
Four New Cases of Hypomyelinating Leukodystrophy Associated with the UFM1 c.-155_-153delTCA Founder Mutation in Pediatric Patients of Roma Descent in Hungary

 , ,
, ,
Abstract
:1. Introduction
2. Patients and Methods
2.1. Patients
2.2. Methods
3. Results
3.1. Case Presentations
3.1.1. Patient 1
3.1.2. Patient 2
3.1.3. Patient 3
3.1.4. Patient 4
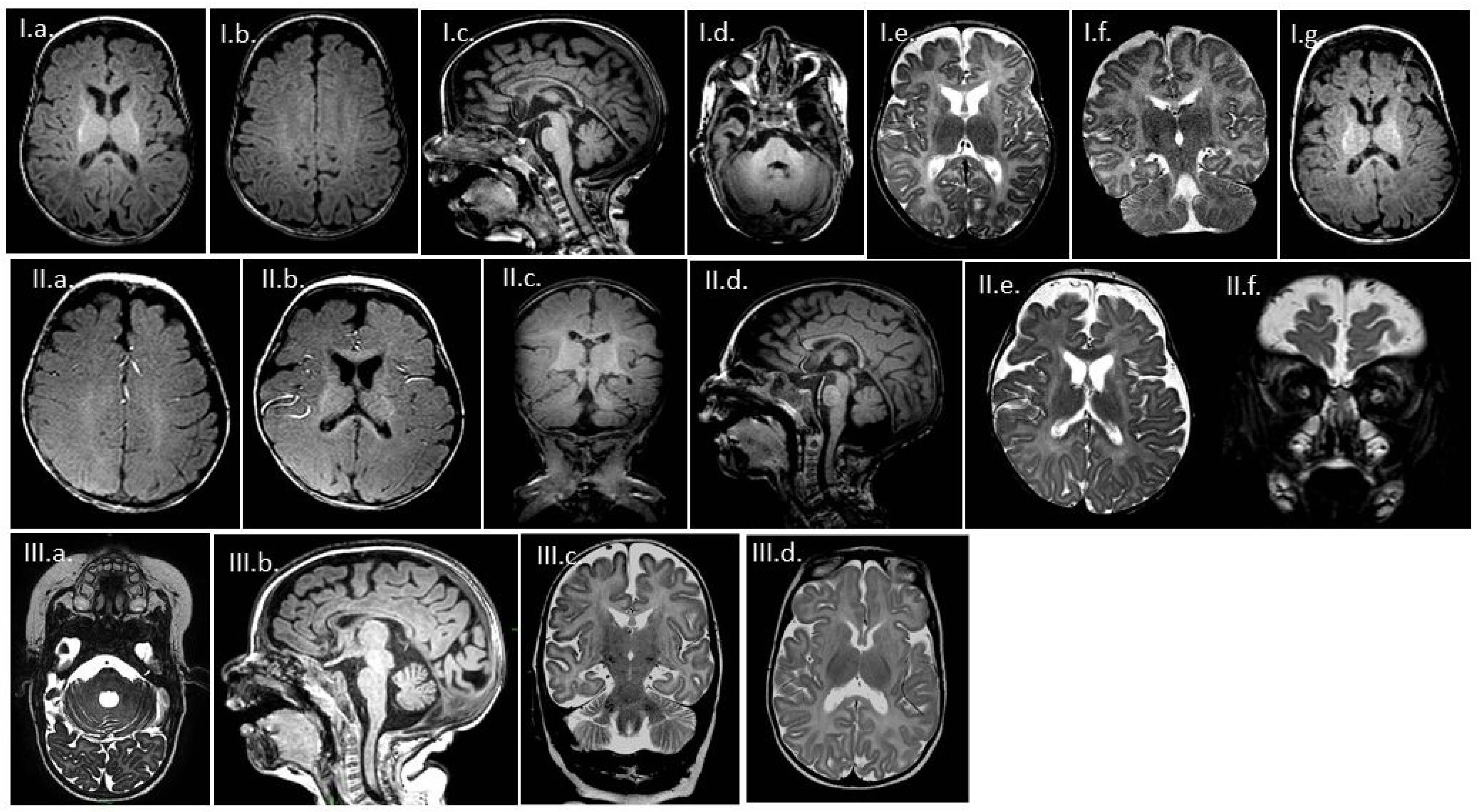
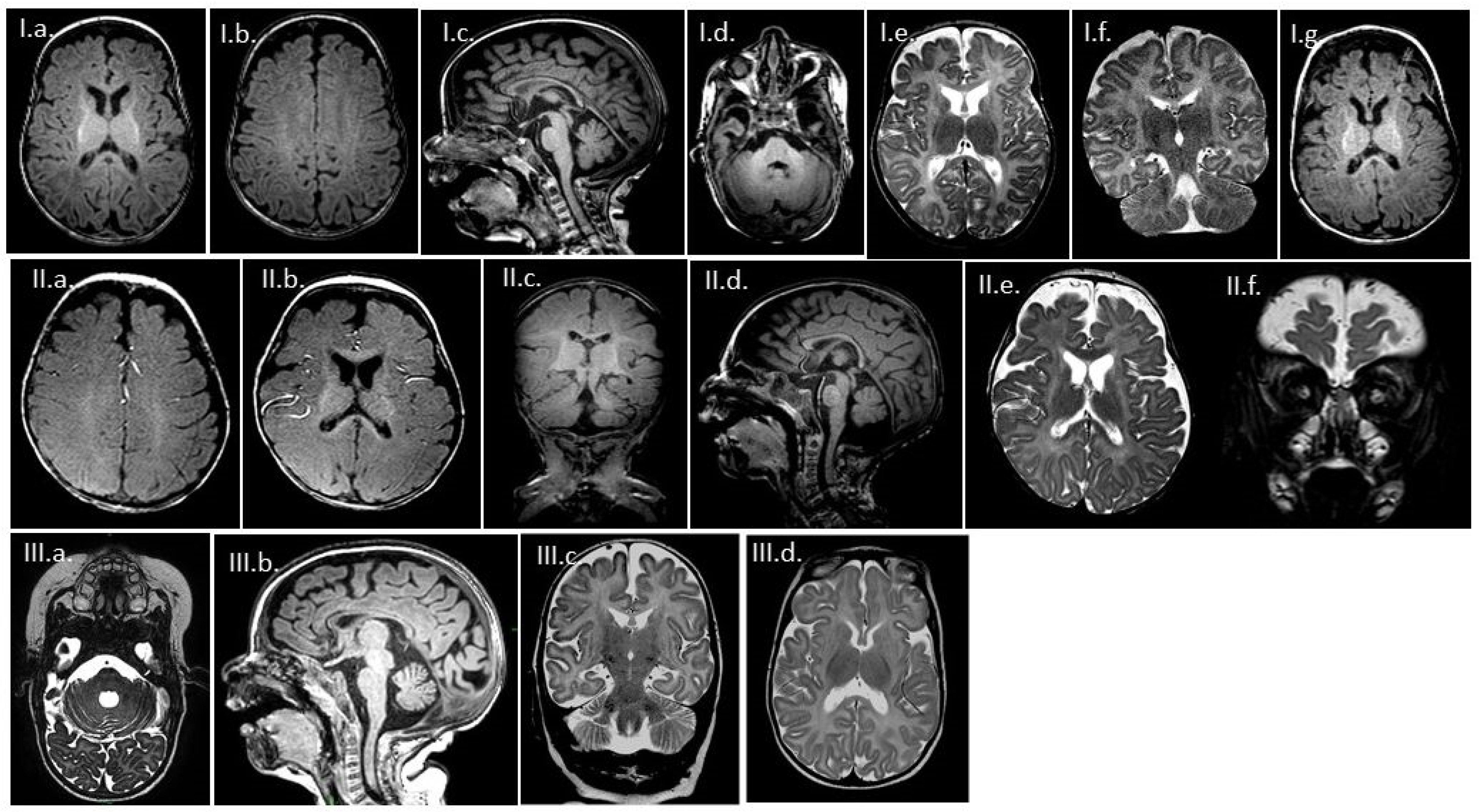
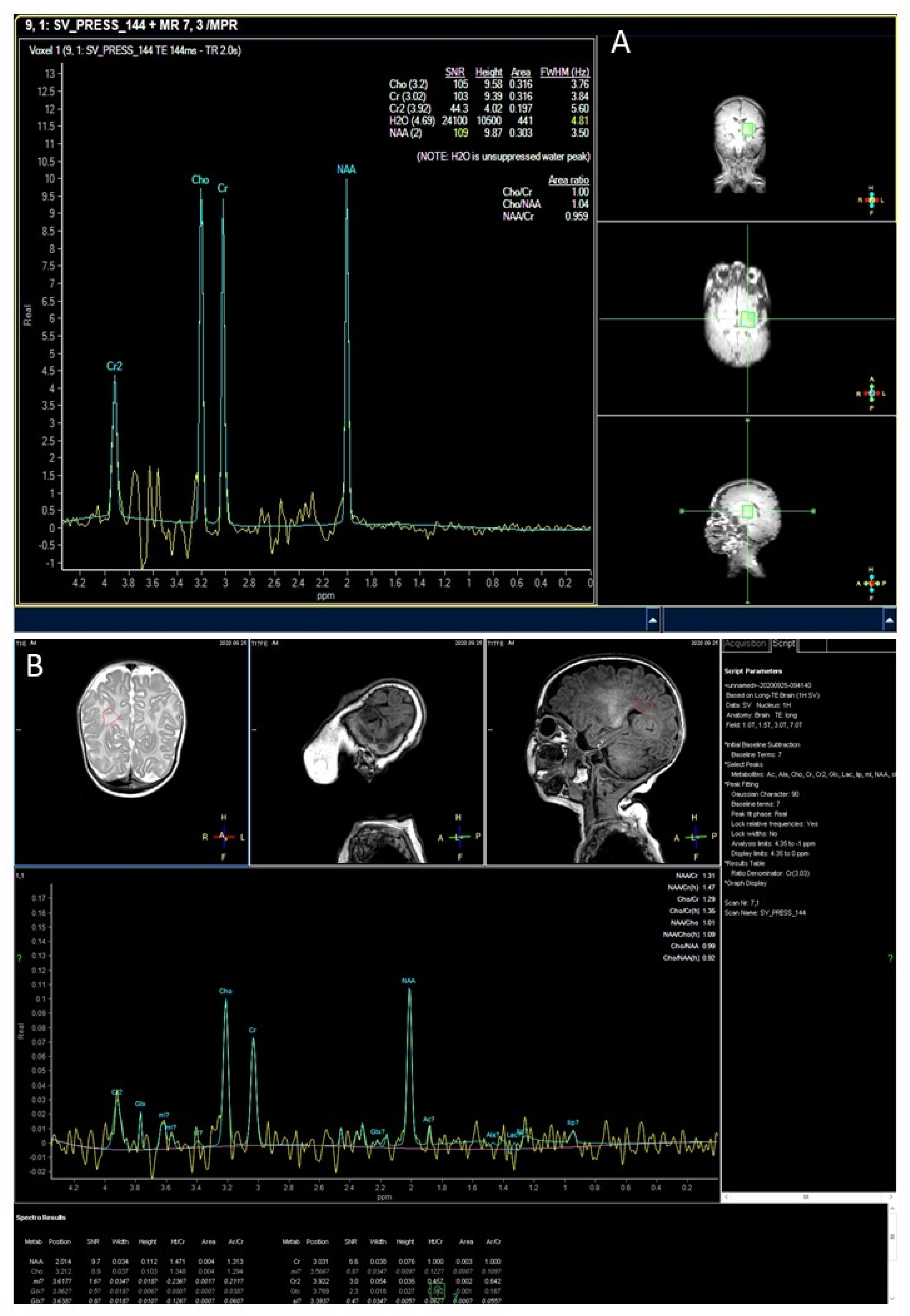
3.2. MR Findings
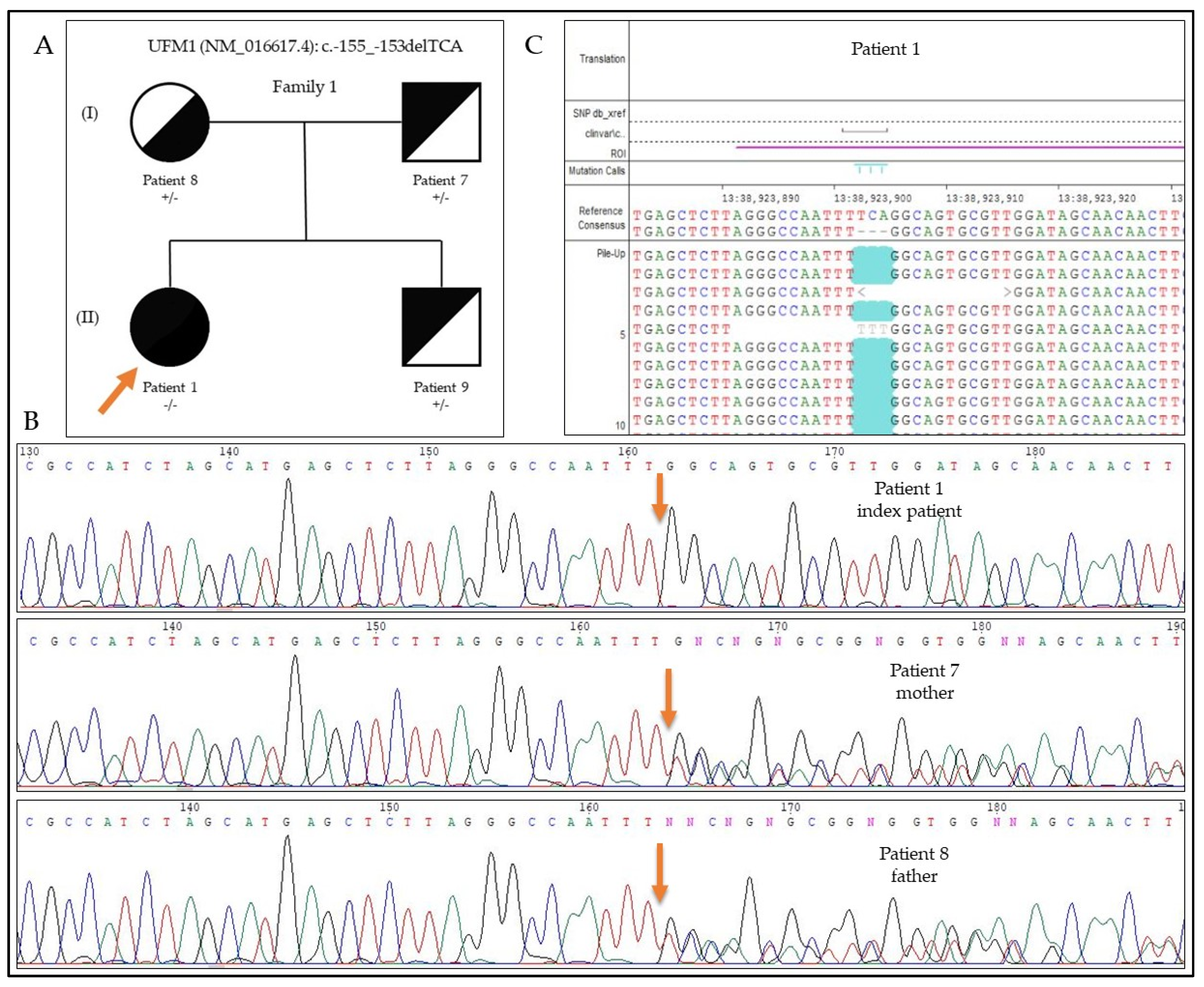
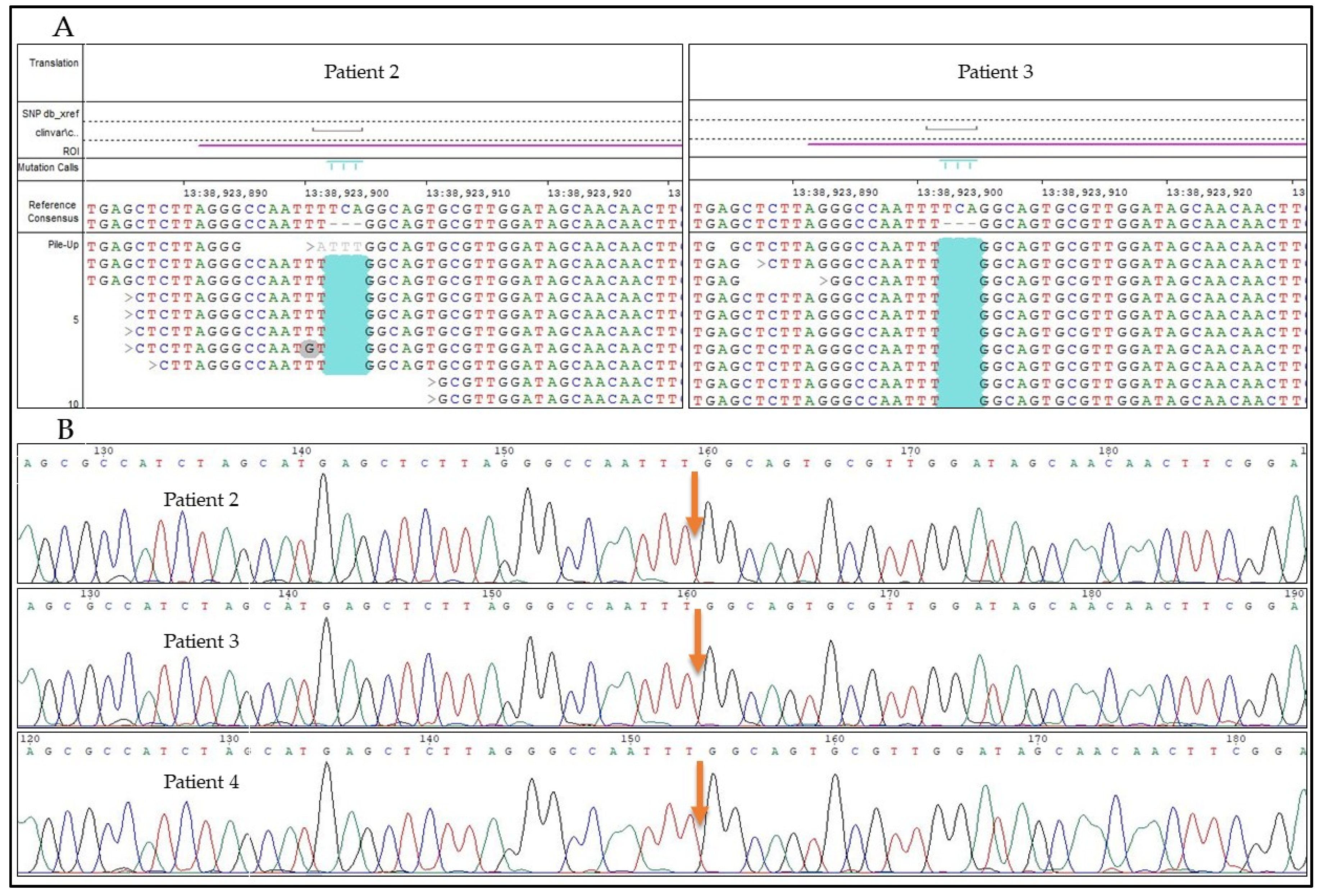
3.3. Results of the Genetic Testing
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wei, Y.; Xu, X. UFMylation: A unique & fashionable modification for life. Genom. Proteom. Bioinform. 2016, 14, 140–146. [Google Scholar] [CrossRef] [Green Version]
- Hershko, A.; Ciechanover, A. The ubiquitin system. Annu. Rev. Biochem. 1998, 67, 425–479. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, M.; Chiba, T.; Tatsumi, K.; Iemura, S.; Tanida, I.; Okazaki, N.; Ueno, T.; Kominami, E.; Natsume, T.; Tanaka, K. A novel protein-conjugating system for Ufm1, a ubiquitin-fold modifier. EMBO J. 2004, 23, 1977–1986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerakis, Y.; Quintero, M.; Li, H.; Hetz, C. The UFMylation system in proteostasis and beyond. Trends Cell Biol. 2019, 29, 974–986. [Google Scholar] [CrossRef] [PubMed]
- Nahorski, M.S.; Maddirevula, S.; Ishimura, R.; Alsahli, S.; Brady, A.F.; Begemann, A.; Mizushima, T.; Guzmán-Vega, F.J.; Obata, M.; Ichimura, Y.; et al. Biallelic UFM1 and UFC1 mutations expand the essential role of ufmylation in brain development. Brain 2018, 141, 1934–1945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inoue, K. PLP1-related inherited dysmyelinating disorders: Pelizaeus-Merzbacher disease and spastic paraplegia type 2. Neurogenetics 2005, 6, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, E.M.C.; Bertini, E.; Kalaydjieva, L.; Morar, B.; Dojčáková, D.; Liu, J.; Vanderver, A.; Curiel, J.; Persoon, C.M.; Diodato, D.; et al. UFM1 founder mutation in the Roma population causes recessive variant of H-ABC. Neurology 2017, 89, 1821–1828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertoli-Avella, A.M.; Beetz, C.; Ameziane, N.; Rocha, M.E.; Guatibonza, P.; Pereira, C.; Calvo, M.; Herrera-Ordonez, N.; Segura-Castel, M.; Diego-Alvarez, D.; et al. Successful application of genome sequencing in a diagnostic setting: 1007 index cases from a clinically heterogeneous cohort. Eur. J. Hum. Genet. 2020, 29, 141–153. [Google Scholar] [CrossRef] [PubMed]
- Cheema, H.; Bertoli-Avella, A.M.; Skrahina, V.; Anjum, M.N.; Waheed, N.; Saeed, A.; Beetz, C.; Perez-Lopez, J.; Rocha, M.E.; Alawbathani, S.; et al. Genomic testing in 1019 individuals from 349 Pakistani families results in high diagnostic yield and clinical utility. NPJ Genom. Med. 2020, 5, 44. [Google Scholar] [CrossRef] [PubMed]
- Bergant, G.; Maver, A.; Peterlin, B. Whole-genome sequencing in diagnostics of selected slovenian undiagnosed patients with rare disorders. Life 2021, 11, 205. [Google Scholar] [CrossRef] [PubMed]
- Richards, C.S.; Bale, S.; Bellissimo, D.B.; Das, S.; Grody, W.W.; Hegde, M.R.; Lyon, E.; Ward, B.E.; Molecular Subcommittee of the ACMG Laboratory Quality Assurance Committee. ACMG recommendations for standards for interpretation and reporting of sequence variations: Revisions 2007. Genet. Med. 2008, 10, 294–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- World Medical Association. World Medical Association declaration of Helsinki: Ethical principles for medical research involving human subjects. JAMA 2013, 310, 2191–2194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Einhorn, Y.; Einhorn, M.; Kamshov, A.; Lev, O.; Trabelsi, A.; Paz-Yaacov, N.; Gross, S.J. Gene-Specific Artificial Intelligence-Based Variant Classification Engine: Results of a Time-Capsule Experiment. 2019. Available online: https://franklin.genoox.com (accessed on 1 August 2021).
- Kugel, H.; Heindel, W.; Roth, B.; Ernst, S.; Lackner, K. Proton MR spectroscopy in infants with cerebral energy deficiency due to hypoxia and metabolic disorders. Acta Radiol. 1998, 39, 701–710. [Google Scholar] [CrossRef] [PubMed]
- Thayyil, S.; Chandrasekaran, M.; Taylor, A.; Bainbridge, A.; Cady, E.B.; Chong, W.K.K.; Murad, S.; Omar, R.Z.; Robertson, N.J. Cerebral magnetic resonance biomarkers in neonatal encephalopathy: A meta-analysis. Pediatrics 2010, 125, e382–e395. [Google Scholar] [CrossRef] [PubMed]
- Joubert, K.; Darvay, S.; Gyenis, G.; Éltető, Ö.; Mag, K.; van’t Hof, M.; Ágfalvi, R. Központi Statisztikai Hivatal Népességtudományi Kutatóintézetének Kutatási Jelentései 83; KSH Népességtudományi Kutatóintézet: Budapest, Hungary, 2006; Volume 83, ISBN 9639597090. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Family | Sample | Zygosity |
|---|---|---|
| F1 | P1 | Homozygous |
| F1 | P7 (mother) | Heterozygous |
| F1 | P8 (father) | Heterozygous |
| F1 | P9 (sibling) | Heterozygous |
| F2 | P2 | Homozygous |
| F3 | P3 | Homozygous |
| F4 | P4 | Homozygous |
| Clinical Features | P1 | P2 | P3 | P4 | Total |
|---|---|---|---|---|---|
| General | |||||
| Gender | F | M | M | F | 1:1 ratio |
| Age of onset/medical attention drawn | 2 weeks | 1 week | 6 months | 3.5 months | Average months 2.5 |
| Growth | |||||
| Failure to thrive | Yes | Yes | Yes | No | 3/4 |
| Short stature | No | Yes | Yes | No | 2/4 |
| Microcephaly | Yes | Yes | Yes | No | 3/4 |
| Central nervous system | |||||
| Global developmental delay (severe) | Yes | Yes | Yes | Yes | 4/4 |
| Intellectual disability (profound) | Yes | Yes | Yes | Yes | 4/4 |
| Axial hypotonia | Yes | N/A | Yes | Yes | 3/4 |
| Spasticity | Yes | Yes | Yes | Yes | 4/4 |
| Regression | Yes | Yes | Yes | Yes | 4/4 |
| Seizures | No | Yes | No | No | 1/4 |
| Abnormal EEG (diffuse cortical dysfunction) | Yes | Yes | Yes | Yes | 4/4 |
| Brain atrophy (cortex/ basal ganglia) | Yes | Yes | Yes | Yes | 4/4 |
| Delayed/absent myelination | Yes | Yes | Yes | Yes | 4/4 |
| Cerebellar hypoplasia | Yes | Yes | Yes | Yes | 4/4 |
| Vegetative functions | |||||
| Feeding difficulty | Yes | Yes | Yes | No | 3/4 |
| Laryngeal stridor | Yes | Yes | Yes | No | 3/4 |
| Bradypnea | Yes | Yes | Yes | No | 3/4 |
| Bradycardia | Yes | No | N/A | No | 1/4 |
| Altered levels of consciousness (somnolence, stupor, irritability) | Yes | Yes | Yes | Yes | 4/4 |
| Age of death (months) | 17 | 38 | 30 | Alive at 17 months | Median survival 28 months |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Szűcs, Z.; Fitala, R.; Nyuzó, Á.R.; Fodor, K.; Czemmel, É.; Vrancsik, N.; Bessenyei, M.; Szabó, T.; Szakszon, K.; Balogh, I. Four New Cases of Hypomyelinating Leukodystrophy Associated with the UFM1 c.-155_-153delTCA Founder Mutation in Pediatric Patients of Roma Descent in Hungary. Genes 2021, 12, 1331. https://doi.org/10.3390/genes12091331
Szűcs Z, Fitala R, Nyuzó ÁR, Fodor K, Czemmel É, Vrancsik N, Bessenyei M, Szabó T, Szakszon K, Balogh I. Four New Cases of Hypomyelinating Leukodystrophy Associated with the UFM1 c.-155_-153delTCA Founder Mutation in Pediatric Patients of Roma Descent in Hungary. Genes. 2021; 12(9):1331. https://doi.org/10.3390/genes12091331
Chicago/Turabian StyleSzűcs, Zsuzsanna, Réka Fitala, Ágnes Renáta Nyuzó, Krisztina Fodor, Éva Czemmel, Nóra Vrancsik, Mónika Bessenyei, Tamás Szabó, Katalin Szakszon, and István Balogh. 2021. "Four New Cases of Hypomyelinating Leukodystrophy Associated with the UFM1 c.-155_-153delTCA Founder Mutation in Pediatric Patients of Roma Descent in Hungary" Genes 12, no. 9: 1331. https://doi.org/10.3390/genes12091331
APA StyleSzűcs, Z., Fitala, R., Nyuzó, Á. R., Fodor, K., Czemmel, É., Vrancsik, N., Bessenyei, M., Szabó, T., Szakszon, K., & Balogh, I. (2021). Four New Cases of Hypomyelinating Leukodystrophy Associated with the UFM1 c.-155_-153delTCA Founder Mutation in Pediatric Patients of Roma Descent in Hungary. Genes, 12(9), 1331. https://doi.org/10.3390/genes12091331