
Complex Interactions in Regulation of Haematopoiesis—An Unexplored Iron Mine


Abstract
1. Introduction
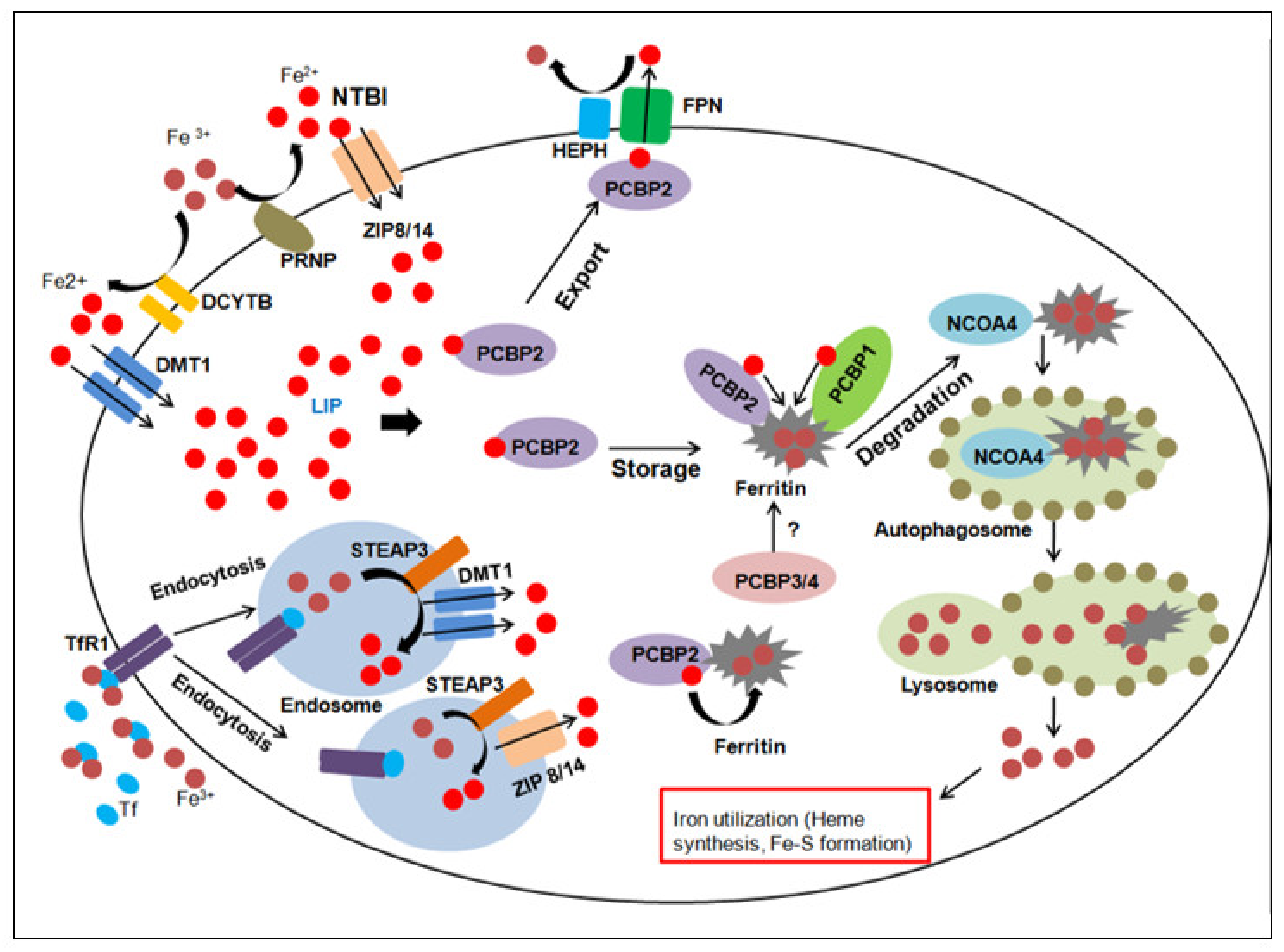
2. Overview of Iron Homeostasis and Metabolism
3. Iron in Haematopoiesis—A Vital Micronutrient
3.1. Role of Iron in Haematopoietic Stem Cell Maintenance and Differentiation
Effects of Iron Deficiency and Overloading on HSC Biology
3.2. Iron in Maintenance of Mesenchymal Stem Cells
3.3. Role of Iron in Erythropoiesis
Effects of Iron Deficiency and Overloading on Erythropoiesis
3.4. Intricate Balance between Iron Levels and Megakaryopoiesis—An Incompletely Understood Phenomenon
3.5. Significance of Iron in Myeloid Lineage
3.5.1. Iron and Macrophage—A Two Way Story
3.5.2. Role of Iron in Regulation of Neutrophil Biology
4. Iron and Immunity
Significance of Iron in Development of the Lymphoid Lineage
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Weissman, I.L. Stem Cells: Units of Development, Units of Regeneration, and Units in Evolution. Cell 2000, 100, 157–168. [Google Scholar] [CrossRef]
- Graf, E.; Mahoney, J.R.; Bryant, R.G.; Eaton, J.W. Iron-Catalyzed Hydroxyl Radical Formation. Stringent Requirement for Free Iron Coordination Site. J. Biol. Chem. 1984, 259, 3620–3624. [Google Scholar] [CrossRef]
- Ye, Z.-W.; Zhang, J.; Townsend, D.M.; Tew, K.D. Oxidative Stress, Redox Regulation and Diseases of Cellular Differentiation. Biochim. Biophys. Acta 2015, 1850, 1607–1621. [Google Scholar] [CrossRef]
- Kulnigg-Dabsch, S.; Schmid, W.; Howaldt, S.; Stein, J.; Mickisch, O.; Waldhör, T.; Evstatiev, R.; Kamali, H.; Volf, I.; Gasche, C. Iron Deficiency Generates Secondary Thrombocytosis and Platelet Activation in IBD: The Randomized, Controlled ThromboVIT Trial. Inflamm. Bowel Dis. 2013, 19, 1609–1616. [Google Scholar] [CrossRef] [PubMed]
- Garrick, M.D.; Dolan, K.G.; Horbinski, C.; Ghio, A.J.; Higgins, D.; Porubcin, M.; Moore, E.G.; Hainsworth, L.N.; Umbreit, J.N.; Conrad, M.E.; et al. DMT1: A Mammalian Transporter for Multiple Metals. Biometals 2003, 16, 41–54. [Google Scholar] [CrossRef]
- McKie, A.T.; Barrow, D.; Latunde-Dada, G.O.; Rolfs, A.; Sager, G.; Mudaly, E.; Mudaly, M.; Richardson, C.; Barlow, D.; Bomford, A.; et al. An Iron-Regulated Ferric Reductase Associated with the Absorption of Dietary Iron. Science 2001, 291, 1755–1759. [Google Scholar] [CrossRef]
- Kruszewski, M. Labile Iron Pool: The Main Determinant of Cellular Response to Oxidative Stress. Mutat. Res./Fundament. Mol. Mech. Mutagenes. 2003, 531, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Addison, G.M.; Beamish, M.R.; Hales, C.N.; Hodgkins, M.; Jacobs, A.; Llewellin, P. An Immunoradiometric Assay for Ferritin in the Serum of Normal Subjects and Patients with Iron Deficiency and Iron Overload. J. Clin. Pathol. 1972, 25, 326–329. [Google Scholar] [CrossRef] [PubMed]
- Donovan, A.; Brownlie, A.; Zhou, Y.; Shepard, J.; Pratt, S.J.; Moynihan, J.; Paw, B.H.; Drejer, A.; Barut, B.; Zapata, A.; et al. Positional Cloning of Zebrafish Ferroportin1 Identifies a Conserved Vertebrate Iron Exporter. Nature 2000, 403, 776–781. [Google Scholar] [CrossRef]
- Vulpe, C.D.; Kuo, Y.-M.; Murphy, T.L.; Cowley, L.; Askwith, C.; Libina, N.; Gitschier, J.; Anderson, G.J. Hephaestin, a Ceruloplasmin Homologue Implicated in Intestinal Iron Transport, Is Defective in the Sla Mouse. Nat. Genet. 1999, 21, 195. [Google Scholar] [CrossRef]
- Cherukuri, S.; Potla, R.; Sarkar, J.; Nurko, S.; Harris, Z.L.; Fox, P.L. Unexpected Role of Ceruloplasmin in Intestinal Iron Absorption. Cell Metab. 2005, 2, 309–319. [Google Scholar] [CrossRef]
- Lambert, L.A. Molecular Evolution of the Transferrin Family and Associated Receptors. Biochim. Biophys. Acta 2012, 1820, 244–255. [Google Scholar] [CrossRef]
- Lambert, L.A.; Mitchell, S.L. Molecular Evolution of the Transferrin Receptor/Glutamate Carboxypeptidase II Family. J. Mol. Evol. 2007, 64, 113–128. [Google Scholar] [CrossRef]
- Patel, M.; Ramavataram, D.V.S.S. Non Transferrin Bound Iron: Nature, Manifestations and Analytical Approaches for Estimation. Indian J. Clin. Biochem. 2012, 27, 322–332. [Google Scholar] [CrossRef]
- Pantopoulos, K. Iron Metabolism and the IRE/IRP Regulatory System: An Update. Ann. N. Y. Acad. Sci. 2004, 1012, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Park, C.H.; Valore, E.V.; Waring, A.J.; Ganz, T. Hepcidin, a Urinary Antimicrobial Peptide Synthesized in the Liver. J. Biol. Chem. 2001, 276, 7806–7810. [Google Scholar] [CrossRef]
- Eide, D.; Broderius, M.; Fett, J.; Guerinot, M.L. A Novel Iron-Regulated Metal Transporter from Plants Identified by Functional Expression in Yeast. Proc. Natl. Acad. Sci. USA 1996, 93, 5624–5628. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, A.K.; Haldar, S.; Qian, J.; Beserra, A.; Suda, S.; Singh, A.; Hopfer, U.; Chen, S.G.; Garrick, M.D.; Turner, J.R.; et al. Prion Protein Functions as a Ferrireductase Partner for ZIP14 and DMT1. Free Radic. Biol. Med. 2015, 84, 322–330. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.-Y.; Jenkitkasemwong, S.; Duarte, S.; Sparkman, B.K.; Shawki, A.; Mackenzie, B.; Knutson, M.D. ZIP8 Is an Iron and Zinc Transporter Whose Cell-Surface Expression Is Up-Regulated by Cellular Iron Loading. J. Biol. Chem. 2012, 287, 34032–34043. [Google Scholar] [CrossRef]
- Fleming, M.D.; Romano, M.A.; Su, M.A.; Garrick, L.M.; Garrick, M.D.; Andrews, N.C. Nramp2 Is Mutated in the Anemic Belgrade (b) Rat: Evidence of a Role for Nramp2 in Endosomal Iron Transport. Proc. Natl. Acad. Sci. USA 1998, 95, 1148–1153. [Google Scholar] [CrossRef]
- Gálvez-Peralta, M.; He, L.; Jorge-Nebert, L.F.; Wang, B.; Miller, M.L.; Eppert, B.L.; Afton, S.; Nebert, D.W. ZIP8 Zinc Transporter: Indispensable Role for Both Multiple-Organ Organogenesis and Hematopoiesis In Utero. PLoS ONE 2012, 7, e36055. [Google Scholar] [CrossRef]
- Shi, H.; Bencze, K.Z.; Stemmler, T.L.; Philpott, C.C. A Cytosolic Iron Chaperone That Delivers Iron to Ferritin. Science 2008, 320, 1207–1210. [Google Scholar] [CrossRef] [PubMed]
- Leidgens, S.; Bullough, K.Z.; Shi, H.; Li, F.; Shakoury-Elizeh, M.; Yabe, T.; Subramanian, P.; Hsu, E.; Natarajan, N.; Nandal, A.; et al. Each Member of the Poly-r(C)-Binding Protein 1 (PCBP) Family Exhibits Iron Chaperone Activity toward Ferritin. J. Biol. Chem. 2013, 288, 17791–17802. [Google Scholar] [CrossRef] [PubMed]
- Yanatori, I.; Yasui, Y.; Tabuchi, M.; Kishi, F. Chaperone Protein Involved in Transmembrane Transport of Iron. Biochem. J. 2014, 462, 25–37. [Google Scholar] [CrossRef] [PubMed]
- Mancias, J.D.; Wang, X.; Gygi, S.P.; Harper, J.W.; Kimmelman, A.C. Quantitative Proteomics Identifies NCOA4 as the Cargo Receptor Mediating Ferritinophagy. Nature 2014, 509, 105. [Google Scholar] [CrossRef] [PubMed]
- Bogdan, A.R.; Miyazawa, M.; Hashimoto, K.; Tsuji, Y. Regulators of Iron Homeostasis: New Players in Metabolism, Cell Death, and Disease. Trends Biochem. Sci. 2016, 41, 274–286. [Google Scholar] [CrossRef] [PubMed]
- Cheshier, S.H.; Morrison, S.J.; Liao, X.; Weissman, I.L. In Vivo Proliferation and Cell Cycle Kinetics of Long-Term Self-Renewing Hematopoietic Stem Cells. Proc. Natl. Acad. Sci. USA 1999, 96, 3120–3125. [Google Scholar] [CrossRef]
- Nakamura, T.; Naguro, I.; Ichijo, H. Iron Homeostasis and Iron-Regulated ROS in Cell Death, Senescence and Human Diseases. Biochim. Biophys. Acta Gen. Subj. 2019, 1863, 1398–1409. [Google Scholar] [CrossRef]
- Pilo, F.; Angelucci, E. A Storm in the Niche: Iron, Oxidative Stress and Haemopoiesis. Blood Rev. 2017. [Google Scholar] [CrossRef] [PubMed]
- Wessling-Resnick, M. Excess Iron: Considerations Related to Development and Early Growth. Am. J. Clin. Nutr. 2017, 106, 1600S–1605S. [Google Scholar] [CrossRef]
- Juntilla, M.M.; Patil, V.D.; Calamito, M.; Joshi, R.P.; Birnbaum, M.J.; Koretzky, G.A. AKT1 and AKT2 Maintain Hematopoietic Stem Cell Function by Regulating Reactive Oxygen Species. Blood 2010, 115, 4030–4038. [Google Scholar] [CrossRef] [PubMed]
- Jang, Y.-Y.; Sharkis, S.J. A Low Level of Reactive Oxygen Species Selects for Primitive Hematopoietic Stem Cells That May Reside in the Low-Oxygenic Niche. Blood 2007, 110, 3056–3063. [Google Scholar] [CrossRef]
- Zhao, R.-Z.; Jiang, S.; Zhang, L.; Yu, Z.-B. Mitochondrial Electron Transport Chain, ROS Generation and Uncoupling (Review). Int. J. Mol. Med. 2019, 44, 3–15. [Google Scholar] [CrossRef]
- Ansó, E.; Weinberg, S.E.; Diebold, L.P.; Thompson, B.J.; Malinge, S.; Schumacker, P.T.; Liu, X.; Zhang, Y.; Shao, Z.; Steadman, M.; et al. The Mitochondrial Respiratory Chain Is Essential for Haematopoietic Stem Cell Function. Nat. Cell Biol. 2017, 19, 614–625. [Google Scholar] [CrossRef]
- Wang, S.; He, X.; Wu, Q.; Jiang, L.; Chen, L.; Yu, Y.; Zhang, P.; Huang, X.; Wang, J.; Ju, Z.; et al. Transferrin Receptor 1-Mediated Iron Uptake Plays an Essential Role in Hematopoiesis. Haematologica 2020, 105, 2071–2082. [Google Scholar] [CrossRef]
- Esposito, B.P.; Breuer, W.; Sirankapracha, P.; Pootrakul, P.; Hershko, C.; Cabantchik, Z.I. Labile Plasma Iron in Iron Overload: Redox Activity and Susceptibility to Chelation. Blood 2003, 102, 2670–2677. [Google Scholar] [CrossRef] [PubMed]
- Angelucci, E.; Cianciulli, P.; Finelli, C.; Mecucci, C.; Voso, M.T.; Tura, S. Unraveling the Mechanisms behind Iron Overload and Ineffective Hematopoiesis in Myelodysplastic Syndromes. Leukemia Res. 2017, 62, 108–115. [Google Scholar] [CrossRef] [PubMed]
- Chai, X.; Li, D.; Cao, X.; Zhang, Y.; Mu, J.; Lu, W.; Xiao, X.; Li, C.; Meng, J.; Chen, J.; et al. ROS-Mediated Iron Overload Injures the Hematopoiesis of Bone Marrow by Damaging Hematopoietic Stem/Progenitor Cells in Mice. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, H.; Espinoza, J.L.; Fujiwara, R.; Rai, S.; Morita, Y.; Ashida, T.; Kanakura, Y.; Matsumura, I. Excessive Reactive Iron Impairs Hematopoiesis by Affecting Both Immature Hematopoietic Cells and Stromal Cells. Cells 2019, 8, 226. [Google Scholar] [CrossRef]
- Ito, K.; Hirao, A.; Arai, F.; Matsuoka, S.; Takubo, K.; Hamaguchi, I.; Nomiyama, K.; Hosokawa, K.; Sakurada, K.; Nakagata, N.; et al. Regulation of Oxidative Stress by ATM Is Required for Self-Renewal of Haematopoietic Stem Cells. Nature 2004, 431, 997–1002. [Google Scholar] [CrossRef] [PubMed]
- Lewandowski, D.; Barroca, V.; Ducongé, F.; Bayer, J.; Van Nhieu, J.T.; Pestourie, C.; Fouchet, P.; Tavitian, B.; Roméo, P.-H. In Vivo Cellular Imaging Pinpoints the Role of Reactive Oxygen Species in the Early Steps of Adult Hematopoietic Reconstitution. Blood 2010, 115, 443–452. [Google Scholar] [CrossRef] [PubMed]
- Kaweme, N.M.; Zhou, S.; Changwe, G.J.; Zhou, F. The Significant Role of Redox System in Myeloid Leukemia: From Pathogenesis to Therapeutic Applications. Biomark. Res. 2020, 8, 63. [Google Scholar] [CrossRef] [PubMed]
- Powell, L.W.; Seckington, R.C.; Deugnier, Y. Haemochromatosis. Lancet 2016, 388, 706–716. [Google Scholar] [CrossRef]
- Sinha, S.; Pereira-Reis, J.; Guerra, A.; Rivella, S.; Duarte, D. The Role of Iron in Benign and Malignant Hematopoiesis. Antioxid. Redox Signal. 2020. [Google Scholar] [CrossRef]
- Friedenstein, A.J.; Piatetzky-Shapiro, I.I.; Petrakova, K.V. Osteogenesis in Transplants of Bone Marrow Cells. J. Embryol. Exp. Morphol. 1966, 16, 381–390. [Google Scholar] [PubMed]
- Lazarus, H.M.; Haynesworth, S.E.; Gerson, S.L.; Rosenthal, N.S.; Caplan, A.I. Ex Vivo Expansion and Subsequent Infusion of Human Bone Marrow-Derived Stromal Progenitor Cells (Mesenchymal Progenitor Cells): Implications for Therapeutic Use. Bone Marrow Transplant. 1995, 16, 557–564. [Google Scholar]
- Pittenger, M.F.; Mackay, A.M.; Beck, S.C.; Jaiswal, R.K.; Douglas, R.; Mosca, J.D.; Moorman, M.A.; Simonetti, D.W.; Craig, S.; Marshak, D.R. Multilineage Potential of Adult Human Mesenchymal Stem Cells. Science 1999, 284, 143–147. [Google Scholar] [CrossRef] [PubMed]
- Mehta, K.J. Role of Iron and Iron-Related Proteins in Mesenchymal Stem Cells: Cellular and Clinical Aspects. J. Cell. Physiol. 2021. [Google Scholar] [CrossRef]
- Borriello, A.; Caldarelli, I.; Speranza, M.C.; Scianguetta, S.; Tramontano, A.; Bencivenga, D.; Stampone, E.; Negri, A.; Nobili, B.; Locatelli, F.; et al. Iron Overload Enhances Human Mesenchymal Stromal Cell Growth and Hampers Matrix Calcification. Biochim. Biophys. Acta 2016, 1860, 1211–1223. [Google Scholar] [CrossRef]
- Yang, F.; Yang, L.; Li, Y.; Yan, G.; Feng, C.; Liu, T.; Gong, R.; Yuan, Y.; Wang, N.; Idiiatullina, E.; et al. Melatonin Protects Bone Marrow Mesenchymal Stem Cells against Iron Overload-Induced Aberrant Differentiation and Senescence. J. Pineal Res. 2017, 63, e12422. [Google Scholar] [CrossRef]
- Park, S.Y.; Jeong, A.-J.; Kim, G.-Y.; Jo, A.; Lee, J.E.; Leem, S.-H.; Yoon, J.-H.; Ye, S.K.; Chung, J.W. Lactoferrin Protects Human Mesenchymal Stem Cells from Oxidative Stress-Induced Senescence and Apoptosis. J. Microbiol. Biotechnol. 2017, 27, 1877–1884. [Google Scholar] [CrossRef] [PubMed]
- Khoshlahni, N.; Mohammadzadeh, M.; Sagha, M. Iron Deficiency Decreases Oxidative Stress and Improves the Viability of the Bone-Marrow-Derived Mesenchymal Stem Cells in Vitro. In Proceedings of the 3rd International Congress of Transfusion Medicine, Evidence-Based Use of Blood Components and Plasma Derived Medicines, Tehran, Iran, 15 December 2015. [Google Scholar]
- Patient, R.K.; McGhee, J.D. The GATA Family (Vertebrates and Invertebrates). Curr. Opin. Genet. Dev. 2002, 12, 416–422. [Google Scholar] [CrossRef]
- Omichinski, J.G.; Trainor, C.; Evans, T.; Gronenborn, A.M.; Clore, G.M.; Felsenfeld, G. A Small Single-“finger” Peptide from the Erythroid Transcription Factor GATA-1 Binds Specifically to DNA as a Zinc or Iron Complex. Proc. Natl. Acad. Sci. USA 1993, 90, 1676–1680. [Google Scholar] [CrossRef]
- Muckenthaler, M.U.; Rivella, S.; Hentze, M.W.; Galy, B. A Red Carpet for Iron Metabolism. Cell 2017, 168, 344–361. [Google Scholar] [CrossRef] [PubMed]
- White, C.; Yuan, X.; Schmidt, P.J.; Bresciani, E.; Samuel, T.K.; Campagna, D.; Hall, C.; Bishop, K.; Calicchio, M.L.; Lapierre, A.; et al. HRG1 Is Essential for Heme Transport from the Phagolysosome of Macrophages during Erythrophagocytosis. Cell Metab. 2013, 17, 261–270. [Google Scholar] [CrossRef]
- Kovtunovych, G.; Ghosh, M.C.; Ollivierre, W.; Weitzel, R.P.; Eckhaus, M.A.; Tisdale, J.F.; Yachie, A.; Rouault, T.A. Wild-Type Macrophages Reverse Disease in Heme Oxygenase 1-Deficient Mice. Blood 2014, 124, 1522–1530. [Google Scholar] [CrossRef] [PubMed]
- Leimberg, M.J.; Prus, E.; Konijn, A.M.; Fibach, E. Macrophages Function as a Ferritin Iron Source for Cultured Human Erythroid Precursors. J. Cell. Biochem. 2008, 103, 1211–1218. [Google Scholar] [CrossRef] [PubMed]
- Camaschella, C.; Nai, A.; Silvestri, L. Iron Metabolism and Iron Disorders Revisited in the Hepcidin Era. Haematologica 2020, 105, 260–272. [Google Scholar] [CrossRef] [PubMed]
- Kim, A.; Nemeth, E. New Insights into Iron Regulation and Erythropoiesis. Curr. Opin. Hematol. 2015, 22, 199–205. [Google Scholar] [CrossRef]
- Myllyharju, J. Prolyl 4-Hydroxylases, Master Regulators of the Hypoxia Response. Acta Physiol. 2013, 208, 148–165. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.L.; Semenza, G.L. Molecular Basis of Hypoxia-Induced Erythropoietin Expression. Curr. Opin. Hematol. 1996, 3, 156–162. [Google Scholar] [CrossRef]
- Warner, M.J.; Kamran, M.T. Iron Deficiency Anemia. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- Pagani, A.; Nai, A.; Silvestri, L.; Camaschella, C. Hepcidin and Anemia: A Tight Relationship. Front. Physiol. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.; Musallam, K.M.; Taher, A.T.; Rivella, S. Ineffective Erythropoiesis, Anemia and Iron Overload. Hematol. Oncol. Clin. 2018, 32, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Nai, A.; Lidonnici, M.R.; Rausa, M.; Mandelli, G.; Pagani, A.; Silvestri, L.; Ferrari, G.; Camaschella, C. The Second Transferrin Receptor Regulates Red Blood Cell Production in Mice. Blood 2015, 125, 1170–1179. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.; Zhao, M.; Rajbhandary, S.; Xie, F.; Chai, X.; Mu, J.; Meng, J.; Liu, Y.; Jiang, Y.; Xu, X.; et al. Free Iron Catalyzes Oxidative Damage to Hematopoietic Cells/Mesenchymal Stem Cells in Vitro and Suppresses Hematopoiesis in Iron Overload Patients. Eur. J. Haematol. 2013, 91, 249–261. [Google Scholar] [CrossRef]
- Song, A.B.; Kuter, D.J.; Al‐Samkari, H. Characterization of the Rate, Predictors, and Thrombotic Complications of Thrombocytosis in Iron Deficiency Anemia. Am. J. Hematol. 2020, 95, 1180–1186. [Google Scholar] [CrossRef]
- Perlman, M.K.; Schwab, J.G.; Nachman, J.B.; Rubin, C.M. Thrombocytopenia in Children with Severe Iron Deficiency. J. Pediatr. Hematol. Oncol. 2002, 24, 380–384. [Google Scholar] [CrossRef] [PubMed]
- Reya, T.; Morrison, S.J.; Clarke, M.F.; Weissman, I.L. Stem Cells, Cancer, and Cancer Stem Cells. Nature 2001, 414, 105. [Google Scholar] [CrossRef]
- Akashi, K.; Traver, D.; Miyamoto, T.; Weissman, I.L. A Clonogenic Common Myeloid Progenitor That Gives Rise to All Myeloid Lineages. Nature 2000, 404, 193. [Google Scholar] [CrossRef]
- Sanjuan-Pla, A.; Macaulay, I.C.; Jensen, C.T.; Woll, P.S.; Luis, T.C.; Mead, A.; Moore, S.; Carella, C.; Matsuoka, S.; Bouriez Jones, T.; et al. Platelet-Biased Stem Cells Reside at the Apex of the Haematopoietic Stem-Cell Hierarchy. Nature 2013, 502, 232–236. [Google Scholar] [CrossRef]
- Jimenez, K.; Khare, V.; Evstatiev, R.; Kulnigg-Dabsch, S.; Jambrich, M.; Strobl, H.; Gasche, C. Increased Expression of HIF2α during Iron Deficiency-Associated Megakaryocytic Differentiation. J. Thromb. Haemost. 2015, 13, 1113–1127. [Google Scholar] [CrossRef]
- Ali, M.; Ali, S.A.; Mansoori, H. Extensive Iron Overload in Bone Marrow: A Cause of Pancytopenia in a Thalassemia Major Patient—A Case Report. Asian J. Transfus. Sci. 2020, 14, 195. [Google Scholar] [CrossRef] [PubMed]
- Dahi, A.A.; Hanafy, E.; Al Pakra, M. Iron Overload and Platelet Function Defects. J. Investig. Med. High Impact Case Rep. 2016, 4. [Google Scholar] [CrossRef]
- Garg, S.K.; Weiner, M.; Karpatkin, S. Thrombocyte and Megathrombocyte Kinetics during Thrombocytosis Induced by Acute and Chronic Blood Loss and by Iron Deficiency Diet. Pathophysiol. Haemostas. Thrombos. 1972, 1, 121–135. [Google Scholar] [CrossRef]
- Ganti, A.K.; Shonka, N.A.; Haire, W.D. Pancytopenia Due to Iron Deficiency Worsened by Iron Infusion: A Case Report. J. Med. Case Rep. 2007, 1, 175. [Google Scholar] [CrossRef] [PubMed]
- Batra, S.; Gupta, A.; Peddint, R. An Unusual Case of Thrombocytopenia. Clin. Pediatr. 2010. [Google Scholar] [CrossRef] [PubMed]
- Cunha, V.; Ferreira, M.; Barosa, R.; Fonseca, A.G.; Delerue, F.; Carvalho, C. Iron-Induced Thrombocytopenia in Severe Iron-Deficiency Anemia. Exp. Rev. Hematol. 2015, 8, 247–251. [Google Scholar] [CrossRef] [PubMed]
- Evstatiev, R.; Bukaty, A.; Jimenez, K.; Kulnigg‐Dabsch, S.; Surman, L.; Schmid, W.; Eferl, R.; Lippert, K.; Scheiber‐Mojdehkar, B.; Kvasnicka, H.M.; et al. Iron Deficiency Alters Megakaryopoiesis and Platelet Phenotype Independent of Thrombopoietin. Am. J. Hematol. 2014, 89, 524–529. [Google Scholar] [CrossRef] [PubMed]
- Beguin, Y.; Loo, M.; R’Zik, S.; Sautois, B.; Lejeune, F.; Rorive, G.; Fillet, G. Effect of Recombinant Human Erythropoietin on Platelets in Patients with Anemia of Renal Failure: Correlation of Platelet Count with Erythropoietic Activity and Iron Parameters. Eur. J. Haematol. 1994, 53, 265–270. [Google Scholar] [CrossRef]
- Broudy, V.C.; Lin, N.L.; Kaushansky, K. Thrombopoietin (c-Mpl Ligand) Acts Synergistically with Erythropoietin, Stem Cell Factor, and Interleukin-11 to Enhance Murine Megakaryocyte Colony Growth and Increases Megakaryocyte Ploidy in Vitro. Blood 1995, 85, 1719–1726. [Google Scholar] [CrossRef] [PubMed]
- Racke, F.K. EPO and TPO Sequences Do Not Explain Thrombocytosis in Iron Deficiency Anemia. J. Pediatr. Hematol. Oncol. 2003, 25, 919. [Google Scholar] [CrossRef]
- Klimchenko, O.; Mori, M.; Distefano, A.; Langlois, T.; Larbret, F.; Lécluse, Y.; Féraud, O.; Vainchenker, W.; Norol, F.; Debili, N. A Common Bipotent Progenitor Generates the Erythroid and Megakaryocyte Lineages in Embryonic Stem Cell-Derived Primitive Hematopoiesis. Blood 2009, 114, 1506–1517. [Google Scholar] [CrossRef]
- Geddis, A.E.; Kaushansky, K. Cross-Reactivity between Erythropoietin and Thrombopoietin at the Level of Mpl Does Not Account for the Thrombocytosis Seen in Iron Deficiency. J. Pediatr. Hematol. Oncol. 2003, 25, 919–920. [Google Scholar] [CrossRef]
- Xavier-Ferrucio, J.; Scanlon, V.; Li, X.; Zhang, P.-X.; Lozovatsky, L.; Ayala-Lopez, N.; Tebaldi, T.; Halene, S.; Cao, C.; Fleming, M.D.; et al. Low Iron Promotes Megakaryocytic Commitment of Megakaryocytic-Erythroid Progenitors in Humans and Mice. Blood 2019, 134, 1547–1557. [Google Scholar] [CrossRef]
- Okabe, Y.; Medzhitov, R. Tissue-Specific Signals Control Reversible Program of Localization and Functional Polarization of Macrophages. Cell 2014, 157, 832–844. [Google Scholar] [CrossRef] [PubMed]
- Im, D.D.; Ho, C.H.; Chan, R.Y. Hypersegmented Neutrophils in an Adolescent Male With Heatstroke. J. Pediatr. Hematol. Oncol. 2015, 37, 488. [Google Scholar] [CrossRef] [PubMed]
- Berrak, S.G.; Angaji, M.; Turkkan, E.; Canpolat, C.; Timur, C.; Eksioglu-Demiralp, E. The Effects of Iron Deficiency on Neutrophil/Monocyte Apoptosis in Children. Cell Prolif. 2007, 40, 741–754. [Google Scholar] [CrossRef] [PubMed]
- Klei, T.R.L.; Meinderts, S.M.; van den Berg, T.K.; van Bruggen, R. From the Cradle to the Grave: The Role of Macrophages in Erythropoiesis and Erythrophagocytosis. Front. Immunol. 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Korolnek, T.; Hamza, I. Macrophages and Iron Trafficking at the Birth and Death of Red Cells. Blood 2015, 125, 2893–2897. [Google Scholar] [CrossRef] [PubMed]
- Gozzelino, R.; Jeney, V.; Soares, M.P. Mechanisms of Cell Protection by Heme Oxygenase-1. Annu. Rev. Pharmacol. Toxicol. 2010, 50, 323–354. [Google Scholar] [CrossRef] [PubMed]
- Haldar, M.; Kohyama, M.; So, A.Y.-L.; Kc, W.; Wu, X.; Briseño, C.G.; Satpathy, A.T.; Kretzer, N.M.; Arase, H.; Rajasekaran, N.S.; et al. Heme-Mediated SPI-C Induction Promotes Monocyte Differentiation into Iron-Recycling Macrophages. Cell 2014, 156, 1223–1234. [Google Scholar] [CrossRef]
- Winn, N.C.; Volk, K.M.; Hasty, A.H. Regulation of Tissue Iron Homeostasis: The Macrophage “Ferrostat”. JCI Insight 2020, 5. [Google Scholar] [CrossRef] [PubMed]
- Cairo, G.; Recalcati, S.; Mantovani, A.; Locati, M. Iron Trafficking and Metabolism in Macrophages: Contribution to the Polarized Phenotype. Trends Immunol. 2011, 32, 241–247. [Google Scholar] [CrossRef]
- Hubler, M.J.; Erikson, K.M.; Kennedy, A.J.; Hasty, A.H. MFehi Adipose Tissue Macrophages Compensate for Tissue Iron Perturbations in Mice. Am. J. Physiol. Cell Physiol. 2018, 315, C319–C329. [Google Scholar] [CrossRef] [PubMed]
- Cai, J.; Zhang, M.; Liu, Y.; Li, H.; Shang, L.; Xu, T.; Chen, Z.; Wang, F.; Qiao, T.; Li, K. Iron Accumulation in Macrophages Promotes the Formation of Foam Cells and Development of Atherosclerosis. Cell Biosci. 2020, 10, 137. [Google Scholar] [CrossRef]
- Reed, J.C. Cytochrome c: Can’t Live with It—Can’t Live without It. Cell 1997, 91, 559–562. [Google Scholar] [CrossRef]
- Abdelmahmuod, E.A.; Yassin, M.A. Iron Deficiency Anemia-Induced Lymphocytopenia in a Young Female. Case Rep. Oncol. 2020, 13, 793–797. [Google Scholar] [CrossRef] [PubMed]
- Renassia, C.; Louis, S.; Cuvellier, S.; Boussetta, N.; Deschemin, J.-C.; Borderie, D.; Bailly, K.; Poupon, J.; Dang, P.M.-C.; El-Benna, J.; et al. Neutrophils from Hereditary Hemochromatosis Patients Are Protected from Iron Excess and Are Primed. Blood Adv. 2020, 4, 3853–3863. [Google Scholar] [CrossRef]
- Cronin, S.J.F.; Woolf, C.J.; Weiss, G.; Penninger, J.M. The Role of Iron Regulation in Immunometabolism and Immune-Related Disease. Front. Mol. Biosci. 2019, 6. [Google Scholar] [CrossRef]
- Xiong, S.; She, H.; Tsukamoto, H. Signaling Role of Iron in NF-Kappa B Activation in Hepatic Macrophages. Comp. Hepatol. 2004, 3, S36. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Shinkai, Y.; Rathbun, G.; Lam, K.-P.; Oltz, E.M.; Stewart, V.; Mendelsohn, M.; Charron, J.; Datta, M.; Young, F.; Stall, A.M.; et al. RAG-2-Deficient Mice Lack Mature Lymphocytes Owing to Inability to Initiate V(D)J Rearrangement. Cell 1992, 68, 855–867. [Google Scholar] [CrossRef]
- Ned, R.M.; Swat, W.; Andrews, N.C. Transferrin Receptor 1 Is Differentially Required in Lymphocyte Development. Blood 2003, 102, 3711–3718. [Google Scholar] [CrossRef] [PubMed]
- Hamill, R.L.; Woods, J.C.; Cook, B.A. Congenital Atransferrinemia. A Case Report and Review of the Literature. Am. J. Clin. Pathol. 1991, 96, 215–218. [Google Scholar] [CrossRef] [PubMed]
- Pinto, J.P.; Arezes, J.; Dias, V.; Oliveira, S.; Vieira, I.; Costa, M.; Vos, M.; Carlsson, A.; Rikers, Y.; Rangel, M.; et al. Physiological Implications of NTBI Uptake by T Lymphocytes. Front. Pharmacol. 2014, 5. [Google Scholar] [CrossRef]
- Kemp, J.D.; Thorson, J.A.; Gomez, F.; Smith, K.M.; Cowdery, J.S.; Ballas, Z.K. Inhibition of Lymphocyte Activation with Anti-Transferrin Receptor Mabs: A Comparison of Three Reagents and Further Studies of Their Range of Effects and Mechanism of Action. Cell. Immunol. 1989, 122, 218–230. [Google Scholar] [CrossRef]
- Li, G.; Pone, E.J.; Tran, D.C.; Patel, P.J.; Dao, L.; Xu, Z.; Casali, P. Iron Inhibits Activation-Induced Cytidine Deaminase Enzymatic Activity and Modulates Immunoglobulin Class Switch DNA Recombination. J. Biol. Chem. 2012, 287, 21520–21529. [Google Scholar] [CrossRef] [PubMed]
- Kohli, R.M.; Maul, R.W.; Guminski, A.F.; McClure, R.L.; Gajula, K.S.; Saribasak, H.; McMahon, M.A.; Siliciano, R.F.; Gearhart, P.J.; Stivers, J.T. Local Sequence Targeting in the AID/APOBEC Family Differentially Impacts Retroviral Restriction and Antibody Diversification. J. Biol. Chem. 2010, 285, 40956–40964. [Google Scholar] [CrossRef]
- Jiang, Y.; Li, C.; Wu, Q.; An, P.; Huang, L.; Wang, J.; Chen, C.; Chen, X.; Zhang, F.; Ma, L.; et al. Iron-Dependent Histone 3 Lysine 9 Demethylation Controls B Cell Proliferation and Humoral Immune Responses. Nat. Commun. 2019, 10, 2935. [Google Scholar] [CrossRef]
- Vanoaica, L.; Richman, L.; Jaworski, M.; Darshan, D.; Luther, S.A.; Kühn, L.C. Conditional Deletion of Ferritin H in Mice Reduces B and T Lymphocyte Populations. PLoS ONE 2014, 9, e89270. [Google Scholar] [CrossRef]
- Darshan, D.; Vanoaica, L.; Richman, L.; Beermann, F.; Kühn, L.C. Conditional Deletion of Ferritin H in Mice Induces Loss of Iron Storage and Liver Damage. Hepatology 2009, 50, 852–860. [Google Scholar] [CrossRef]
- Anani, M.M.; Omar, H.H.; El-kelani, A.; Hashem, A.A. Impact of Iron Deficiency Anemia on CD4 and CD8-T Lymphocytes among Preschool-School Children. Comp. Clin. Pathol. 2017, 26, 1063–1068. [Google Scholar] [CrossRef]
- Dal-Bianco, A.; Grabner, G.; Kronnerwetter, C.; Weber, M.; Höftberger, R.; Berger, T.; Auff, E.; Leutmezer, F.; Trattnig, S.; Lassmann, H.; et al. Slow Expansion of Multiple Sclerosis Iron Rim Lesions: Pathology and 7 T Magnetic Resonance Imaging. Acta Neuropathol. 2017, 133, 25–42. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Zhou, Y.; He, H.; Chen, W.; Lenahan, C.; Li, X.; Deng, Y.; Shao, A.; Huang, J. Crosstalk between Macrophages, T Cells, and Iron Metabolism in Tumor Microenvironment. Oxid. Med. Cell Longev. 2021, 2021. [Google Scholar] [CrossRef]


{kind=link}
| Affected Cells | Physiological Effects | Disease/Health Effects | Ref |
|---|---|---|---|
| HSCs | ROS production causes injury and apoptosis | Aberrant ROS levels observed in malignancies such as MDS, AML. | [38,39,41,42] |
| Reduced proliferation, self renewal and apoptosis. | Iron overloading due to reduced expression of FBXL5; may contribute to disease pathology in MDS patients. | [44] | |
| HSPCs | Elevated ROS levels induces increased expression of p38 MAPK | Observed in MDS patients, affected with iron overloading. Progression to AML may occur. | [42] |
| BM-MSCs | ROS production may promote cell cycle arrest and inhibit proliferation. | Differentiation into osteoblasts affected, resulting in low bone mineral density. This may account for haematopoietic niche defects and dysfunctions in iron overloading syndromes. | [48,49] |
| Alternatively, increased expression of cyclin proteins and activation of MAPK signalling may promote proliferation. | [49] | ||
| Osteoblastic commitment and matrix calcification hindered. | [49] | ||
| Erythrocytes | Impaired hepcidin synthesis results in iron overloading, in response to chronic stress/ineffective erythropoiesis. | Observed in patients suffering from β thalassemia | [65] |
| Increased apoptosis of HSCs (by ROS-induced activation of p53) and reduced erythroblast differentiation due to oxidative stress, observed in BM cells of patients affected with iron overloading. | Detrimental to normal haematopoiesis, leads to anaemia. | [67] | |
| Megakaryocytes | Iron overloading due to chronic blood transfusions affects platelet functions. | Dysfunctional platelets observed in patients affected with Diamond-Blackfan anaemia. | [75] |
| Platelet counts affected in patient suffering from β thalassemia major | Pancytopenia, including thrombocytopenia observed in the patient. | [74] | |
| Macrophages | Storage of excess intracellular iron in ferritin renders macrophages bactericidal. | These macrophages are polarised towards a pro-inflammatory phenotype. | [95] |
| Macrophage iron accumulation in Fpn-deficient mice models increases ROS production and systemic inflammation. | These changes may be responsible for progression of atherosclerosis. | [97] | |
| Neutrophils | Iron overloading severely affects their phagocytic and bactericidal activities. | Observed in β thalassemia major and patients with chronically transfused haemodialysis. | [100] |
| Absence of accumulation of intracellular iron in certain diseases protects neutrophils and primes them towards phagocytosis. | Observed in mice models of HH and affected patients. | [100] | |
| Lymphocytes | T cell counts affected in patients suffering from iron overloading syndromes | CD8+ T cells decline in HH patients while CD4+ T cells decline and CD8+ T cells increase in patients affected with β thalassemia major. | [101] |
| Iron deposition has been associated with neurodegeneration, inflammation, abnormal cell proliferation and tumour metastasis. | Hallmark of neuroinflammatory diseases such as MS and multiple types of cancers, where host immune responses are aberrant/compromised. | [114,115] |
| Affected Cells | Physiological Effects | Disease/Health Effects | Ref |
|---|---|---|---|
| HSCs | Iron-deficient Tfr1 knockout mice display impaired differentiation of most lineages. | Cellular iron deficiency attenuates lineage commitment and regeneration potential of HSCs, leading to postnatal lethality. | [35] |
| HSPCs | Loss of Tfr1 dose not seem to be indispensable for production of HSPCs in mice fetal liver. | Cellular iron deficiency in Tfr1 knockout mice affects differentiation and regenerative capacity of HSPCs. | [35] |
| BM-MSCs | Iron deficiency induced by chelators like DFO protects BM-MScs from oxidative stress and increases viability. | [52] | |
| Iron chelators may protect BM-MSCs from toxicity by decreasing levels of ROS and inhibiting certain intracellular signaling pathways (p38 MAPK) | [48] | ||
| Mitochondrial fragmentation in iron overloaded BM-MSCs may be reduced by iron chelators such as DFO and NAC. | Observed in MDS patients. | [48] | |
| Erythrocytes | Hypochromic, microcytic erythrocytes typically observed in IDA patients | IDA accounts for 50% of the global burden of anemia. | [63] |
| Aberrant overexpression of hepcidin despite low iron stores and serum iron levels, observed in certain anemias. | Leads to iron restricted erythropoiesis in patients affected with IRIDA and anemia of inflammation, respectively | [64] | |
| Differentiation and maturation of erythroid progenitors are inhibited in presence of iron deficiency, in an EPO responsive manner | Development of anemia occurs in iron deprived mice, which is reversed by application of isocitrate. | [59] | |
| Megakaryocytes | Increase in megakaryocytic progenitor populations, greater states of ploidy proplatelet like structures reported in in vitro studies | May explain reactive thrombocytosis observed in some patients affected with mild to moderate IDA. | [73] |
| Iron may influence platelet biogenesis by regulating formation of certain precursor cell types. This function is affected in severe iron deficiency. | May account for occurences of thrombocytopenia, reported in some severe IDA patients | [76] | |
| Macrophages | Excess iron recycling and low ferritin content mobilises macrophages towards tissue repair and regeneration | These macrophages possess anti-inflammatory properties | [94] |
| Neutrophils | Decreased neutrophil apoptosis observed in some children affected with IDA. | May lead to autoimmune disorders/malignancies in the future. | [89] |
| Contrary reports of neutropenia, observed in a patient affected with severe IDA. | May be responsible for increased risk of infections. | [99] | |
| IDA may be partly responsible for neutrophil hypersegmentation. | [88] | ||
| Lymphocytes T | IDA is known to affect lymphocyte counts. | Decreased CD4+ T cells and increased CD8+ counts, reported in symptomatic as well asymptomatic IDA cases. | [113] |
| Lymphocytopenia reported in an adolescent IDA patient. | May pose risk for future infections. | [99] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
De, R.; Prakash, K.U.; Edison, E.S. Complex Interactions in Regulation of Haematopoiesis—An Unexplored Iron Mine. Genes 2021, 12, 1270. https://doi.org/10.3390/genes12081270
De R, Prakash KU, Edison ES. Complex Interactions in Regulation of Haematopoiesis—An Unexplored Iron Mine. Genes. 2021; 12(8):1270. https://doi.org/10.3390/genes12081270
Chicago/Turabian StyleDe, Ranita, Kulkarni Uday Prakash, and Eunice S. Edison. 2021. "Complex Interactions in Regulation of Haematopoiesis—An Unexplored Iron Mine" Genes 12, no. 8: 1270. https://doi.org/10.3390/genes12081270
APA StyleDe, R., Prakash, K. U., & Edison, E. S. (2021). Complex Interactions in Regulation of Haematopoiesis—An Unexplored Iron Mine. Genes, 12(8), 1270. https://doi.org/10.3390/genes12081270