.; Bates, P.A. First isolation of Leishmania from Northern Thailand: Case report, identification as Leishmania martiniquensis and phylogenetic position within the Leishmania enriettii complex. PLoS Negl. Trop. Dis. 2014, 8, e3339. [Google Scholar] [CrossRef]
- Shaw, J.J. A possible vector of Endotrypanum schaudinni of the sloth Choloepus hoffmanni, in Panama. Nature 1964, 201, 417–418. [Google Scholar] [CrossRef]
- Shaw, J.J.; de Rosa, A.T.; Cruz, A.C.R.; Vasconcelos, P.F.C. Brazilian phlebotomines as hosts and vectors of viruses, bacteria, fungi, protozoa (excluding those belonging to the genus Leishmania) and nematodes. In Brazilian Sand Flies; Rangel, E.F., Shaw, J.J., Eds.; Springer International Publishing AG: Basel, Switzerland, 2018; pp. 417–441. [Google Scholar]
- Shaw, J.J. The behaviour of Endotrypanum schaudinni (Kinetoplastidae:Trypanosomatidae) in three species of laboratory-bred neotropical sandflies (Diptera:Psychodidae) and its influence on the classification of the genus Leishmania. In Parasitological Topics. A Presentation Volume to P. C. C. Garnham, F. R. S., on the Occasion of His 80th Birthday; Canning, E.U., Ed.; Allen Press: Lawrence, KS, USA, 1981; pp. 232–241. [Google Scholar]
- Franco, A.M.; Tesh, R.B.; Guzman, H.; Deane, M.P.; Grimaldi Junior, G. Development of Endotrypanum (Kinetoplastida:Trypanosomatidae) in experimentally infected phlebotomine sand flies (Diptera:Psychodidae). J. Med. Entomol. 1997, 34, 189–192. [Google Scholar] [CrossRef] [PubMed]
- Katakura, K.; Mimori, T.; Furuya, M.; Uezato, H.; Nonaka, S.; Okamoto, M.; Gomez, L.E.; Hashiguchi, Y. Identification of Endotrypanum species from a sloth, a squirrel and Lutzomyia sandflies in Ecuador by PCR amplification and sequencing of the mini-exon gene. J. Vet. Med. Sci. 2003, 65, 649–653. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Christensen, H.A.; Herrer, A. Neotropical sand flies (Diptera: Psychodidae), invertebrate hosts of Endotrypanum schaudinni (Kinetoplastida: Trypanosomatidae). J. Med. Entomol. 1976, 13, 299–303. [Google Scholar] [CrossRef]
- Thies, S.F.; Bronzoni, R.V.M.; Michalsky, E.M.; Santos, E.S.D.; Silva, D.; Dias, E.S.; Damazo, A.S. Aspects on the ecology of phlebotomine sand flies and natural infection by Leishmania hertigi in the Southeastern Amazon Basin of Brazil. Acta Trop. 2018, 177, 37–43. [Google Scholar] [CrossRef]
- Barratt, J.; Kaufer, A.; Peters, B.; Craig, D.; Lawrence, A.; Roberts, T.; Lee, R.; McAuliffe, G.; Stark, D.; Ellis, J. Isolation of novel trypanosomatid, Zelonia australiensis sp. nov. (Kinetoplastida: Trypanosomatidae) provides support for a Gondwanan origin of dixenous parasitism in the Leishmaniinae. PLoS Negl. Trop. Dis. 2017, 11, e0005215. [Google Scholar] [CrossRef]
- Harkins, K.M.; Schwartz, R.S.; Cartwright, R.A.; Stone, A.C. Phylogenomic reconstruction supports supercontinent origins for Leishmania. Infect. Genet. Evol. J. Mol. Epidemiol. Evol. Genet. Infect. Dis. 2016, 38, 101–109. [Google Scholar] [CrossRef] [PubMed]
- O’Leary, M.A.; Bloch, J.I.; Flynn, J.J.; Gaudin, T.J.; Giallombardo, A.; Giannini, N.P.; Goldberg, S.L.; Kraatz, B.P.; Luo, Z.X.; Meng, J.; et al. The placental mammal ancestor and the post-K-Pg radiation of placentals. Science 2013, 339, 662–667. [Google Scholar] [CrossRef]
- Lopes, A.H.; Iovannisci, D.; Petrillo-Peixoto, M.; McMahon-Pratt, D.; Beverley, S.M. Evolution of nuclear DNA and the occurrence of sequences related to new small chromosomal DNAs in the trypanosomatid genus Endotrypanum. Mol. Biochem. Parasitol. 1990, 40, 151–161. [Google Scholar] [CrossRef]
- Maslov, D.A.; Lukeš, J.; Jirků, M.; Simpson, L. Phylogeny of trypanosomes as inferred from the small and large subunit rRNAs: Implications for the evolution of parasitism in the trypanosomatid protozoa. Mol. Biochem. Parasitol. 1996, 75, 197–205. [Google Scholar] [CrossRef]
- Maslov, D.A.; Yurchenko, V.Y.; Jirků, M.; Lukeš, J. Two new species of trypanosomatid parasites isolated from Heteroptera in Costa Rica. J. Eukaryot. Microbiol. 2010, 57, 177–188. [Google Scholar] [CrossRef] [PubMed]
- Yurchenko, V.; Votýpka, J.; Tesařová, M.; Klepetková, H.; Kraeva, N.; Jirků, M.; Lukeš, J. Ultrastructure and molecular phylogeny of four new species of monoxenous trypanosomatids from flies (Diptera: Brachycera) with redefinition of the genus Wallaceina. Folia Parasitol. 2014, 61, 97–112. [Google Scholar] [CrossRef]
- Losev, A.; Grybchuk-Ieremenko, A.; Kostygov, A.Y.; Lukes, J.; Yurchenko, V. Host specificity, pathogenicity, and mixed infections of trypanoplasms from freshwater fishes. Parasitol. Res. 2015, 114, 1071–1078. [Google Scholar] [CrossRef] [PubMed]
- Kostygov, A.Y.; Grybchuk-Ieremenko, A.; Malysheva, M.N.; Frolov, A.O.; Yurchenko, V. Molecular revision of the genus Wallaceina. Protist 2014, 165, 594–604. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed on 8 March 2021).
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. J. Comput. Mol. Cell Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed]
- Laetsch, D.R.; Blaxter, M.L. BlobTools: Interrogation of genome assemblies. F1000Research 2017, 6, 1287. [Google Scholar] [CrossRef]
- Mikheenko, A.; Prjibelski, A.; Saveliev, V.; Antipov, D.; Gurevich, A. Versatile genome assembly evaluation with QUAST-LG. Bioinformatics 2018, 34, i142–i150. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef]
- Steinbiss, S.; Silva-Franco, F.; Brunk, B.; Foth, B.; Hertz-Fowler, C.; Berriman, M.; Otto, T.D. Companion: A web server for annotation and analysis of parasite genomes. Nucleic Acids Res. 2016, 44, W29–W34. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, R.M.; Seppey, M.; Simão, F.A.; Manni, M.; Ioannidis, P.; Klioutchnikov, G.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO applications from quality assessments to gene prediction and phylogenomics. Mol. Biol. Evol. 2018, 35, 543–548. [Google Scholar] [CrossRef] [PubMed]
- Smit, A.F.A.; Hubley, R.; Green, P. RepeatMasker Open-4.0. Available online: http://www.repeatmasker.org (accessed on 19 March 2021).
- Soderlund, C.; Nelson, W.; Shoemaker, A.; Paterson, A. SyMAP: A system for discovering and viewing syntenic regions of FPC maps. Genome Res. 2006, 16, 1159–1168. [Google Scholar] [CrossRef]
- Tamazian, G.; Dobrynin, P.; Krasheninnikova, K.; Komissarov, A.; Koepfli, K.P.; O’Brien, S.J. Chromosomer: A reference-based genome arrangement tool for producing draft chromosome sequences. Gigascience 2016, 5, 38. [Google Scholar] [CrossRef]
- Quinlan, A.R. BEDTools: The swiss-army tool for genome feature analysis. Curr. Protoc. Bioinform. 2014, 47, 11–12. [Google Scholar] [CrossRef] [PubMed]
- Wickham, H.; François, R.; Henry, L.; Müller, K. Dplyr: A Grammar of Data Manipulation. R Package Version 1.0.2; 2020. [Google Scholar]
- Ginestet, C. ggplot2: Elegant graphics for data analysis. J. R. Stat. Soc. 2011, 174, 245. [Google Scholar] [CrossRef]
- Lipovetsky, S. Statistical inference via data science: A modern dive into R and the tidyverse. Technometrics 2020, 62, 283. [Google Scholar] [CrossRef]
- Boyd, Z.; Hughes, J. WeeSAM: Script for Parsing SAM/BAM Files for Coverage Statistics; 2018. [Google Scholar]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed]
- Rimmer, A.; Phan, H.; Mathieson, I.; Iqbal, Z.; Twigg, S.R.; Wilkie, A.O.; McVean, G.; Lunter, G. Integrating mapping-, assembly- and haplotype-based approaches for calling variants in clinical sequencing applications. Nat. Genet. 2014, 46, 912–918. [Google Scholar] [CrossRef]
- Emms, D.M.; Kelly, S. OrthoFinder: Phylogenetic orthology inference for comparative genomics. Genome Biol. 2019, 20, 238. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Capella-Gutiérrez, S.; Silla-Martinez, J.M.; Gabaldon, T. trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef]
- Eddy, S.R. Accelerated profile HMM searches. PLoS Comput. Biol. 2011, 7, e1002195. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Lartillot, N.; Rodrigue, N.; Stubbs, D.; Richer, J. PhyloBayes MPI: Phylogenetic reconstruction with infinite mixtures of profiles in a parallel environment. Syst. Biol. 2013, 62, 611–615. [Google Scholar] [CrossRef]
- Yu, G.C.; Smith, D.K.; Zhu, H.C.; Guan, Y.; Lam, T.T.Y. ggtree: An R package for visualization and annotation of phylogenetic trees with their covariates and other associated data. Methods Ecol. Evol. 2017, 8, 28–36. [Google Scholar] [CrossRef]
- Csűrös, M. Count: Evolutionary analysis of phylogenetic profiles with parsimony and likelihood. Bioinformatics 2010, 26, 1910–1912. [Google Scholar] [CrossRef]
- Kanehisa, M.; Sato, Y.; Morishima, K. BlastKOALA and GhostKOALA: KEGG Tools for Functional Characterization of Genome and Metagenome Sequences. J. Mol. Biol. 2016, 428, 726–731. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.; Binns, D.; Chang, H.Y.; Fraser, M.; Li, W.; McAnulla, C.; McWilliam, H.; Maslen, J.; Mitchell, A.; Nuka, G.; et al. InterProScan 5: Genome-scale protein function classification. Bioinformatics 2014, 30, 1236–1240. [Google Scholar] [CrossRef]
- Lex, A.; Gehlenborg, N.; Strobelt, H.; Vuillemot, R.; Pfister, H. UpSet: Visualization of Intersecting Sets. IEEE Trans. Vis. Comput. Graph. 2014, 20, 1983–1992. [Google Scholar] [CrossRef]
- Opperdoes, F.R.; Butenko, A.; Flegontov, P.; Yurchenko, V.; Lukeš, J. Comparative metabolism of free-living Bodo saltans and parasitic trypanosomatids. J. Eukaryot. Microbiol. 2016, 63, 657–678. [Google Scholar] [CrossRef]
- Binns, D.; Dimmer, E.; Huntley, R.; Barrell, D.; O’Donovan, C.; Apweiler, R. QuickGO: A web-based tool for Gene Ontology searching. Bioinformatics 2009, 25, 3045–3046. [Google Scholar] [CrossRef]
- Finn, R.D.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Mistry, J.; Mitchell, A.L.; Potter, S.C.; Punta, M.; Qureshi, M.; Sangrador-Vegas, A.; et al. The Pfam protein families database: Towards a more sustainable future. Nucleic Acids Res. 2016, 44, D279–D285. [Google Scholar] [CrossRef]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Soding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef] [PubMed]
- Rambaut, A. FigTree v.1.4.4. Available online: http://tree.bio.ed.ac.uk/software/figtree/ (accessed on 19 March 2021).
- Krogh, A.; Larsson, B.; von Heijne, G.; Sonnhammer, E.L. Predicting transmembrane protein topology with a hidden Markov model: Application to complete genomes. J. Mol. Biol. 2001, 305, 567–580. [Google Scholar] [CrossRef]
- Gerlt, J.A.; Bouvier, J.T.; Davidson, D.B.; Imker, H.J.; Sadkhin, B.; Slater, D.R.; Whalen, K.L. Enzyme Function Initiative-Enzyme Similarity Tool (EFI-EST): A web tool for generating protein sequence similarity networks. Biochim. Biophys. Acta 2015, 1854, 1019–1037. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Butenko, A.; Hammond, M.; Field, M.C.; Ginger, M.L.; Yurchenko, V.; Lukeš, J. Reductionist pathways for parasitism in euglenozoans? Expanded datasets provide new insights. Trends Parasitol. 2021, 37, 100–116. [Google Scholar] [CrossRef]
- Jackson, A.P. The evolution of amastin surface glycoproteins in trypanosomatid parasites. Mol. Biol. Evol. 2010, 27, 33–45. [Google Scholar] [CrossRef]
- Zhang, J.; Nielsen, R.; Yang, Z. Evaluation of an improved branch-site likelihood method for detecting positive selection at the molecular level. Mol. Biol. Evol. 2005, 22, 2472–2479. [Google Scholar] [CrossRef] [PubMed]
- Huerta-Cepas, J.; Serra, F.; Bork, P. ETE 3: Reconstruction, analysis, and visualization of phylogenomic data. Mol. Biol. Evol. 2016, 33, 1635–1638. [Google Scholar] [CrossRef]
- Alexa, A.; Rahnenfuhrer, J.; Lengauer, T. Improved scoring of functional groups from gene expression data by decorrelating GO graph structure. Bioinformatics 2006, 22, 1600–1607. [Google Scholar] [CrossRef]
- Flegontov, P.; Butenko, A.; Firsov, S.; Kraeva, N.; Eliáš, M.; Field, M.C.; Filatov, D.; Flegontova, O.; Gerasimov, E.S.; Hlaváčová, J.; et al. Genome of Leptomonas pyrrhocoris: A high-quality reference for monoxenous trypanosomatids and new insights into evolution of Leishmania. Sci. Rep. 2016, 6, 23704. [Google Scholar] [CrossRef]
- Sloan, M.A.; Brooks, K.; Otto, T.D.; Sanders, M.J.; Cotton, J.A.; Ligoxygakis, P. Transcriptional and genomic parallels between the monoxenous parasite Herpetomonas muscarum and Leishmania. PLoS Genet. 2019, 15, e1008452. [Google Scholar] [CrossRef] [PubMed]
- Szöör, B. Trypanosomatid protein phosphatases. Mol. Biochem. Parasitol. 2010, 173, 53–63. [Google Scholar] [CrossRef]
- Soulat, D.; Bogdan, C. Function of macrophage and parasite phosphatases in leishmaniasis. Front. Immunol. 2017, 8, 1838. [Google Scholar] [CrossRef]
- Orr, G.A.; Werner, C.; Xu, J.; Bennett, M.; Weiss, L.M.; Takvorkan, P.; Tanowitz, H.B.; Wittner, M. Identification of novel serine/threonine protein phosphatases in Trypanosoma cruzi: A potential role in control of cytokinesis and morphology. Infect. Immun. 2000, 68, 1350–1358. [Google Scholar] [CrossRef]
- Kraeva, N.; Leštinová, T.; Ishemgulova, A.; Majerová, K.; Butenko, A.; Vaselek, S.; Bespyatykh, J.; Charyyeva, A.; Spitzová, T.; Kostygov, A.Y.; et al. LmxM.22.0250-encoded dual specificity protein/lipid phosphatase impairs Leishmania mexicana virulence in vitro. Pathogens 2019, 8, 241. [Google Scholar] [CrossRef]
- Qureshi, R.; Jakkula, P.; Sagurthi, S.R.; Qureshi, I.A. Protein phosphatase 1 of Leishmania donovani exhibits conserved catalytic residues and pro-inflammatory response. Biochem. Biophys. Res. Commun. 2019, 516, 770–776. [Google Scholar] [CrossRef]
- Kaufer, A.; Stark, D.; Ellis, J. Evolutionary insight into the Trypanosomatidae using alignment-free phylogenomics of the kinetoplast. Pathogens 2019, 8, 157. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, A.; Krieger, M.A. Genomic and phylogenetic evidence of VIPER retrotransposon domestication in trypanosomatids. Mem. Do Inst. Oswaldo Cruz 2016, 111, 765–769. [Google Scholar] [CrossRef]
- Kelly, F.D.; Sanchez, M.A.; Landfear, S.M. Touching the surface: Diverse roles for the flagellar membrane in kinetoplastid parasites. Microbiol. Mol. Biol. Rev. Mmbr. 2020, 84, e00079-19. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Lei, J.; Darby, A.C.; Kadowaki, T. Trypanosomatid parasite dynamically changes the transcriptome during infection and modifies honey bee physiology. Commun. Biol. 2020, 3, 51. [Google Scholar] [CrossRef]
- Rochette, A.; McNicoll, F.; Girard, J.; Breton, M.; Leblanc, E.; Bergeron, M.G.; Papadopoulou, B. Characterization and developmental gene regulation of a large gene family encoding amastin surface proteins in Leishmania spp. Mol. Biochem. Parasitol. 2005, 140, 205–220. [Google Scholar] [CrossRef]
- Myler, P.J.; Lodes, M.J.; Merlin, G.; de Vos, T.; Stuart, K.D. An amplified DNA element in Leishmania encodes potential integral membrane and nucleotide-binding proteins. Mol. Biochem. Parasitol. 1994, 66, 11–20. [Google Scholar] [CrossRef]
- Ravooru, N.; Paul, O.S.; Nagendra, H.G.; Sathyanarayanan, N. Data enabled prediction analysis assigns folate/biopterin transporter (BT1) family to 36 hypothetical membrane proteins in Leishmania donovani. Bioinformation 2019, 15, 697–708. [Google Scholar] [CrossRef]
- Ouellette, M.; Drummelsmith, J.; El-Fadili, A.; Kundig, C.; Richard, D.; Roy, G. Pterin transport and metabolism in Leishmania and related trypanosomatid parasites. Int. J. Parasitol. 2002, 32, 385–398. [Google Scholar] [CrossRef]
- Ouameur, A.A.; Girard, I.; Legare, D.; Ouellette, M. Functional analysis and complex gene rearrangements of the folate/biopterin transporter (FBT) gene family in the protozoan parasite Leishmania. Mol. Biochem. Parasitol. 2008, 162, 155–164. [Google Scholar] [CrossRef]
- Singer, S.J. The structure and insertion of integral proteins in membranes. Annu. Rev. Cell Biol. 1990, 6, 247–296. [Google Scholar] [CrossRef]
- El-Sayed, N.M.; Myler, P.J.; Blandin, G.; Berriman, M.; Crabtree, J.; Aggarwal, G.; Caler, E.; Renauld, H.; Worthey, E.A.; Hertz-Fowler, C.; et al. Comparative genomics of trypanosomatid parasitic protozoa. Science 2005, 309, 404–409. [Google Scholar] [CrossRef] [PubMed]
- Opperdoes, F.R.; Coombs, G.H. Metabolism of Leishmania: Proven and predicted. Trends Parasitol. 2007, 23, 149–158. [Google Scholar] [CrossRef]
- Opperdoes, F.; Michels, P.A. The metabolic repertoire of Leishmania and implications for drug discovery. In Leishmania: After the Genome; Myler, P., Fasel, N., Eds.; Caister Academic Press: Norfolk, UK, 2008; pp. 123–158. [Google Scholar]
- Škodová-Sveráková, I.; Záhonová, K.; Bučková, B.; Füssy, Z.; Yurchenko, V.; Lukeš, J. Catalase and ascorbate peroxidase in euglenozoan protists. Pathogens 2020, 9, 317. [Google Scholar] [CrossRef]
- Kraeva, N.; Horáková, E.; Kostygov, A.; Kořený, L.; Butenko, A.; Yurchenko, V.; Lukeš, J. Catalase in Leishmaniinae: With me or against me? Infect. Genet. Evol. J. Mol. Epidemiol. Evol. Genet. Infect. Dis. 2017, 50, 121–127. [Google Scholar] [CrossRef]
- Turnock, D.C.; Ferguson, M.A. Sugar nucleotide pools of Trypanosoma brucei, Trypanosoma cruzi, and Leishmania major. Eukaryot. Cell 2007, 6, 1450–1463. [Google Scholar] [CrossRef]
- Boitz, J.M.; Yates, P.A.; Kline, C.; Gaur, U.; Wilson, M.E.; Ullman, B.; Roberts, S.C. Leishmania donovani ornithine decarboxylase is indispensable for parasite survival in the mammalian host. Infect. Immun. 2009, 77, 756–763. [Google Scholar] [CrossRef] [PubMed]
- Moreira, D.S.; Xavier, M.V.; Murta, S.M.F. Ascorbate peroxidase overexpression protects Leishmania braziliensis against trivalent antimony effects. Mem. Do Inst. Oswaldo Cruz 2018, 113, e180377. [Google Scholar] [CrossRef] [PubMed]
- Sunter, J.D.; Yanase, R.; Wang, Z.; Catta-Preta, C.M.C.; Moreira-Leite, F.; Myšková, J.; Pružinová, K.; Volf, P.; Mottram, J.C.; Gull, K. Leishmania flagellum attachment zone is critical for flagellar pocket shape, development in the sand fly, and pathogenicity in the host. Proc. Natl. Acad. Sci. USA 2019, 116, 6351–6360. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, S.M.; Russell, D.G.; Kirchhoff, L.V.; Donelson, J.E. A differentially expressed gene family encoding “amastin”, a surface protein of Trypanosoma cruzi amastigotes. J. Biol. Chem. 1994, 269, 20509–20516. [Google Scholar] [CrossRef]
- Wu, Y.; El Fakhry, Y.; Sereno, D.; Tamar, S.; Papadopoulou, B. A new developmentally regulated gene family in Leishmania amastigotes encoding a homolog of amastin surface proteins. Mol. Biochem. Parasitol. 2000, 110, 345–357. [Google Scholar] [CrossRef]
- Coughlin, B.C.; Teixeira, S.M.; Kirchhoff, L.V.; Donelson, J.E. Amastin mRNA abundance in Trypanosoma cruzi is controlled by a 3′-untranslated region position-dependent cis-element and an untranslated region-binding protein. J. Biol. Chem. 2000, 275, 12051–12060. [Google Scholar] [CrossRef]
- Stober, C.B.; Lange, U.G.; Roberts, M.T.; Alcami, A.; Blackwell, J.M. IL-10 from regulatory T cells determines vaccine efficacy in murine Leishmania major infection. J. Immunol. 2005, 175, 2517–2524. [Google Scholar] [CrossRef]
- Ribeiro, P.A.F.; Vale, D.L.; Dias, D.S.; Lage, D.P.; Mendonca, D.V.C.; Ramos, F.F.; Carvalho, L.M.; Carvalho, A.; Steiner, B.T.; Roque, M.C.; et al. Leishmania infantum amastin protein incorporated in distinct adjuvant systems induces protection against visceral leishmaniasis. Cytokine 2020, 129, 155031. [Google Scholar] [CrossRef]
- Pérez-Díaz, L.; Silva, T.C.; Teixeira, S.M. Involvement of an RNA binding protein containing Alba domain in the stage-specific regulation of beta-amastin expression in Trypanosoma cruzi. Mol. Biochem. Parasitol. 2017, 211, 1–8. [Google Scholar] [CrossRef]
- Butenko, A.; Opperdoes, F.R.; Flegontova, O.; Horak, A.; Hampl, V.; Keeling, P.; Gawryluk, R.M.R.; Tikhonenkov, D.; Flegontov, P.; Lukeš, J. Evolution of metabolic capabilities and molecular features of diplonemids, kinetoplastids, and euglenids. BMC Biol. 2020, 18, 23. [Google Scholar] [CrossRef]
- Raymond, F.; Boisvert, S.; Roy, G.; Ritt, J.F.; Legare, D.; Isnard, A.; Stanke, M.; Olivier, M.; Tremblay, M.J.; Papadopoulou, B.; et al. Genome sequencing of the lizard parasite Leishmania tarentolae reveals loss of genes associated to the intracellular stage of human pathogenic species. Nucleic Acids Res. 2012, 40, 1131–1147. [Google Scholar] [CrossRef]
- Wilson, V.; Southgate, B. Lizard Leishmania. In Biology of Kinetoplastida; Lumsden, W., Evans, D.A., Eds.; Academic Press: New York, NY, USA, 1979; pp. 242–268. [Google Scholar]
- Ovezmukhammedov, A.; Saf’ianova, V.M. Taxonomic problems of the Leishmania of reptiles. Parazitologiia 1989, 23, 334–343. (In Russian) [Google Scholar]
- de Paiva, R.M.; Grazielle-Silva, V.; Cardoso, M.S.; Nakagaki, B.N.; Mendonca-Neto, R.P.; Canavaci, A.M.; Souza Melo, N.; Martinelli, P.M.; Fernandes, A.P.; daRocha, W.D.; et al. Amastin knockdown in Leishmania braziliensis affects parasite-macrophage interaction and results in impaired viability of intracellular amastigotes. PLoS Pathog. 2015, 11, e1005296. [Google Scholar] [CrossRef] [PubMed]
- de Menezes, J.P.; Saraiva, E.M.; da Rocha-Azevedo, B. The site of the bite: Leishmania interaction with macrophages, neutrophils and the extracellular matrix in the dermis. Parasites Vectors 2016, 9, 264. [Google Scholar] [CrossRef] [PubMed]
- Pinheiro, L.J.; Paranaiba, L.F.; Alves, A.F.; Parreiras, P.M.; Gontijo, N.F.; Soares, R.P.; Tafuri, W.L. Salivary gland extract modulates the infection of two Leishmania enriettii strains by interfering with macrophage differentiation in the model of Cavia porcellus. Front. Microbiol. 2018, 9, 969. [Google Scholar] [CrossRef] [PubMed]
- Jain, M.; Dole, V.S.; Myler, P.J.; Stuart, K.D.; Madhubala, R. Role of biopterin transporter (BT1) gene on growth and infectivity of Leishmania. Am. J. Biochem. Biotechnol. 2007, 3, 199–206. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Albanaz, A.T.S.; Gerasimov, E.S.; Shaw, J.J.; Sádlová, J.; Lukeš, J.; Volf, P.; Opperdoes, F.R.; Kostygov, A.Y.; Butenko, A.; Yurchenko, V. Genome Analysis of Endotrypanum and Porcisia spp., Closest Phylogenetic Relatives of Leishmania, Highlights the Role of Amastins in Shaping Pathogenicity. Genes 2021, 12, 444. https://doi.org/10.3390/genes12030444
Albanaz ATS, Gerasimov ES, Shaw JJ, Sádlová J, Lukeš J, Volf P, Opperdoes FR, Kostygov AY, Butenko A, Yurchenko V. Genome Analysis of Endotrypanum and Porcisia spp., Closest Phylogenetic Relatives of Leishmania, Highlights the Role of Amastins in Shaping Pathogenicity. Genes. 2021; 12(3):444. https://doi.org/10.3390/genes12030444
Chicago/Turabian StyleAlbanaz, Amanda T. S., Evgeny S. Gerasimov, Jeffrey J. Shaw, Jovana Sádlová, Julius Lukeš, Petr Volf, Fred R. Opperdoes, Alexei Y. Kostygov, Anzhelika Butenko, and Vyacheslav Yurchenko. 2021. "Genome Analysis of Endotrypanum and Porcisia spp., Closest Phylogenetic Relatives of Leishmania, Highlights the Role of Amastins in Shaping Pathogenicity" Genes 12, no. 3: 444. https://doi.org/10.3390/genes12030444
APA StyleAlbanaz, A. T. S., Gerasimov, E. S., Shaw, J. J., Sádlová, J., Lukeš, J., Volf, P., Opperdoes, F. R., Kostygov, A. Y., Butenko, A., & Yurchenko, V. (2021). Genome Analysis of Endotrypanum and Porcisia spp., Closest Phylogenetic Relatives of Leishmania, Highlights the Role of Amastins in Shaping Pathogenicity. Genes, 12(3), 444. https://doi.org/10.3390/genes12030444