
Alternative Splicing and Hypoxia Puzzle in Alzheimer’s and Parkinson’s Diseases


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
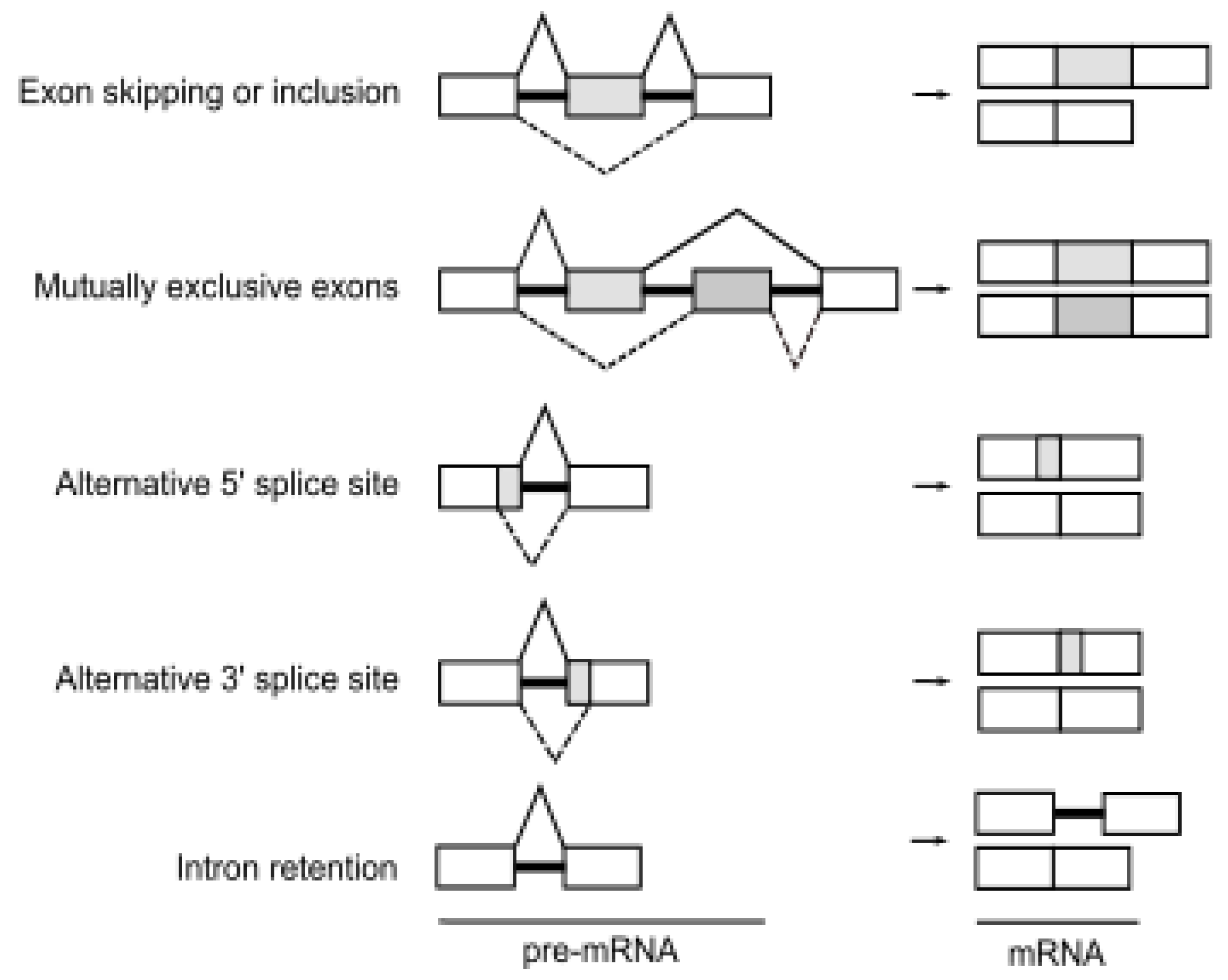
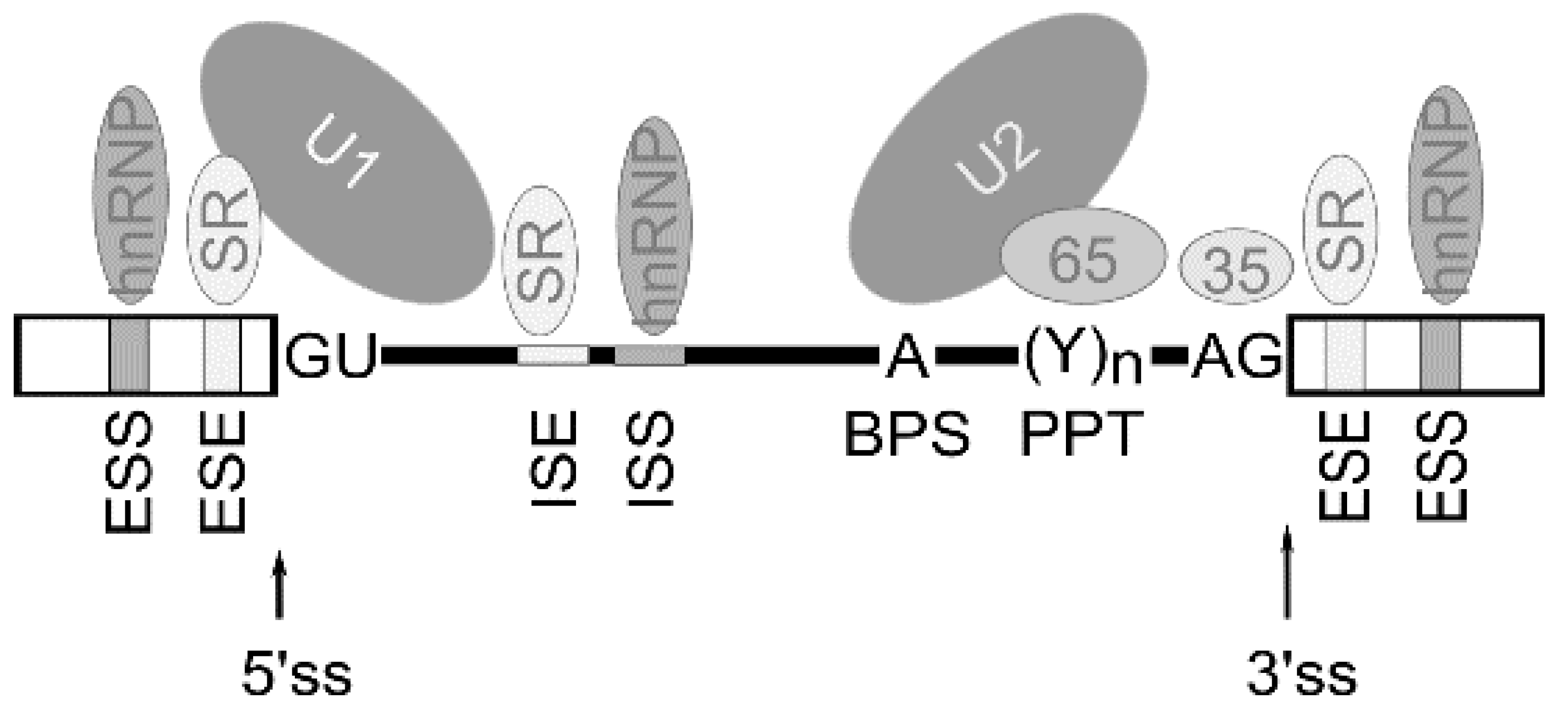
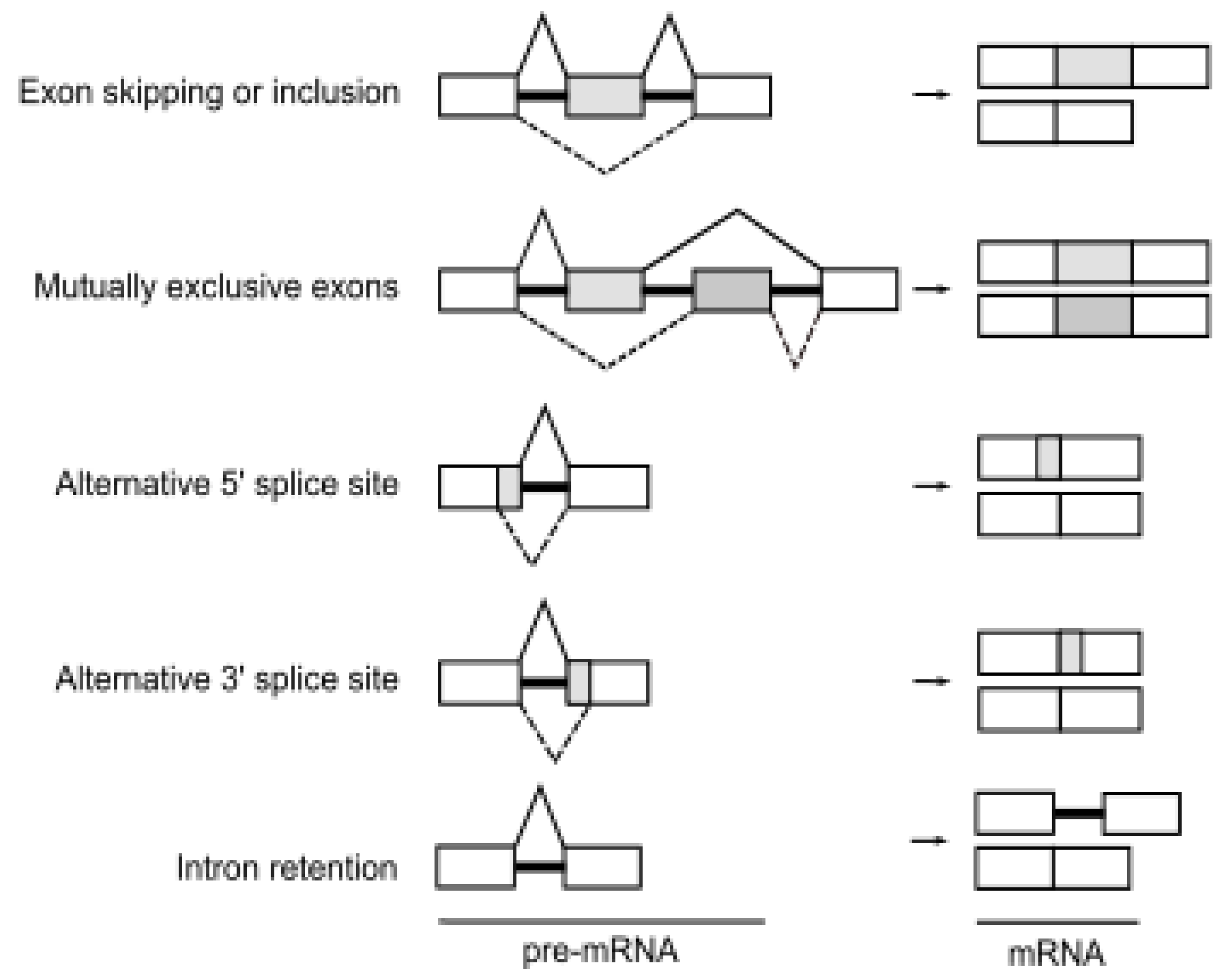
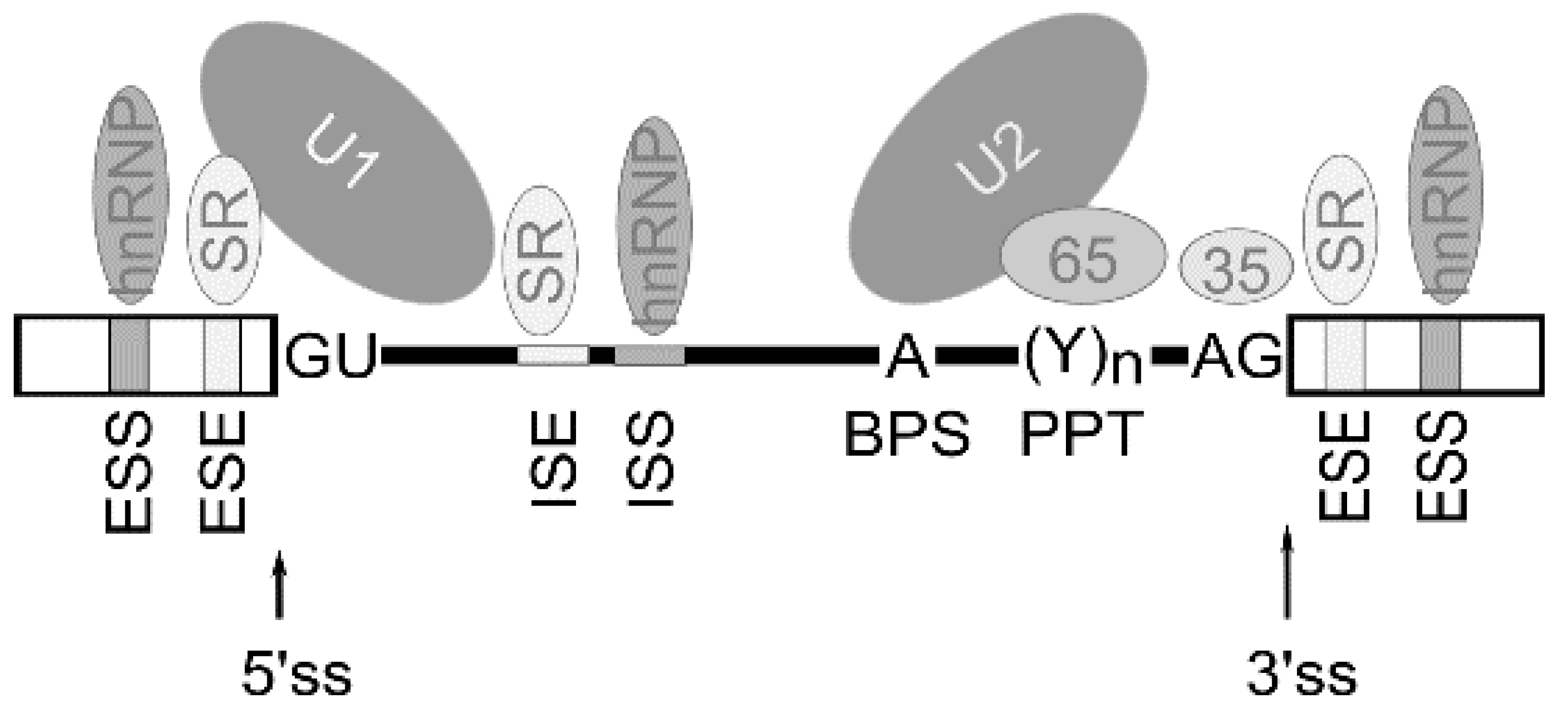
2. Alternative Pre-mRNA Splicing
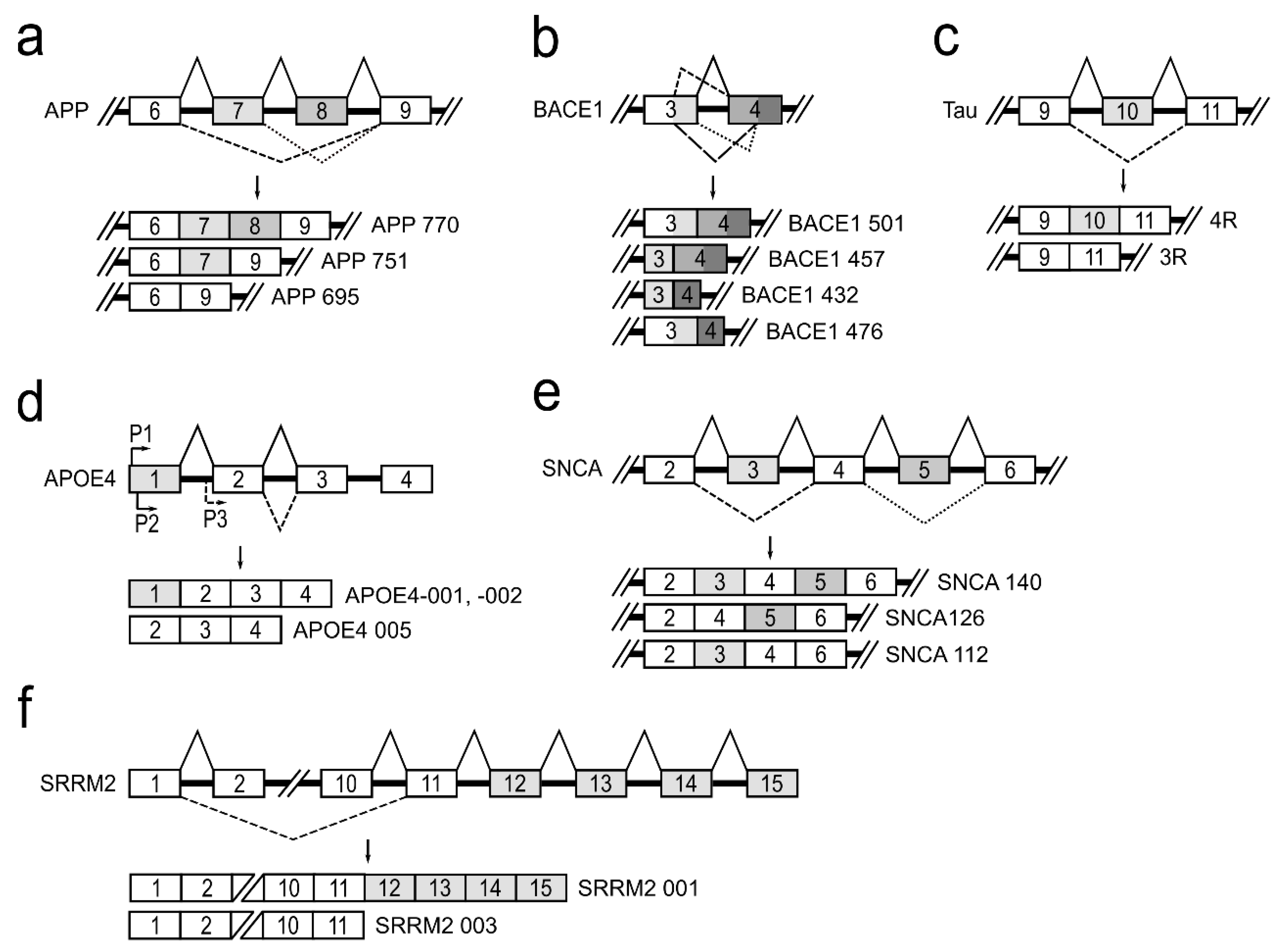
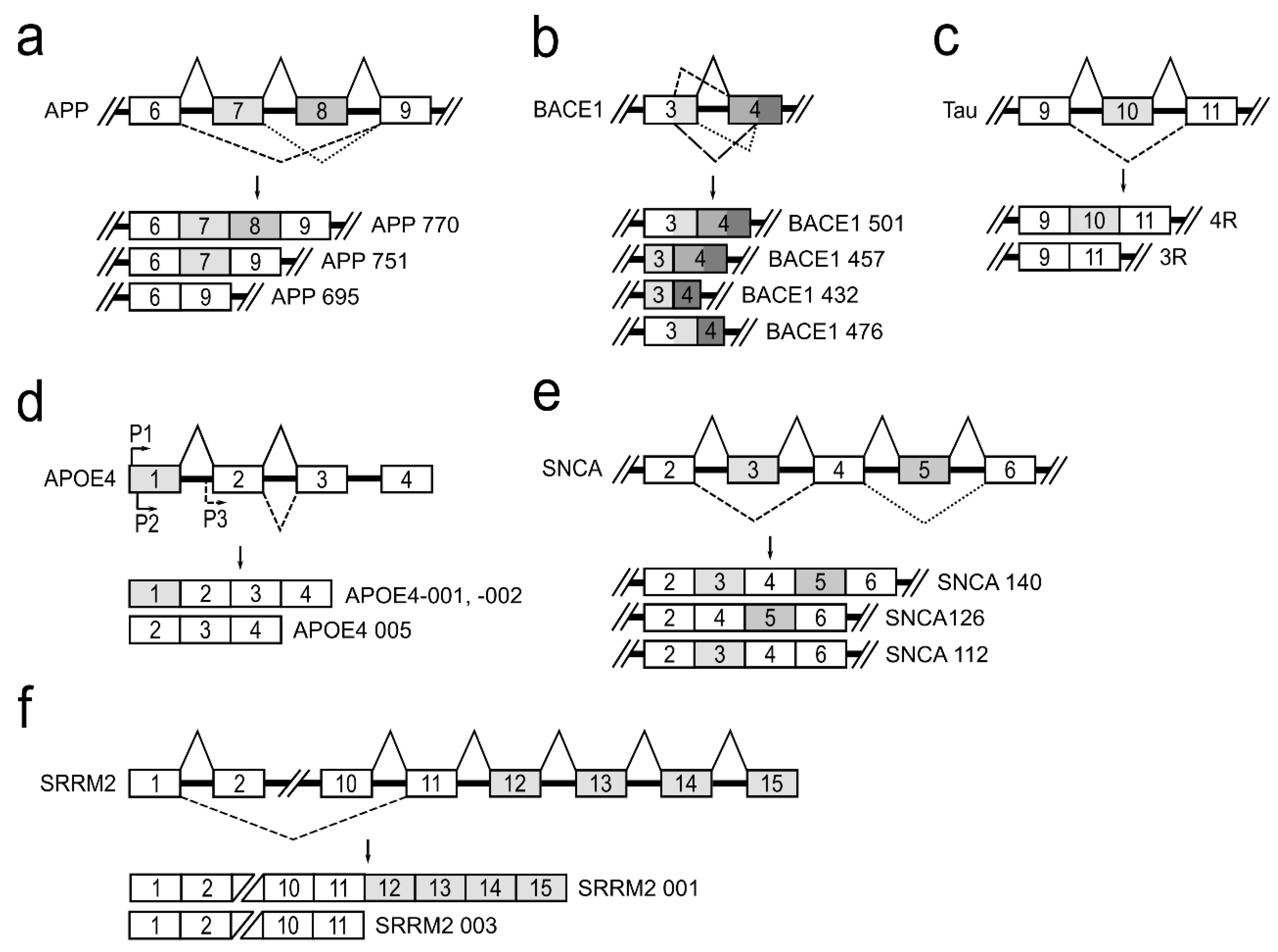
3. Alternative Splicing of Genes Associated with Neurologic Conditions
4. Hypoxia, Splicing, and Neurodegeneration
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Perrone, B.; La Cognata, V.; Sprovieri, T.; Ungaro, C.; Conforti, F.L.; Ando, S.; Cavallaro, S. Alternative Splicing of ALS Genes: Misregulation and Potential Therapies. Cell. Mol. Neurobiol. 2020, 40, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Park, J.W.; Graveley, B.R. Complex alternative splicing. Adv. Exp. Med. Biol. 2007, 623, 50–63. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Liu, J.; Huang, B.O.; Xu, Y.M.; Li, J.; Huang, L.F.; Lin, J.; Zhang, J.; Min, Q.H.; Yang, W.M.; et al. Mechanism of alternative splicing and its regulation. Biomed. Rep. 2015, 3, 152–158. [Google Scholar] [CrossRef] [Green Version]
- Bowler, E.; Oltean, S. Alternative Splicing in Angiogenesis. Int. J. Mol. Sci. 2019, 20, 2067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biamonti, G.; Amato, A.; Belloni, E.; Di Matteo, A.; Infantino, L.; Pradella, D.; Ghigna, C. Alternative splicing in Alzheimer’s disease. Aging Clin. Exp. Res. 2019, 33, 747–758. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Wang, L.; Chen, D.; Li, F. The Function of Pre-mRNA Alternative Splicing in Mammal Spermatogenesis. Int. J. Biol. Sci. 2020, 16, 38–48. [Google Scholar] [CrossRef]
- Nik, S.; Bowman, T.V. Splicing and neurodegeneration: Insights and mechanisms. Wiley Interdiscip. Rev. RNA 2019, 10, e1532. [Google Scholar] [CrossRef]
- Pankratz, N.; Foroud, T. Genetics of Parkinson disease. Genet. Med. Off. J. Am. Coll. Med. Genet. 2007, 9, 801–811. [Google Scholar] [CrossRef] [Green Version]
- Johnson, V.E.; Stewart, W.; Smith, D.H. Traumatic brain injury and amyloid-β pathology: A link to Alzheimer’s disease? Nat. Rev. Neurosci. 2010, 11, 361–370. [Google Scholar] [CrossRef] [Green Version]
- Chin-Chan, M.; Navarro-Yepes, J.; Quintanilla-Vega, B. Environmental pollutants as risk factors for neurodegenerative disorders: Alzheimer and Parkinson diseases. Front. Cell. Neurosci. 2015, 9, 124. [Google Scholar] [CrossRef] [Green Version]
- DeTure, M.A.; Dickson, D.W. The neuropathological diagnosis of Alzheimer’s disease. Mol. Neurodegener. 2019, 14, 32. [Google Scholar] [CrossRef] [Green Version]
- Barker, W.W.; Luis, C.A.; Kashuba, A.; Luis, M.; Harwood, D.G.; Loewenstein, D.; Waters, C.; Jimison, P.; Shepherd, E.; Sevush, S.; et al. Relative frequencies of Alzheimer disease, Lewy body, vascular and frontotemporal dementia, and hippocampal sclerosis in the State of Florida Brain Bank. Alzheimer Dis. Assoc. Disord. 2002, 16, 203–212. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Niu, L.; Li, S.; Le, W. Pathological Impacts of Chronic Hypoxia on Alzheimer’s Disease. ACS Chem. Neurosci. 2019, 10, 902–909. [Google Scholar] [CrossRef]
- Antony, P.M.; Diederich, N.J.; Kruger, R.; Balling, R. The hallmarks of Parkinson’s disease. FEBS J. 2013, 280, 5981–5993. [Google Scholar] [CrossRef] [Green Version]
- Sanders, L.H.; Greenamyre, J.T. Regulation of complex I by Engrailed is complex too. Nat. Neurosci. 2011, 14, 1221–1222. [Google Scholar] [CrossRef] [PubMed]
- Jakubauskiene, E.; Janaviciute, V.; Peciuliene, I.; Soderkvist, P.; Kanopka, A. G/A polymorphism in intronic sequence affects the processing of MAO-B gene in patients with Parkinson disease. FEBS Lett. 2012, 586, 3698–3704. [Google Scholar] [CrossRef] [Green Version]
- Love, J.E.; Hayden, E.J.; Rohn, T.T. Alternative Splicing in Alzheimer’s Disease. J. Parkinsons Dis. Alzheimers Dis. 2015, 2. [Google Scholar] [CrossRef]
- La Cognata, V.; D’Agata, V.; Cavalcanti, F.; Cavallaro, S. Splicing: Is there an alternative contribution to Parkinson’s disease? Neurogenetics 2015, 16, 245–263. [Google Scholar] [CrossRef] [Green Version]
- Will, C.L.; Luhrmann, R. Spliceosome structure and function. Cold Spring Harb. Perspect. Biol. 2011, 3, a003707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakayama, K.; Kataoka, N. Regulation of Gene Expression under Hypoxic Conditions. Int. J. Mol. Sci. 2019, 20, 3278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.K.; Pham, M.H.C.; Ko, K.S.; Rhee, B.D.; Han, J. Alternative splicing isoforms in health and disease. Pflug. Arch. Eur. J. Physiol. 2018, 470, 995–1016. [Google Scholar] [CrossRef] [PubMed]
- Yeo, G.; Holste, D.; Kreiman, G.; Burge, C.B. Variation in alternative splicing across human tissues. Genome Biol. 2004, 5, R74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porter, R.S.; Jaamour, F.; Iwase, S. Neuron-specific alternative splicing of transcriptional machineries: Implications for neurodevelopmental disorders. Mol. Cell Neurosci. 2018, 87, 35–45. [Google Scholar] [CrossRef]
- Tollervey, J.R.; Wang, Z.; Hortobagyi, T.; Witten, J.T.; Zarnack, K.; Kayikci, M.; Clark, T.A.; Schweitzer, A.C.; Rot, G.; Curk, T.; et al. Analysis of alternative splicing associated with aging and neurodegeneration in the human brain. Genome Res. 2011, 21, 1572–1582. [Google Scholar] [CrossRef] [Green Version]
- Ray, P.; Torck, A.; Quigley, L.; Wangzhou, A.; Neiman, M.; Rao, C.; Lam, T.; Kim, J.Y.; Kim, T.H.; Zhang, M.Q.; et al. Comparative transcriptome profiling of the human and mouse dorsal root ganglia: An RNA-seq-based resource for pain and sensory neuroscience research. Pain 2018, 159, 1325–1345. [Google Scholar] [CrossRef]
- Imbimbo, B.P.; Lombard, J.; Pomara, N. Pathophysiology of Alzheimer’s disease. Neuroimaging Clin. N. Am. 2005, 15, 727–753. [Google Scholar] [CrossRef]
- Nalivaeva, N.N.; Turner, A.J. The amyloid precursor protein: A biochemical enigma in brain development, function and disease. FEBS Lett. 2013, 587, 2046–2054. [Google Scholar] [CrossRef] [Green Version]
- Matsui, T.; Ingelsson, M.; Fukumoto, H.; Ramasamy, K.; Kowa, H.; Frosch, M.P.; Irizarry, M.C.; Hyman, B.T. Expression of APP pathway mRNAs and proteins in Alzheimer’s disease. Brain Res. 2007, 1161, 116–123. [Google Scholar] [CrossRef]
- Menendez-Gonzalez, M.; Perez-Pinera, P.; Martinez-Rivera, M.; Calatayud, M.T.; Blazquez Menes, B. APP processing and the APP-KPI domain involvement in the amyloid cascade. Neurodegener. Dis. 2005, 2, 277–283. [Google Scholar] [CrossRef]
- Donev, R.; Newall, A.; Thome, J.; Sheer, D. A role for SC35 and hnRNPA1 in the determination of amyloid precursor protein isoforms. Mol. Psychiatry 2007, 12, 681–690. [Google Scholar] [CrossRef] [PubMed]
- Mills, J.D.; Janitz, M. Alternative splicing of mRNA in the molecular pathology of neurodegenerative diseases. Neurobiol. Aging 2012, 33, 1012.e11–1012.e24. [Google Scholar] [CrossRef]
- Cole, S.L.; Vassar, R. The Basic Biology of BACE1: A Key Therapeutic Target for Alzheimer’s Disease. Curr. Genom. 2007, 8, 509–530. [Google Scholar] [CrossRef] [Green Version]
- Tanahashi, H.; Tabira, T. Three novel alternatively spliced isoforms of the human β-site amyloid precursor protein cleaving enzyme (BACE) and their effect on amyloid β-peptide production. Neurosci. Lett. 2001, 307, 9–12. [Google Scholar] [CrossRef]
- Mowrer, K.R.; Wolfe, M.S. Promotion of BACE1 mRNA alternative splicing reduces amyloid β-peptide production. J. Biol. Chem. 2008, 283, 18694–18701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mowrer, K.R.; Wolfe, M.S. Identification of a cis-acting element involved in the regulation of BACE1 mRNA alternative splicing. J. Neurochem. 2009, 109, 1008–1016. [Google Scholar] [CrossRef] [PubMed]
- Bodendorf, U.; Fischer, F.; Bodian, D.; Multhaup, G.; Paganetti, P. A splice variant of β-secretase deficient in the amyloidogenic processing of the amyloid precursor protein. J. Biol. Chem. 2001, 276, 12019–12023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Annies, M.; Stefani, M.; Hueber, A.; Fischer, F.; Paganetti, P. Splicing of intron 3 of human BACE requires the flanking introns 2 and 4. Biochem. Biophys. Res. Commun. 2009, 388, 434–438. [Google Scholar] [CrossRef] [PubMed]
- Park, S.A.; Ahn, S.I.; Gallo, J.M. Tau mis-splicing in the pathogenesis of neurodegenerative disorders. BMB Rep. 2016, 49, 405–413. [Google Scholar] [CrossRef] [Green Version]
- Goode, B.L.; Chau, M.; Denis, P.E.; Feinstein, S.C. Structural and functional differences between 3-repeat and 4-repeat tau isoforms. Implications for normal tau function and the onset of neurodegenetative disease. J. Biol. Chem. 2000, 275, 38182–38189. [Google Scholar] [CrossRef] [Green Version]
- Niblock, M.; Gallo, J.M. Tau alternative splicing in familial and sporadic tauopathies. Biochem. Soc. Trans. 2012, 40, 677–680. [Google Scholar] [CrossRef]
- Liu, F.; Gong, C.X. Tau exon 10 alternative splicing and tauopathies. Mol. Neurodegener. 2008, 3, 8. [Google Scholar] [CrossRef] [Green Version]
- Andreadis, A. Tau gene alternative splicing: Expression patterns, regulation and modulation of function in normal brain and neurodegenerative diseases. Biochim. Biophys. Acta 2005, 1739, 91–103. [Google Scholar] [CrossRef] [Green Version]
- D’Souza, I.; Schellenberg, G.D. Regulation of tau isoform expression and dementia. BBA-Mol. Basis. Dis. 2005, 1739, 104–115. [Google Scholar] [CrossRef] [Green Version]
- Qian, W.; Liu, F. Regulation of alternative splicing of tau exon 10. Neurosci. Bull. 2014, 30, 367–377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Twine, N.A.; Janitz, K.; Wilkins, M.R.; Janitz, M. Whole transcriptome sequencing reveals gene expression and splicing differences in brain regions affected by Alzheimer’s disease. PLoS ONE 2011, 6, e16266. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.J.; Hauser, M.A.; Scott, W.K.; Martin, E.R.; Booze, M.W.; Qin, X.J.; Walter, J.W.; Nance, M.A.; Hubble, J.P.; Koller, W.C.; et al. Apolipoprotein E controls the risk and age at onset of Parkinson disease. Neurology 2004, 62, 2005–2009. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Walker, D.; Bernardo, A.; Brodbeck, J.; Balestra, M.E.; Huang, Y. Intron-3 retention/splicing controls neuronal expression of apolipoprotein E in the CNS. J. Neurosci. Off. J. Soc. Neurosci. 2008, 28, 1452–1459. [Google Scholar] [CrossRef] [PubMed]
- Rhinn, H.; Fujita, R.; Qiang, L.; Cheng, R.; Lee, J.H.; Abeliovich, A. Integrative genomics identifies APOE epsilon4 effectors in Alzheimer’s disease. Nature 2013, 500, 45–50. [Google Scholar] [CrossRef]
- Mills, J.D.; Sheahan, P.J.; Lai, D.; Kril, J.J.; Janitz, M.; Sutherland, G.T. The alternative splicing of the apolipoprotein E gene is unperturbed in the brains of Alzheimer’s disease patients. Mol. Biol. Rep. 2014, 41, 6365–6376. [Google Scholar] [CrossRef]
- Gamez-Valero, A.; Beyer, K. Alternative Splicing of α- and β-Synuclein Genes Plays Differential Roles in Synucleinopathies. Genes 2018, 9, 63. [Google Scholar] [CrossRef] [Green Version]
- Shehadeh, L.A.; Yu, K.; Wang, L.; Guevara, A.; Singer, C.; Vance, J.; Papapetropoulos, S. SRRM2, a potential blood biomarker revealing high alternative splicing in Parkinson’s disease. PLoS ONE 2010, 5, e9104. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, H.; Xia, D.; Kanekiyo, T.; Kelleher, R.J., III; Shen, J. Familial frontotemporal dementia-associated presenilin-1 c.548G>T mutation causes decreased mRNA expression and reduced presenilin function in knock-in mice. J. Neurosci. Off. J. Soc. Neurosci. 2012, 32, 5085–5096. [Google Scholar] [CrossRef] [Green Version]
- Braggin, J.E.; Bucks, S.A.; Course, M.M.; Smith, C.L.; Sopher, B.; Osnis, L.; Shuey, K.D.; Domoto-Reilly, K.; Caso, C.; Kinoshita, C.; et al. Alternative splicing in a presenilin 2 variant associated with Alzheimer disease. Ann. Clin. Transl. Neurol. 2019, 6, 762–777. [Google Scholar] [CrossRef]
- Luzzi, S.; Colleoni, L.; Corbetta, P.; Baldinelli, S.; Fiori, C.; Girelli, F.; Silvestrini, M.; Caroppo, P.; Giaccone, G.; Tagliavini, F.; et al. Missense mutation in GRN gene affecting RNA splicing and plasma progranulin level in a family affected by frontotemporal lobar degeneration. Neurobiol. Aging 2017, 54, 214.e1–214.e6. [Google Scholar] [CrossRef]
- Maruszak, A.; Safranow, K.; Gustaw, K.; Kijanowska-Haladyna, B.; Jakubowska, K.; Olszewska, M.; Styczynska, M.; Berdynski, M.; Tysarowski, A.; Chlubek, D.; et al. PIN1 gene variants in Alzheimer’s disease. BMC Med. Genet. 2009, 10, 115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Douglas, A.G.; Wood, M.J. RNA splicing: Disease and therapy. Brief. Funct. Genom. 2011, 10, 151–164. [Google Scholar] [CrossRef] [Green Version]
- Peers, C.; Dallas, M.L.; Boycott, H.E.; Scragg, J.L.; Pearson, H.A.; Boyle, J.P. Hypoxia and neurodegeneration. Ann. N. Y. Acad. Sci. 2009, 1177, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. HIF-1: Upstream and downstream of cancer metabolism. Curr. Opin. Genet. Dev. 2010, 20, 51–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merelli, A.; Rodriguez, J.C.G.; Folch, J.; Regueiro, M.R.; Camins, A.; Lazarowski, A. Understanding the Role of Hypoxia Inducible Factor During Neurodegeneration for New Therapeutics Opportunities. Curr. Neuropharmacol. 2018, 16, 1484–1498. [Google Scholar] [CrossRef]
- Kanopka, A. Cell survival: Interplay between hypoxia and pre-mRNA splicing. Exp. Cell Res. 2017, 356, 187–191. [Google Scholar] [CrossRef]
- Semenza, G.L. HIF-1 mediates metabolic responses to intratumoral hypoxia and oncogenic mutations. J. Clin. Investig. 2013, 123, 3664–3671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sena, J.A.; Wang, L.; Heasley, L.E.; Hu, C.J. Hypoxia regulates alternative splicing of HIF and non-HIF target genes. Mol. Cancer Res. MCR 2014, 12, 1233–1243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirschfeld, M.; zur Hausen, A.; Bettendorf, H.; Jager, M.; Stickeler, E. Alternative splicing of Cyr61 is regulated by hypoxia and significantly changed in breast cancer. Cancer Res. 2009, 69, 2082–2090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peciuliene, I.; Vilys, L.; Jakubauskiene, E.; Zaliauskiene, L.; Kanopka, A. Hypoxia alters splicing of the cancer associated Fas gene. Exp. Cell Res. 2019, 380, 29–35. [Google Scholar] [CrossRef]
- Jha, N.K.; Jha, S.K.; Sharma, R.; Kumar, D.; Ambasta, R.K.; Kumar, P. Hypoxia-Induced Signaling Activation in Neurodegenerative Diseases: Targets for New Therapeutic Strategies. J. Alzheimers Dis. JAD 2018, 62, 15–38. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; He, G.; Qing, H.; Zhou, W.; Dobie, F.; Cai, F.; Staufenbiel, M.; Huang, L.E.; Song, W. Hypoxia facilitates Alzheimer’s disease pathogenesis by up-regulating BACE1 gene expression. Proc. Natl. Acad. Sci. USA 2006, 103, 18727–18732. [Google Scholar] [CrossRef] [Green Version]
- Kanda, A.; Ebihara, S.; Arai, H.; Takeda, A.; Sasaki, H. Parkinson’s disease and impaired chemosensitivity to hypoxia. Lancet 2000, 356, 2100. [Google Scholar] [CrossRef]
- Roggla, G.; Weber, W.; Roggla, M. Parkinson’s disease and impaired chemosensitivity to hypoxia. Lancet 2000, 356, 2099. [Google Scholar] [CrossRef]
- Andrzejewski, K.; Jampolska, M.; Zaremba, M.; Joniec-Maciejak, I.; Boguszewski, P.M.; Kaczynska, K. Respiratory pattern and phrenic and hypoglossal nerve activity during normoxia and hypoxia in 6-OHDA-induced bilateral model of Parkinson’s disease. J. Physiol. Sci. 2020, 70, 16. [Google Scholar] [CrossRef] [Green Version]
- Snyder, B.; Shell, B.; Cunningham, J.T.; Cunningham, R.L. Chronic intermittent hypoxia induces oxidative stress and inflammation in brain regions associated with early-stage neurodegeneration. Physiol. Rep. 2017, 5, e13258. [Google Scholar] [CrossRef]
- Kim, S.M.; Kim, H.; Lee, J.S.; Park, K.S.; Jeon, G.S.; Shon, J.; Ahn, S.W.; Kim, S.H.; Lee, K.M.; Sung, J.J.; et al. Intermittent hypoxia can aggravate motor neuronal loss and cognitive dysfunction in ALS mice. PLoS ONE 2013, 8, e81808. [Google Scholar] [CrossRef] [Green Version]
- Guglielmotto, M.; Aragno, M.; Autelli, R.; Giliberto, L.; Novo, E.; Colombatto, S.; Danni, O.; Parola, M.; Smith, M.A.; Perry, G.; et al. The up-regulation of BACE1 mediated by hypoxia and ischemic injury: Role of oxidative stress and HIF1alpha. J. Neurochem. 2009, 108, 1045–1056. [Google Scholar] [CrossRef]
- Jakubauskiene, E.; Vilys, L.; Peciuliene, I.; Kanopka, A. The role of hypoxia on Alzheimer’s disease-related APP and Tau mRNA formation. Gene 2021, 766, 145146. [Google Scholar] [CrossRef] [PubMed]
- Naro, C.; Sette, C. Phosphorylation-mediated regulation of alternative splicing in cancer. Int. J. Cell Biol. 2013, 2013, 151839. [Google Scholar] [CrossRef]
- Aubol, B.E.; Fattet, L.; Adams, J.A. A conserved sequence motif bridges two protein kinases for enhanced phosphorylation and nuclear function of a splicing factor. FEBS J. 2021, 288, 566–581. [Google Scholar] [CrossRef]
- Jakubauskiene, E.; Vilys, L.; Makino, Y.; Poellinger, L.; Kanopka, A. Increased Serine-Arginine (SR) Protein Phosphorylation Changes Pre-mRNA Splicing in Hypoxia. J. Biol. Chem. 2015, 290, 18079–18089. [Google Scholar] [CrossRef] [Green Version]
- Bowler, E.; Porazinski, S.; Uzor, S.; Thibault, P.; Durand, M.; Lapointe, E.; Rouschop, K.M.A.; Hancock, J.; Wilson, I.; Ladomery, M. Hypoxia leads to significant changes in alternative splicing and elevated expression of CLK splice factor kinases in PC3 prostate cancer cells. BMC Cancer 2018, 18, 355. [Google Scholar] [CrossRef] [PubMed]
- Vilys, L.; Peciuliene, I.; Jakubauskiene, E.; Zinkeviciute, R.; Makino, Y.; Kanopka, A. U2AF—Hypoxia-induced fas alternative splicing regulator. Exp. Cell Res. 2021, 399, 112444. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jakubauskienė, E.; Kanopka, A. Alternative Splicing and Hypoxia Puzzle in Alzheimer’s and Parkinson’s Diseases. Genes 2021, 12, 1272. https://doi.org/10.3390/genes12081272
Jakubauskienė E, Kanopka A. Alternative Splicing and Hypoxia Puzzle in Alzheimer’s and Parkinson’s Diseases. Genes. 2021; 12(8):1272. https://doi.org/10.3390/genes12081272
Chicago/Turabian StyleJakubauskienė, Eglė, and Arvydas Kanopka. 2021. "Alternative Splicing and Hypoxia Puzzle in Alzheimer’s and Parkinson’s Diseases" Genes 12, no. 8: 1272. https://doi.org/10.3390/genes12081272
APA StyleJakubauskienė, E., & Kanopka, A. (2021). Alternative Splicing and Hypoxia Puzzle in Alzheimer’s and Parkinson’s Diseases. Genes, 12(8), 1272. https://doi.org/10.3390/genes12081272