
Ocular Involvement in Hereditary Amyloidosis


 ,
,  ,
,  ,
, 

Abstract
:1. Introduction
2. Hereditary ATTR Amyloidosis


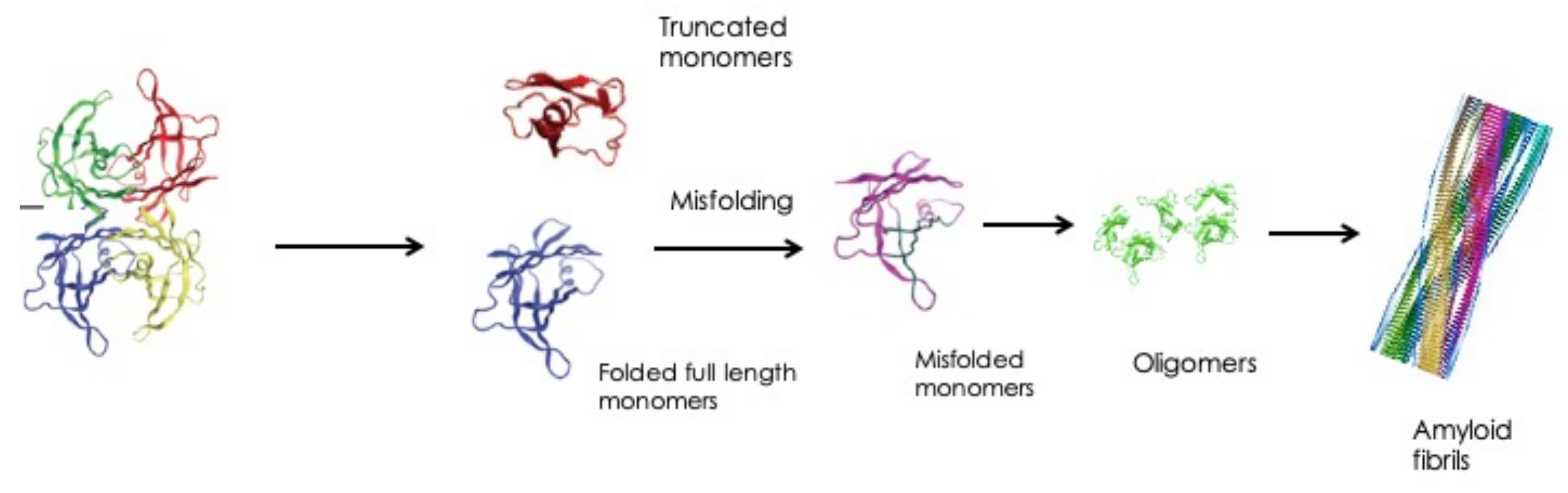
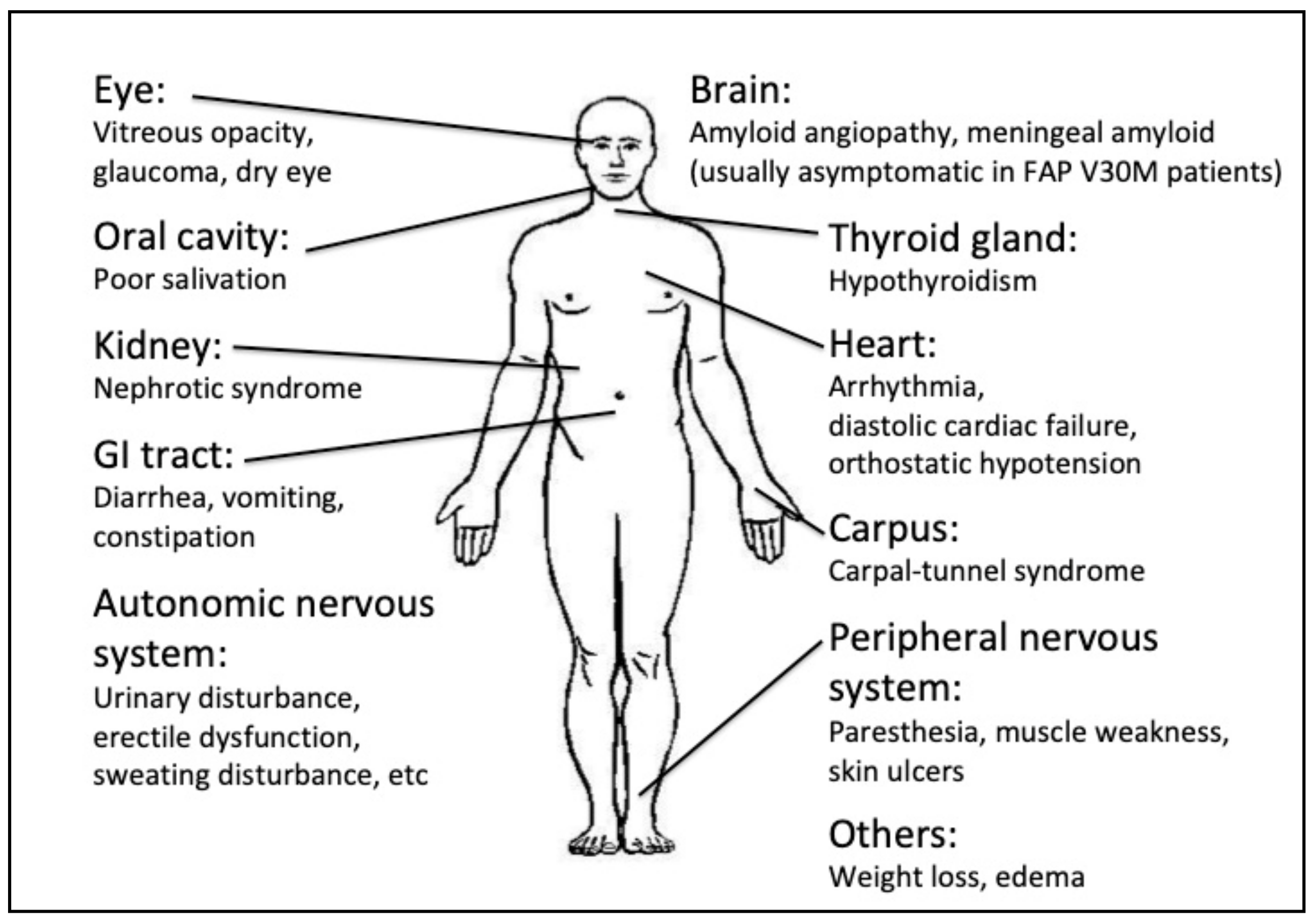
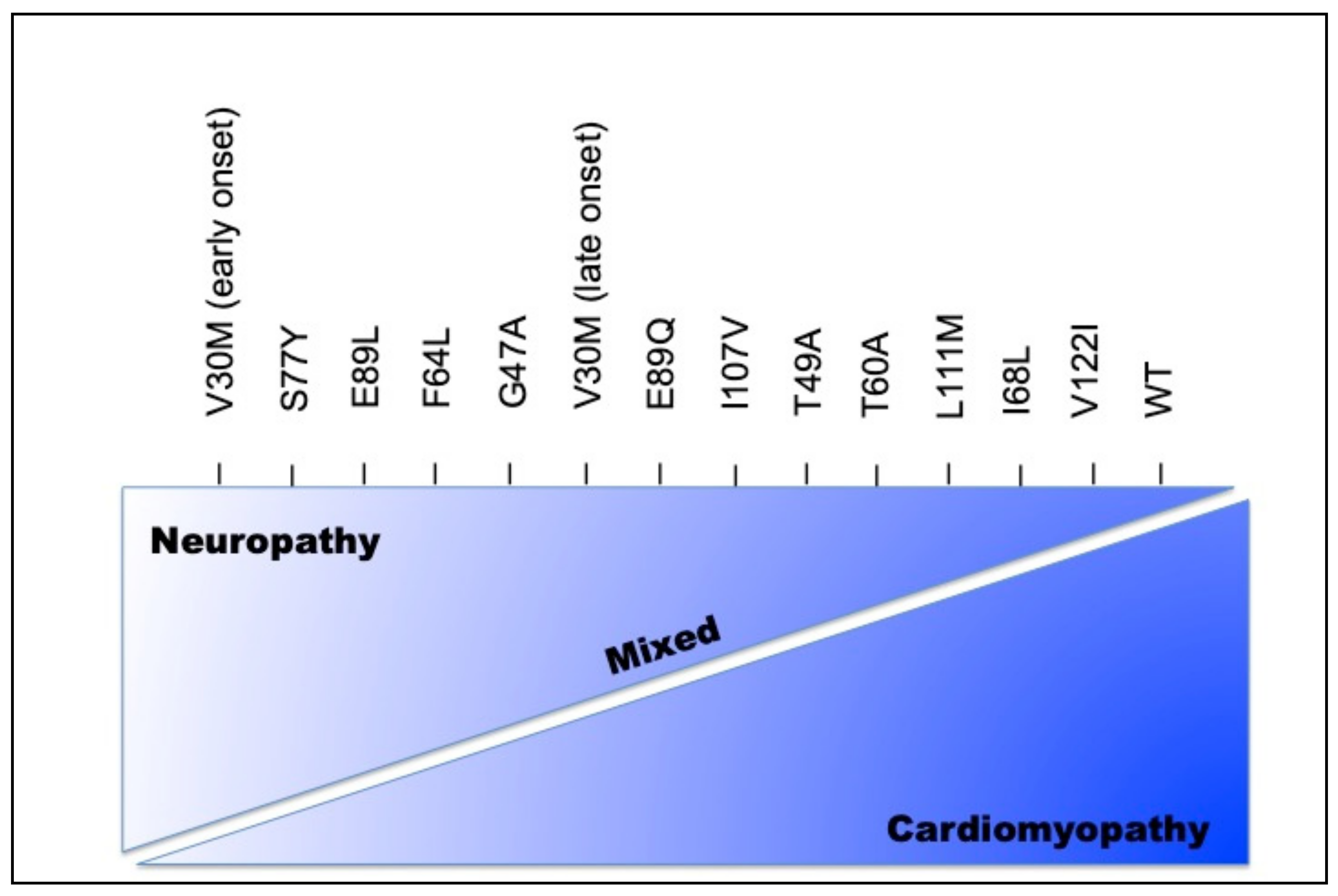
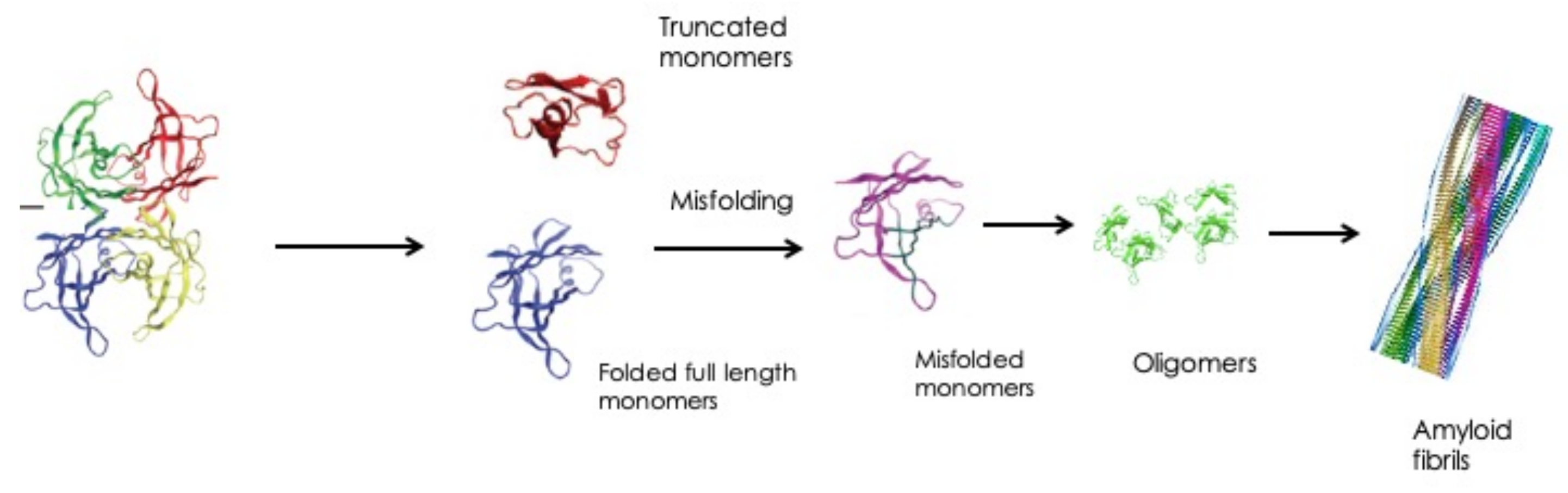
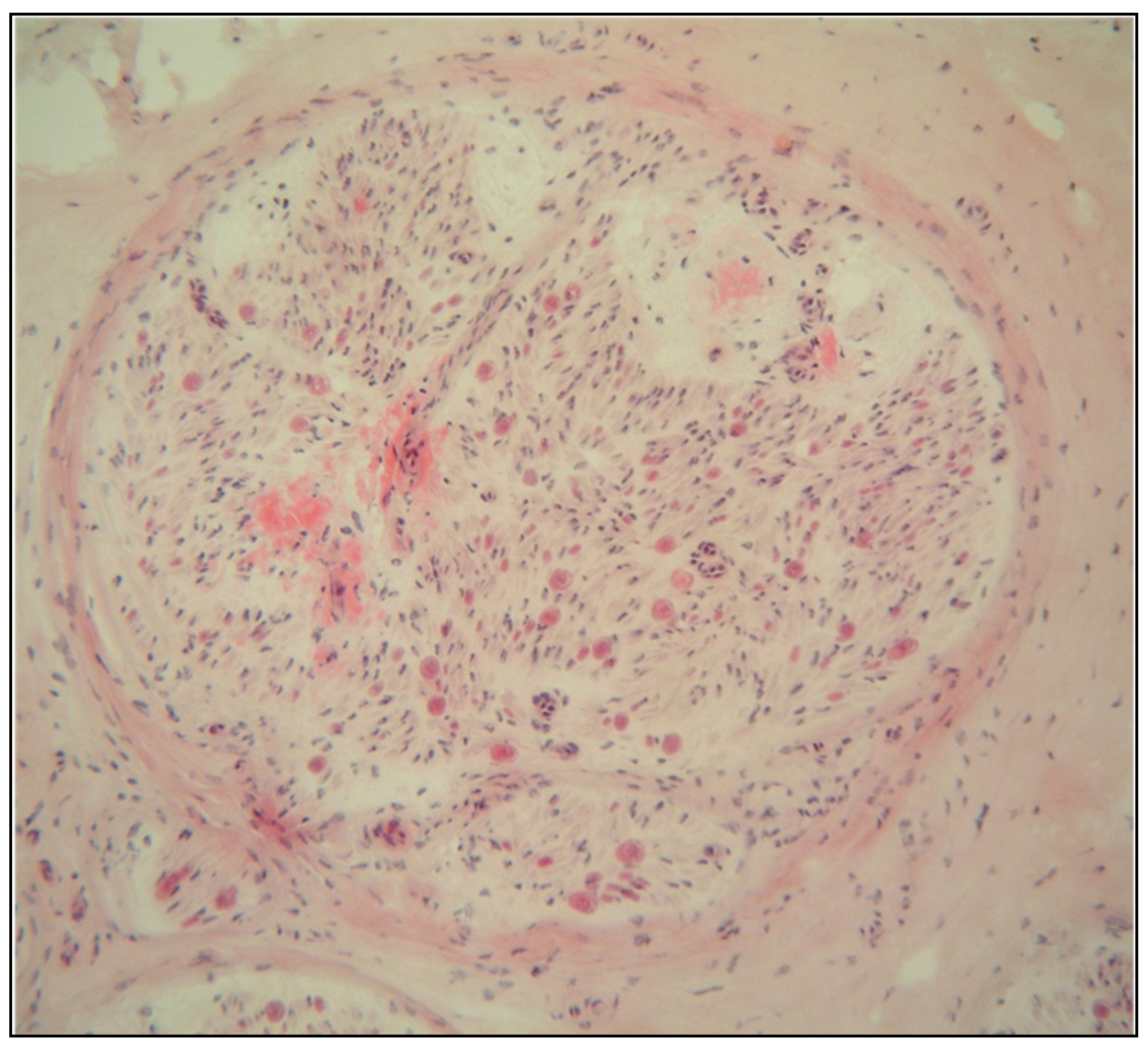
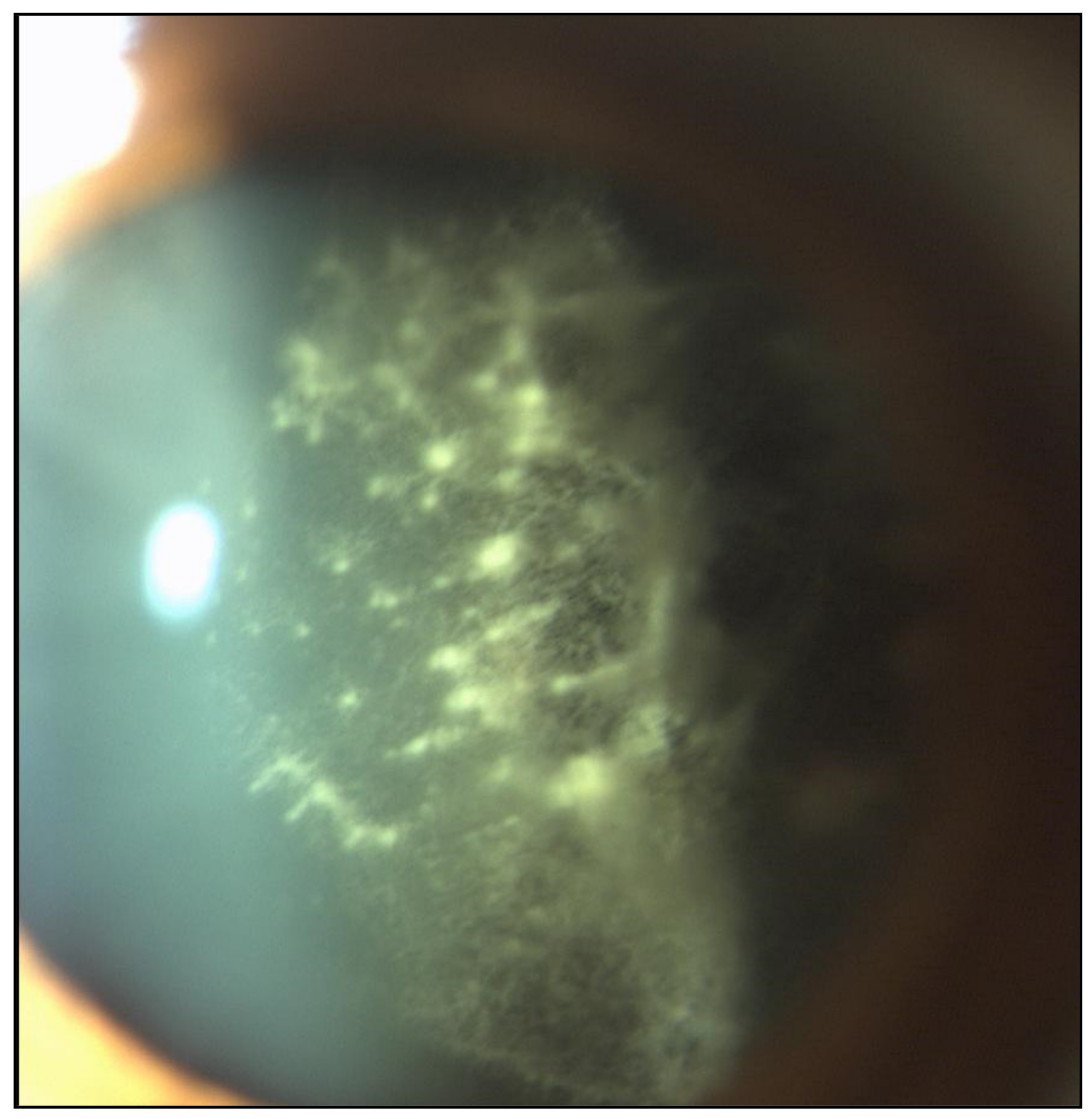
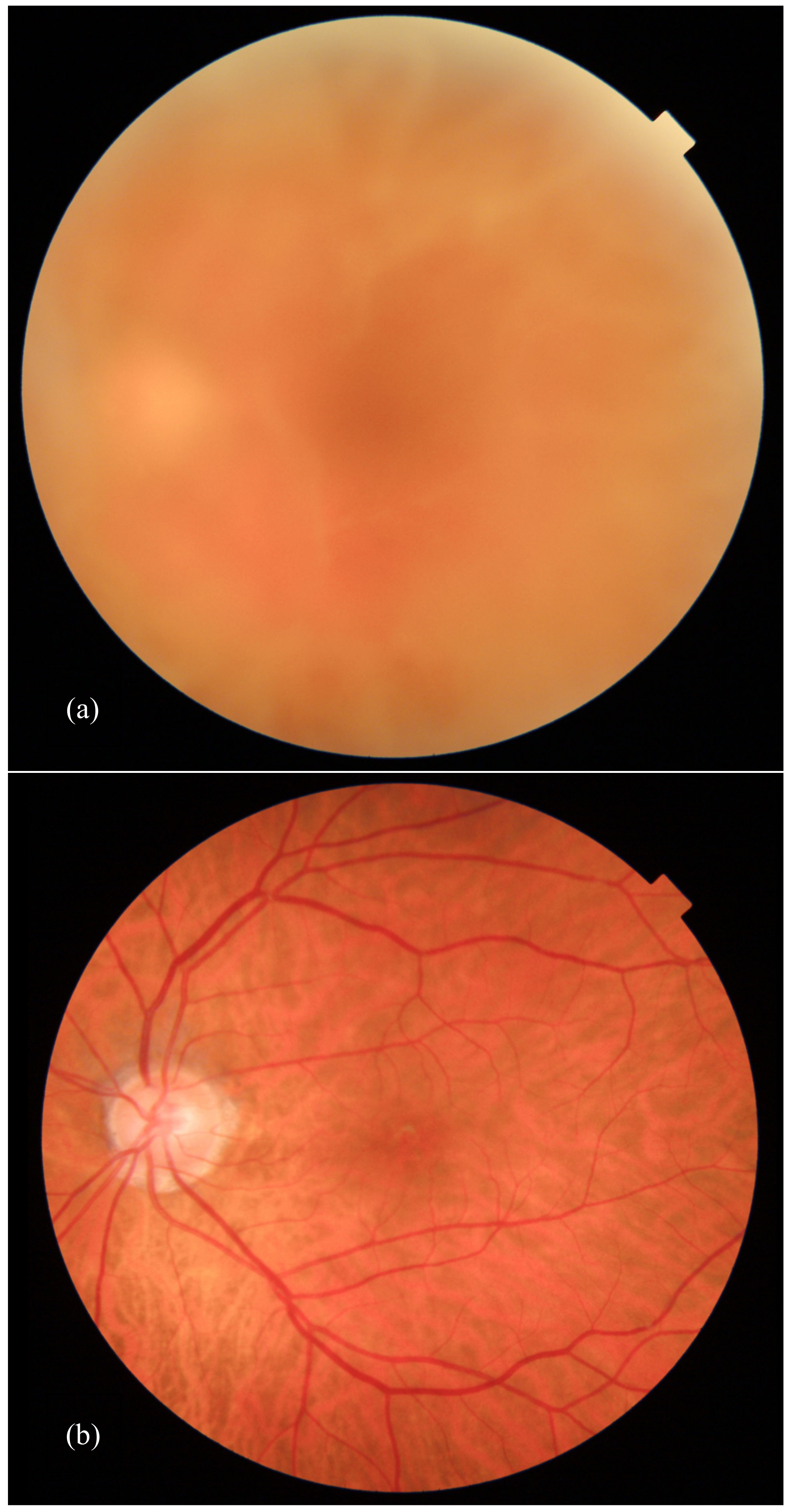
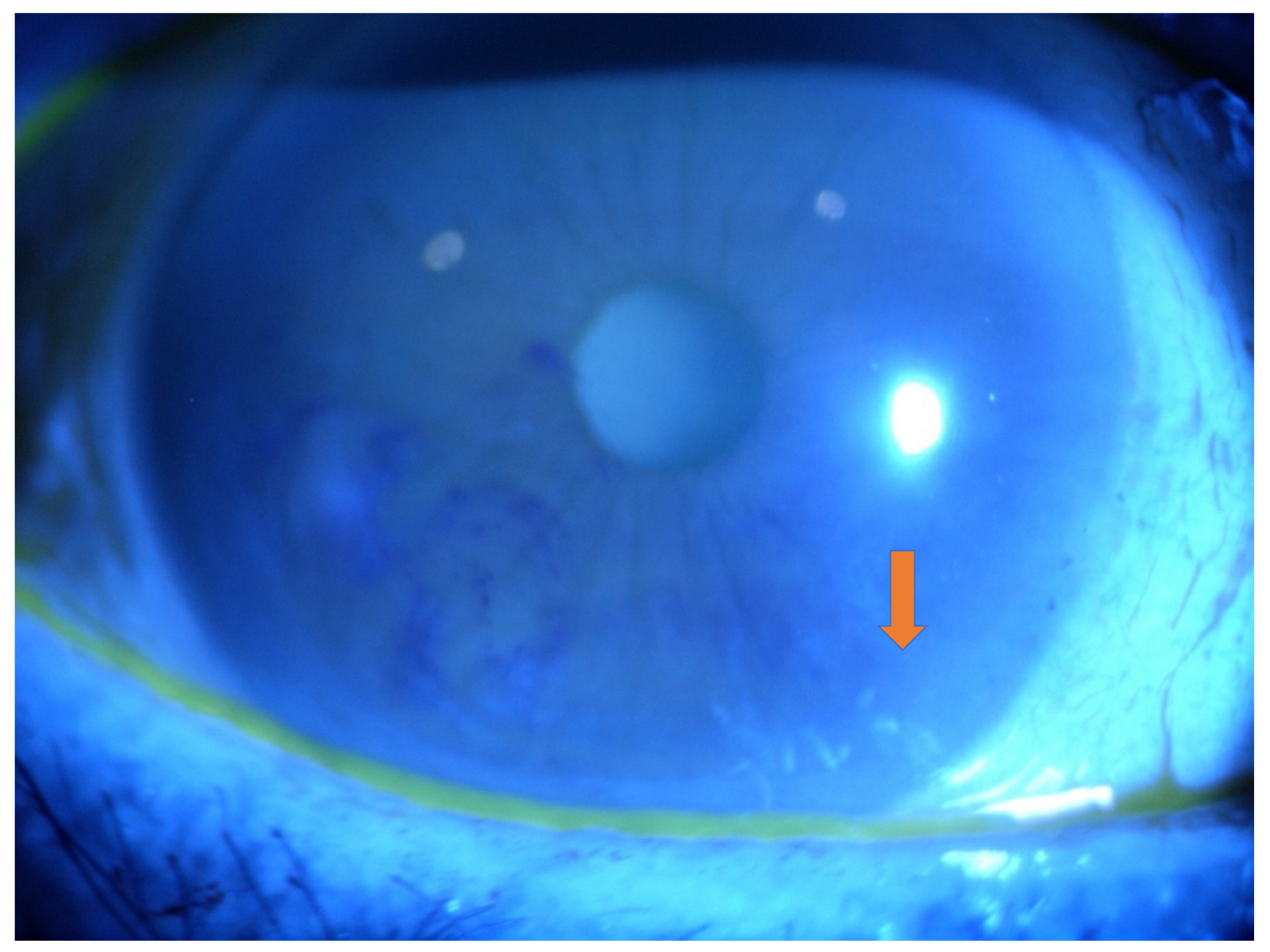
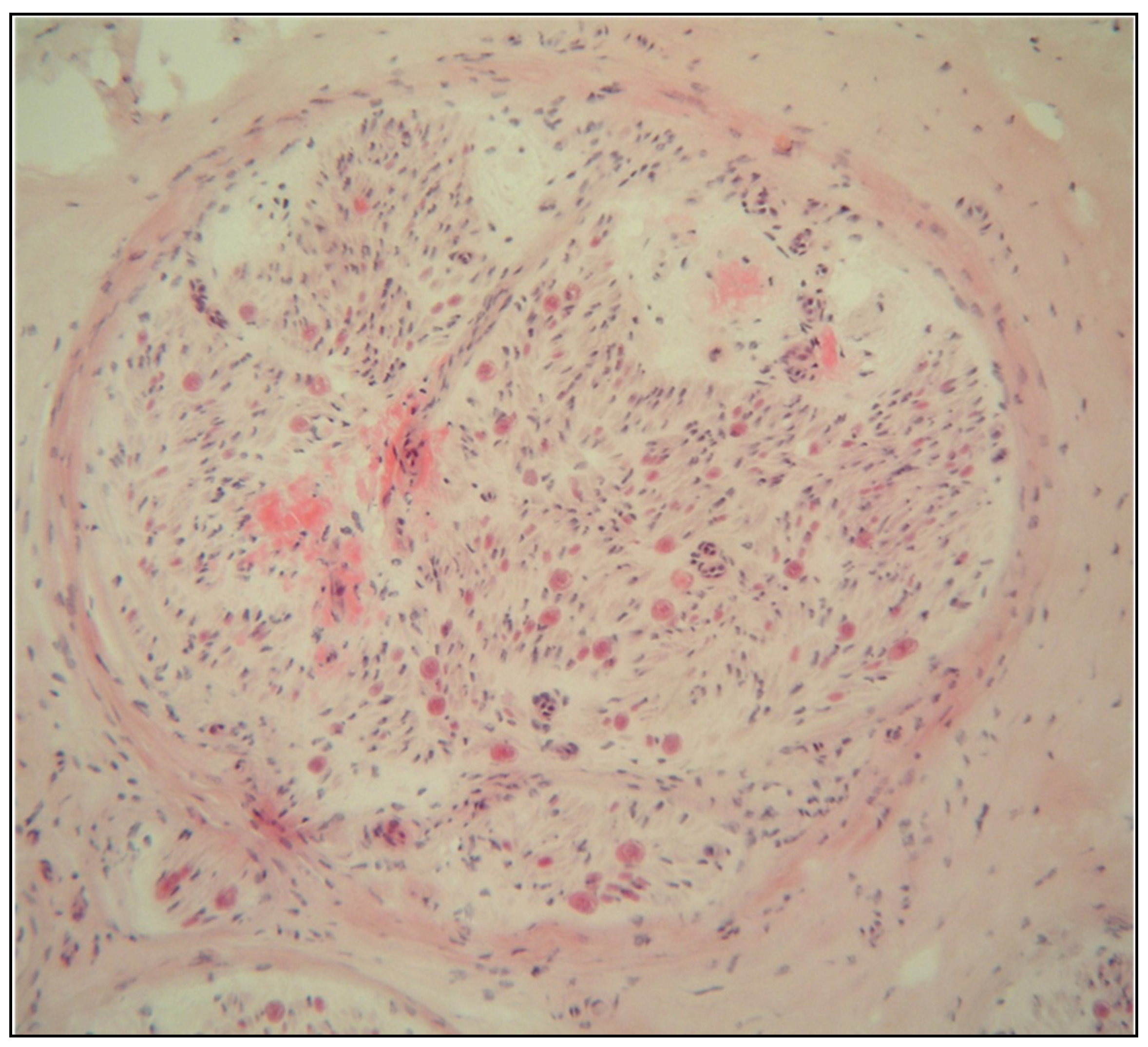
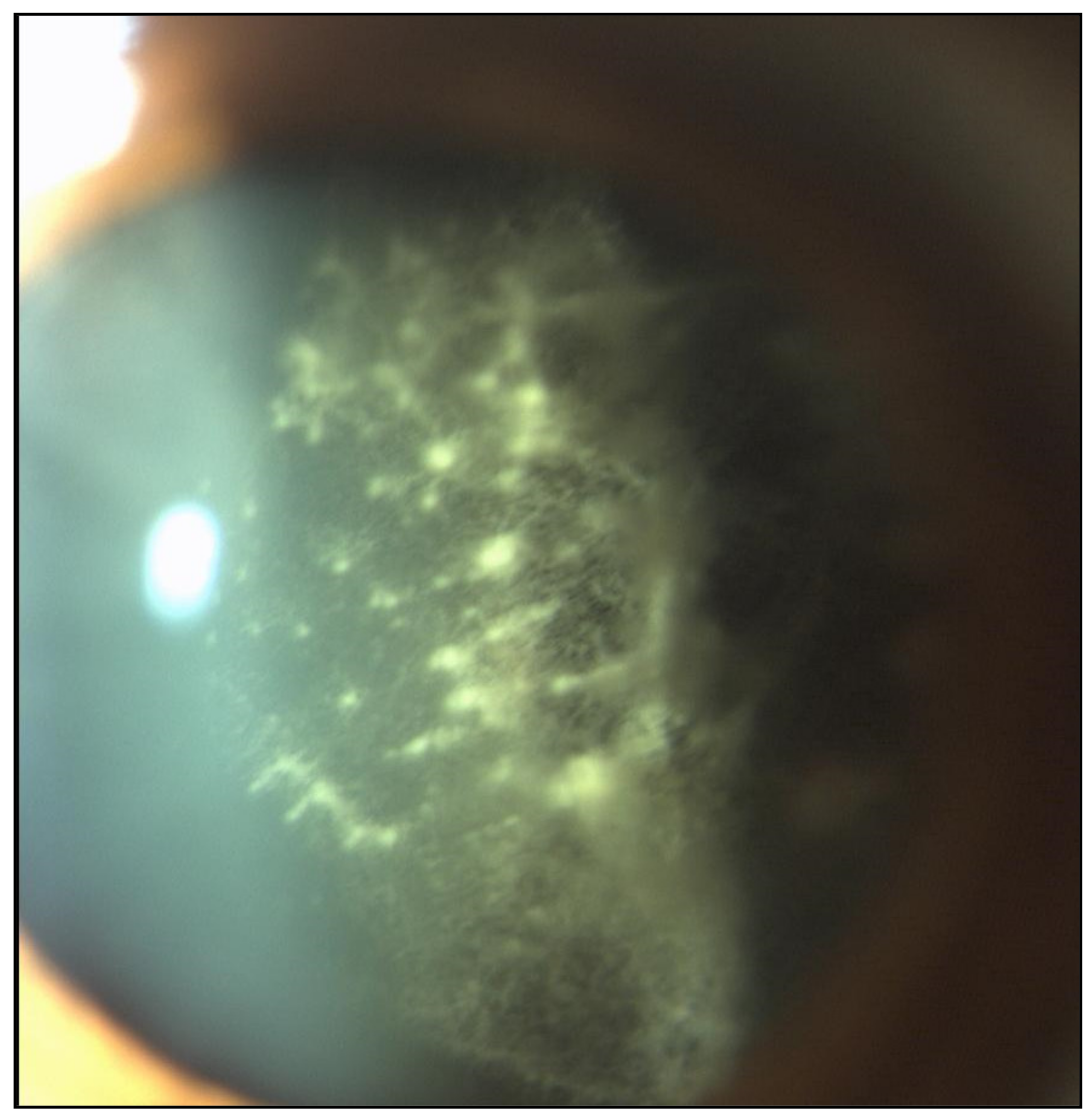
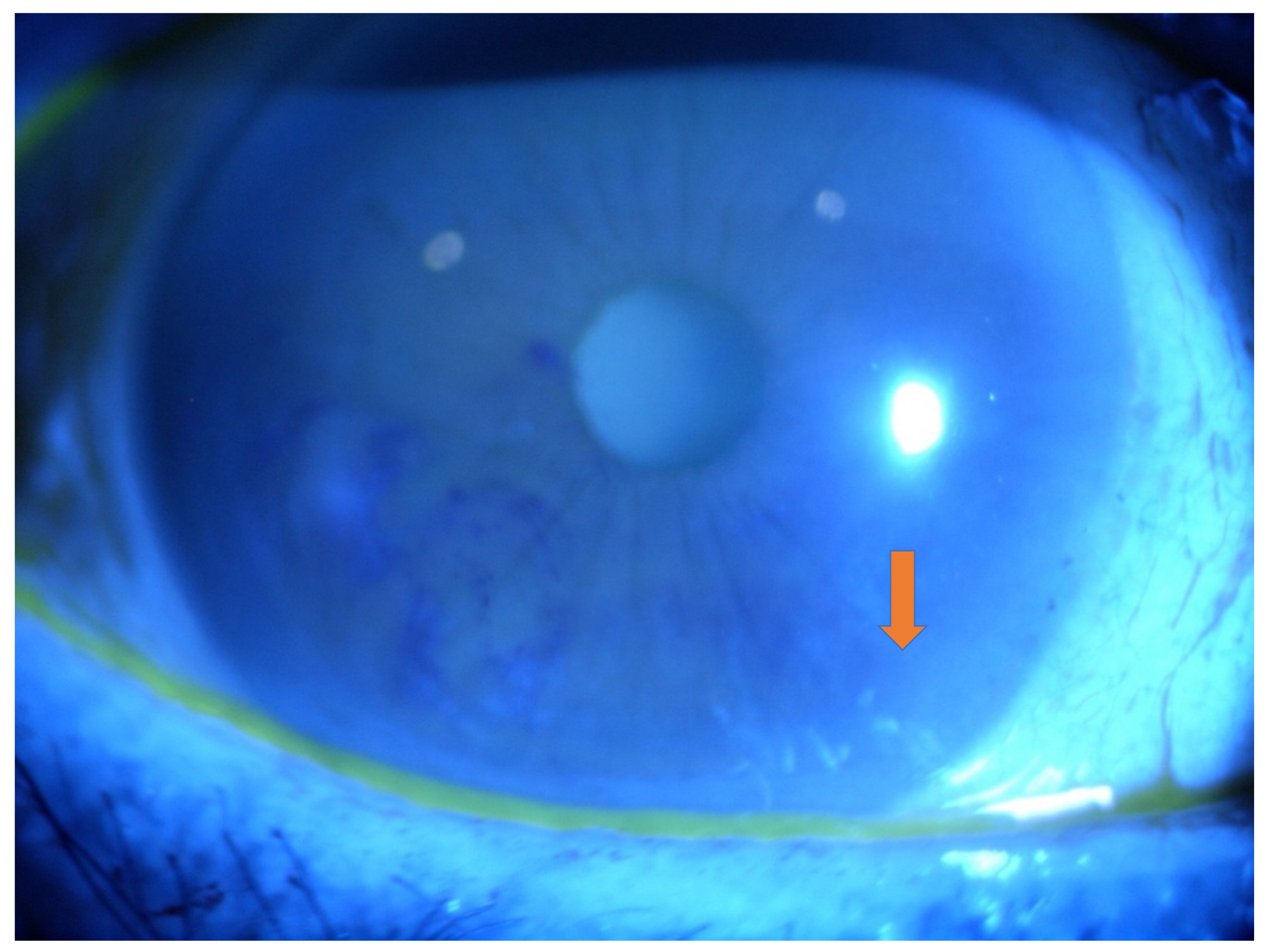
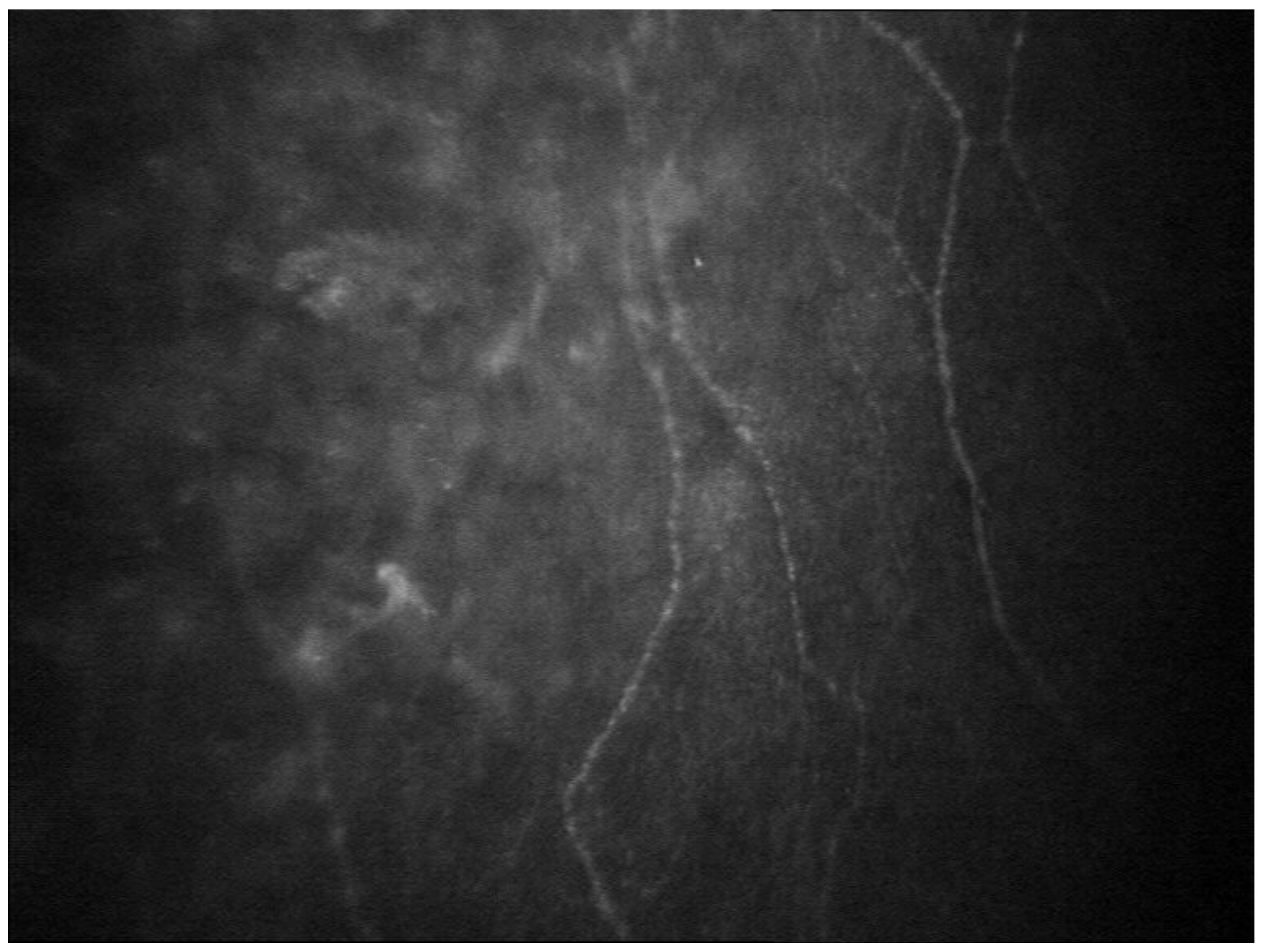
2.1. Eye Involvement in Hereditary TTR Amyloidosis
Ocular Manifestations and Their Treatment

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mutation | |
|---|---|
| Cys10Arg [33] | Leu55Gln [34] |
| Ser23Asn [35] | Leu55Arg [36] |
| Val30Met [37] | Leu55Pro [38] |
| Val30Gly [39] | Leu58Arg [40] |
| Phe33Cys [41] | Phe64Ser [42] |
| Phe33Ile [43] | Tyr69His [44] |
| Arg34Gly [45] | Lys70Asn [46] |
| Lys35Thr [36] | Val71Ala [47] |
| Ala36Pro [48] | Gly83Arg [49] |
| Trp41Leu [50] | Ile84Asn [51] |
| Thr49Ala [52] | Ile84Ser [53] |
| Gly53Ala [54] | Ala97Ser [55] |
| Glu54Gly [56] | Tyr114Cys [57] |
| Glu54Lys [58] | Val122Ala [59] |
- (a)
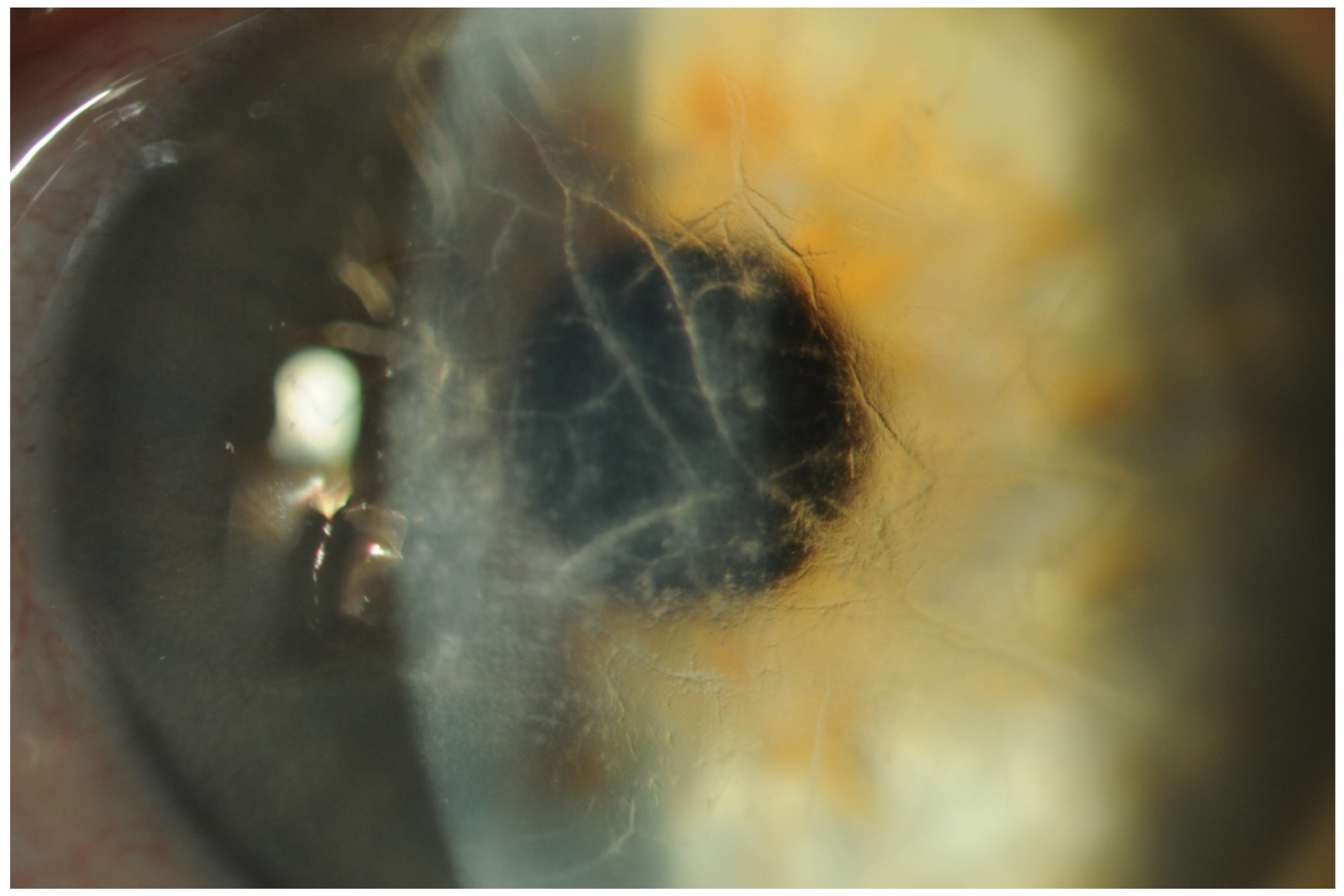
- Vitreous opacities
- (b)
- Chronic Open-Angle Glaucoma (COAG)
- (c)
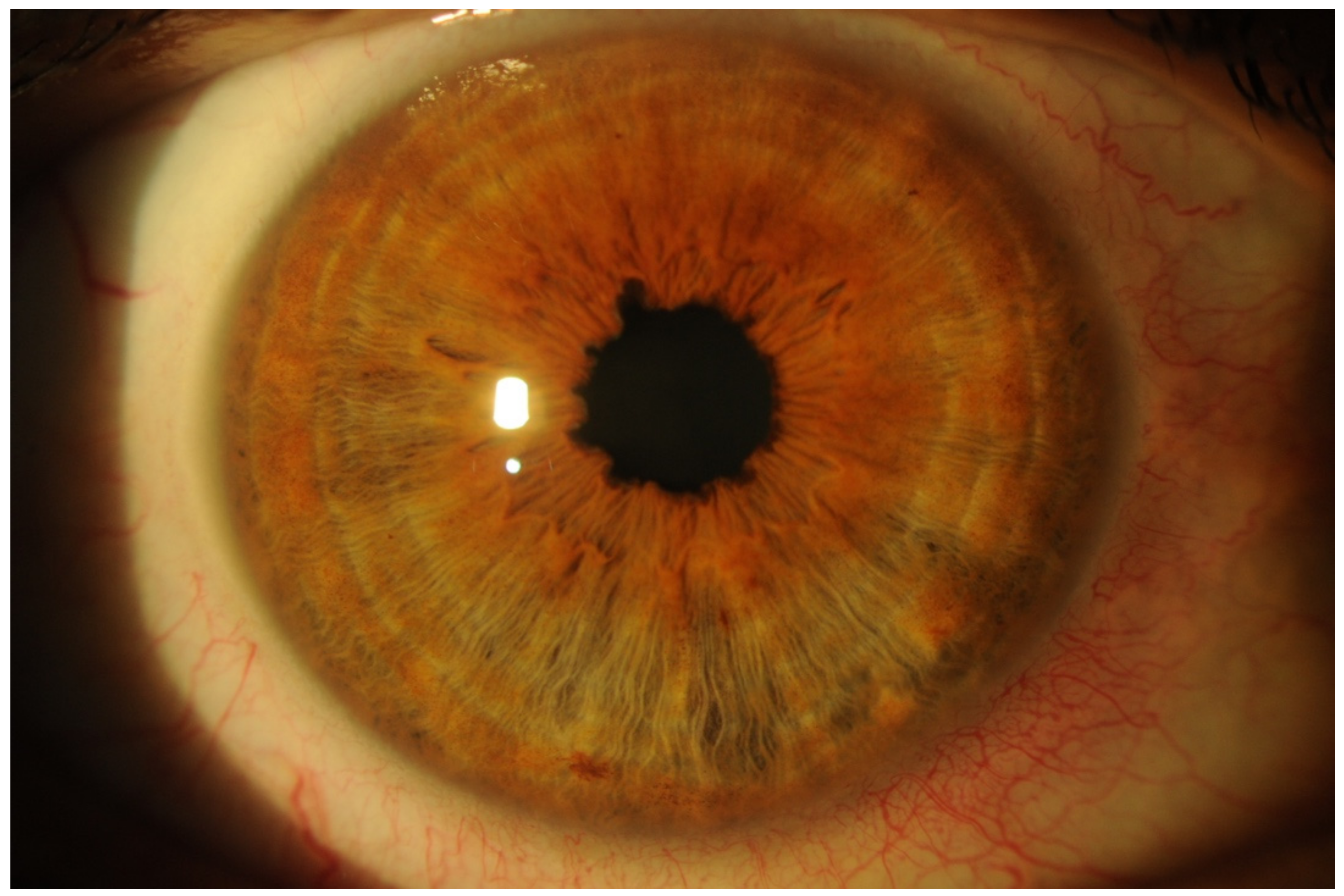
- Abnormal Conjunctival Vessels (ACVs)
- (d)
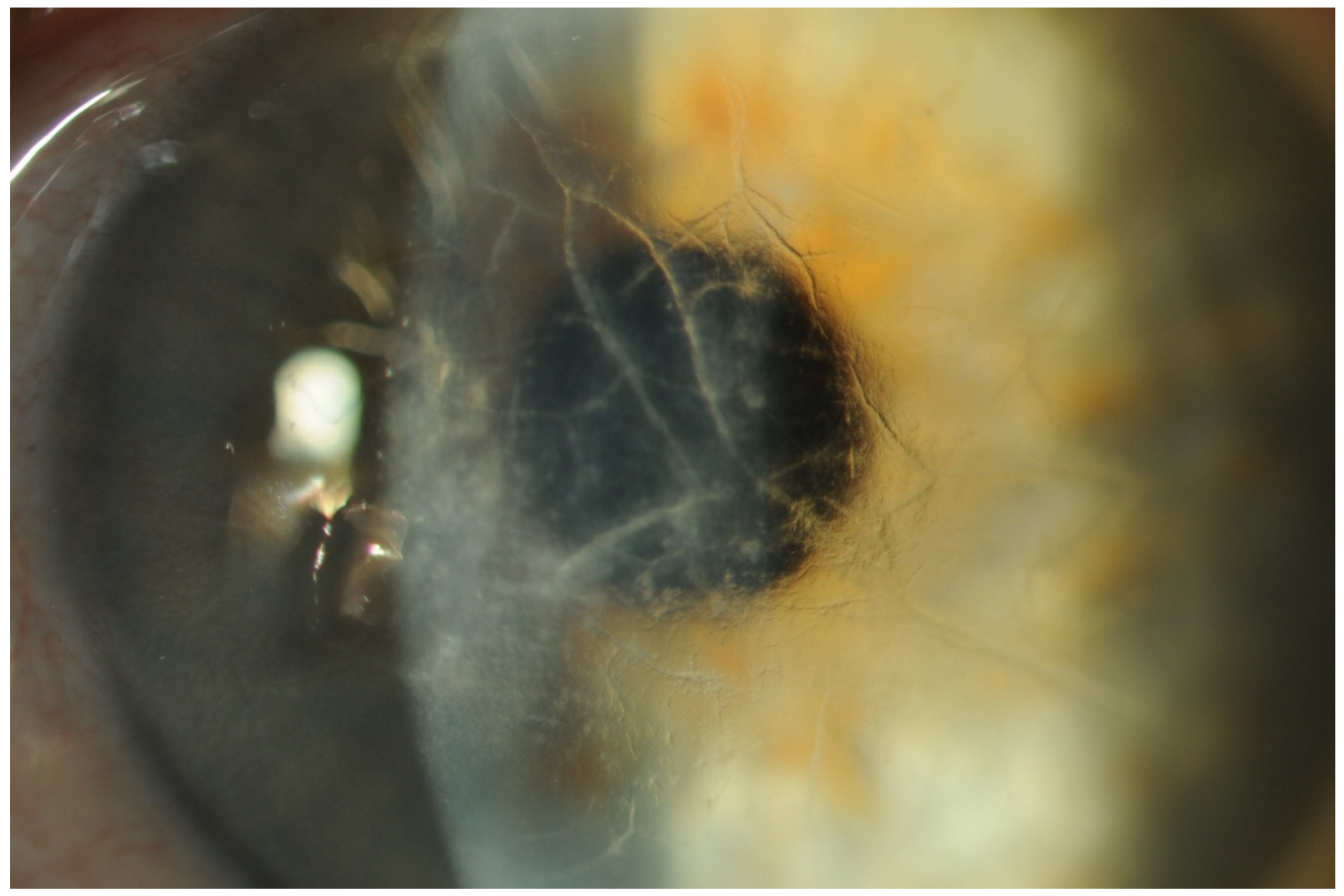
- Keratoconjunctivitis Sicca (KCS) and Corneal Neuropathy
- (e)
- Accommodation Defects
- (f)
- Chorioretinal Vascular Changes
- (g)
- Pupillary Abnormalities
- (h)
- Optic Neuropathy
- (i)
- Ocular Adnexal Amyloidosis (OAA)
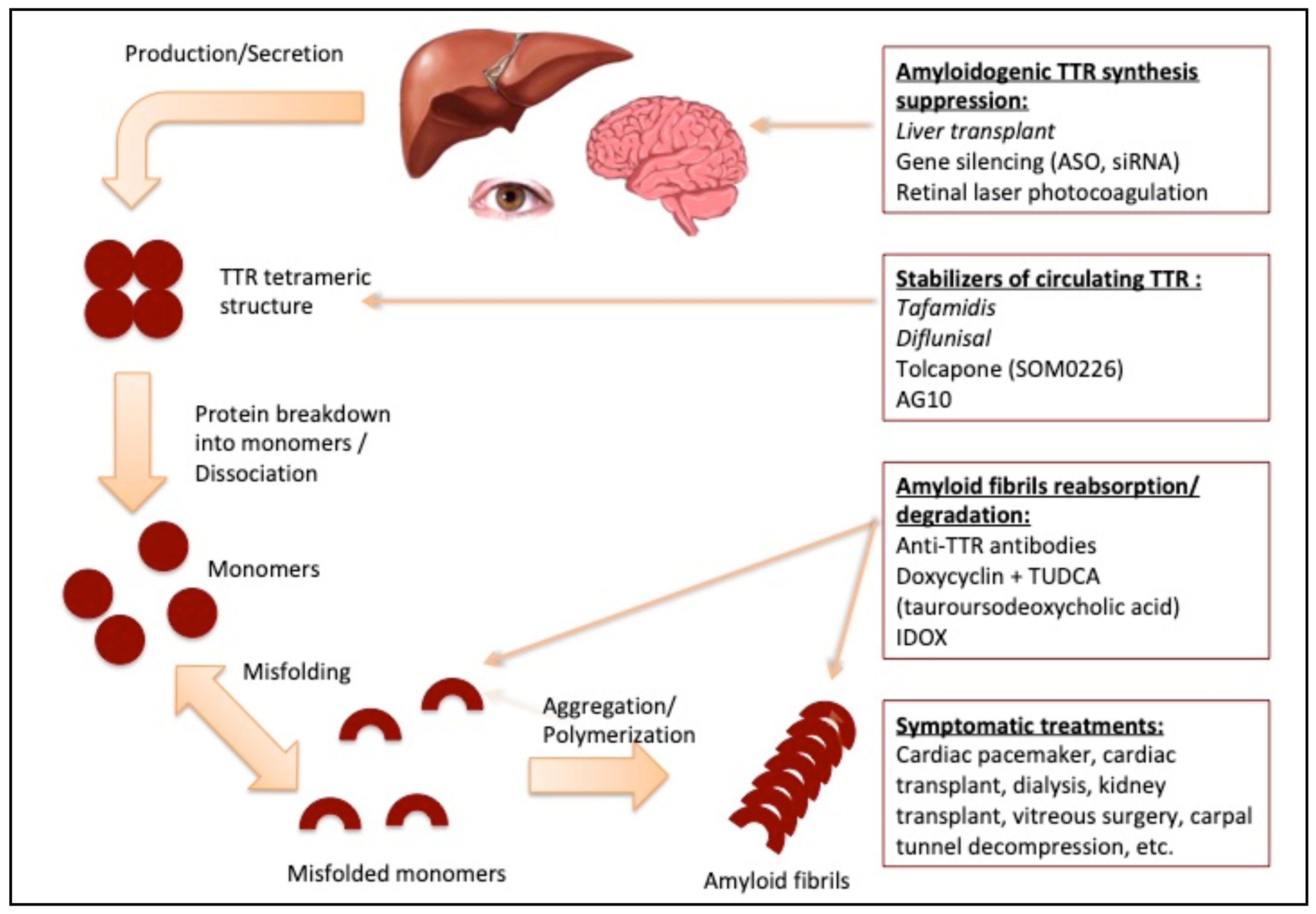
2.2. Systemic Treatment
- (a)
- Liver Transplantation
- (b)
- Gene Silencing Agents
- (c)
- TTR Stabilizers
3. Hereditary Gelsolin Amyloidosis
3.1. Clinical Manifestations
3.1.1. Ocular Involvement
3.1.2. Other Clinical Manifestations
3.2. Diagnosis
3.3. Treatment
4. Keratoepithelin Amyloidosis
5. Lactoferrin Amyloidosis
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Methods of Literature Searching
References
- Reynolds, M.M.; Veverka, K.K.; Gertz, M.A.; Dispenzieri, A.; Zeldenrust, S.R.; Leung, N.; Pulido, J.S. Ocular Manifestations of Familial Transthyretin Amyloidosis. Am. J. Ophthalmol. 2017, 183, 156–162. [Google Scholar] [CrossRef] [PubMed]
- De Larrea, C.F.; Verga, L.; Morbini, P.; Klersy, C.; Lavatelli, F.; Foli, A.; Obici, L.; Milani, P.; Capello, G.L.; Paulli, M.; et al. A practical approach to the diagnosis of systemic amyloidoses. Blood 2015, 125, 2239–2244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venkatesh, P.; Selvan, H.; Singh, S.B.; Gupta, D.; Kashyap, S.; Temkar, S.; Gogia, V.; Tripathy, K.; Chawla, R.; Vohra, R. Vitreous Amyloidosis: Ocular, Systemic, and Genetic Insights. Ophthalmology 2017, 124, 1014–1022. [Google Scholar] [CrossRef] [PubMed]
- Sipe, J.D.; Cohen, A.S. Review: History of the Amyloid Fibril. J. Struct. Biol. 2000, 130, 88–98. [Google Scholar] [CrossRef] [Green Version]
- Cohen, A.S.; Calkins, E. Electron Microscopic Observations on a Fibrous Component in Amyloid of Diverse Origins. Nature 1959, 183, 1202–1203. [Google Scholar] [CrossRef]
- Benson, M.D.; Buxbaum, J.N.; Eisenberg, D.S.; Merlini, G.; Saraiva, M.J.M.; Sekijima, Y.; Sipe, J.D.; Westermark, P. Amyloid nomenclature 2020: Update and recommendations by the International Society of Amyloidosis (ISA) nomenclature committee. Amyloid 2020, 27, 217–222. [Google Scholar] [CrossRef]
- Tripathy, K.; Chawla, R.; Selvan, H.; Venkatesh, P. Ocular Manifestations of Familial Transthyretin Amyloidosis. Am. J. Ophthalmol. 2018, 186, 169–170. [Google Scholar] [CrossRef]
- Ueda, M.; Ando, Y. Recent advances in transthyretin amyloidosis therapy. Transl. Neurodegener. 2014, 3, 19. [Google Scholar] [CrossRef] [Green Version]
- Rapezzi, C.; Quarta, C.C.; Obici, L.; Perfetto, F.; Longhi, S.; Salvi, F.; Biagini, E.; Lorenzini, M.; Grigioni, F.; Leone, O.; et al. Disease profile and differential diagnosis of hereditary transthyretin-related amyloidosis with exclusively cardiac phenotype: An Italian perspective. Eur. Heart J. 2013, 34, 520–528. [Google Scholar] [CrossRef] [Green Version]
- Marcoux, J.; Mangione, P.P.; Porcari, R.; Degiacomi, M.T.; Verona, G.; Taylor, G.W.; Giorgetti, S.; Raimondi, S.; Sanglier-Cianférani, S.; Benesch, J.L.; et al. A novel mechano-enzymatic cleavage mechanism underlies transthyretin amyloidogenesis. EMBO Mol Med. 2015, 7, 1337–1349. [Google Scholar] [CrossRef]
- Dasari, A.K.R.; Arreola, J.; Michael, B.; Griffin, R.G.; Kelly, J.W.; Lim, K.H. Disruption of the CD Loop by Enzymatic Cleavage Promotes the Formation of Toxic Transthyretin Oligomers through a Common Transthyretin Misfolding Pathway. Biochemistry 2020, 59, 2319–2327. [Google Scholar] [CrossRef]
- Reuber, M.; Mayor, R. Recent progress in the understanding and treatment of nonepileptic seizures. Curr. Opin. Psychiatry 2012, 25, 244–250. [Google Scholar] [CrossRef]
- Beirão, M.; Matos, E.; Reis, R.; Beirão, I.; Costa, P.P.; Torres, P. No ocular involvement in familial amyloidotic polyneuropathy ATTR V30M domino liver recipients. Transpl. Int. 2012, 19, 13506129. [Google Scholar] [CrossRef]
- Haraoka, K.; Ando, Y.; Ando, E.; Sandgren, O.; Hirata, A.; Nakamura, M.; Terazaki, H.; Tajiri, T.; Tanoue, Y.; Sun, X.; et al. Amyloid deposition in ocular tissues of patients with familial amyloidotic polyneuropathy (FAP). Amyloid 2002, 9, 183–189. [Google Scholar] [CrossRef]
- Ong, D.E.; Davis, J.T.; O’Day, W.T.; Bok, D. Synthesis and secretion of retinol-binding protein and transthyretin by cultured retinal pigment epithelium. Biochemistry 1994, 33, 1835–1842. [Google Scholar] [CrossRef]
- Schreiber, G. The Evolution of Transthyretin Synthesis in the Choroid Plexus. Clin. Chem. Lab. Med. 2002, 40, 1200–1210. [Google Scholar] [CrossRef]
- Adams, D.; Théaudin, M.; Cauquil, C.; Algalarrondo, V.; Slama, M. FAP Neuropathy and Emerging Treatments. Curr. Neurol. Neurosci. Rep. 2014, 14, 11910. [Google Scholar] [CrossRef]
- Leung, N.; Nasr, S.H.; Sethi, S. How I diagnose amyloidosis. Blood 2012. [Google Scholar] [CrossRef] [Green Version]
- Hara, R.; Kawaji, T.; Ando, E.; Ohya, Y.; Ando, Y.; Tanihara, H. Impact of Liver Transplantation on Transthyretin-Related Ocular Amyloidosis in Japanese Patients. Arch. Ophthalmol. 2010, 128, 206–210. [Google Scholar] [CrossRef] [Green Version]
- You, J. Vitrectomy for vitreous amyloidosis. Int. J. Ophthalmol. 2011, 4, 307–310. [Google Scholar] [CrossRef]
- Yoshinaga, T.; Yazaki, M.; Kametani, F.; Sekijima, Y.; Iesato, Y.; Miyahara, T.; Tsuchiya-Suzuki, A.; Sano, K.; Higuchi, K.; Ikeda, S.-I. Marked biochemical difference in amyloid proportion between intra- and extraocular tissues in a liver-transplanted patient with hereditary ATTR amyloidosis. Amyloid 2017, 24, 17–23. [Google Scholar] [CrossRef] [Green Version]
- Ando, E.; Ando, Y.; Haraoka, K. Ocular amyloid involvement after liver transplantation for polyneuropathy. Ann. Intern. Med. 2001, 135, 931–932. [Google Scholar] [CrossRef]
- Munar-Qués, M.; Salvá-Ladaria, L.; Mulet-Perera, P.; Solé, M.; López-Andreu, F.R.; Saraiva, M.J. Vitreous amyloidosis after liver transplantation in patients with familial amyloid polyneuropathy: Ocular synthesis of mutant transthyretin. Amyloid 2000, 7, 266–269. [Google Scholar] [CrossRef]
- Liepnieks, J.J.; Phan, A.-D.T.; Wise, R.J.; Hrisomalos, F.N.; Benson, M.D. Biochemical characterization of vitreous amyloid formed after liver transplantation. Amyloid 2016, 23, 136–137. [Google Scholar] [CrossRef]
- Martins, A.C.; Rosa, A.M.; Costa, E.; Tavares, C.; Quadrado, J.C.; Murta, J. Ocular Manifestations and Therapeutic Options in Patients with Familial Amyloid Polyneuropathy: A Systematic Review. BioMed Res. Int. 2015, 2015, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Kawaji, T.; Ando, Y.; Hara, R.; Tanihara, H. Novel Therapy for Transthyretin–related Ocular Amyloidosis: A Pilot Study of Retinal Laser Photocoagulation. Ophthalmology 2010, 117, 552–555. [Google Scholar] [CrossRef]
- Sandgren, O.; Kjellgren, D.; Suhr, O.B. Ocular manifestations in liver transplant recipients with familial amyloid polyneuropathy. Acta Ophthalmol. 2008, 86, 520–524. [Google Scholar] [CrossRef]
- Beirão, J.M.; Malheiro, J.; Lemos, C.; Beirão, I.; Costa, P.; Torres, P. Ophthalmological manifestations in hereditary transthyretin (ATTR V30M) carriers: A review of 513 cases. Amyloid 2014, 22, 117–122. [Google Scholar] [CrossRef] [Green Version]
- Ando, E.; Ando, Y.; Okamura, R.; Uchino, M.; Ando, M.; Negi, A. Ocular manifestations of familial amyloidotic polyneuropathy type I: Long term follow up. Br. J. Ophthalmol. 1997, 81, 295–298. [Google Scholar] [CrossRef] [Green Version]
- Rousseau, A.; Terrada, C.; Touhami, S.; Barreau, E.; Rothschild, P.-R.; Valleix, S.; Benoudiba, F.; Errera, M.-H.; Cauquil, C.; Guiochon-Mantel, A.; et al. Angiographic Signatures of the Predominant Form of Familial Transthyretin Amyloidosis (Val30Met Mutation). Am. J. Ophthalmol. 2018, 192, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Beirão, J.M.; Malheiro, J.; Lemos, C.; Matos, E.; Beirão, I.; Pinho-Costa, P.; Torres, P. Impact of liver transplantation on the natural history of oculopathy in Portuguese patients with transthyretin (V30M) amyloidosis. Amyloid 2014, 22, 31–35. [Google Scholar] [CrossRef] [PubMed]
- Rowczenio, D.M.; Noor, I.; Gillmore, J.D.; Lachmann, H.; Whelan, C.; Hawkins, P.N.; Obici, L.; Westermark, P.; Grateau, G.; Wechalekar, A.D. Online Registry for Mutations in Hereditary Amyloidosis Including Nomenclature Recommendations. Hum. Mutat. 2014, 35, E2403–E2412. [Google Scholar] [CrossRef]
- Uemichi, T.; Murrell, J.R.; Zeldenrust, S.; Benson, M.D. A new mutant transthyretin (Arg 10) associated with familial amyloid polyneuropathy. J. Med. Genet. 1992, 29, 888–891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yazaki, M.; Varga, J.; Dyck, P.J.B.; Benson, M.D. A new transthyretin variant Leu55Gln in a patient with systemic amyloidosis. Amyloid 2002, 9, 268–271. [Google Scholar] [CrossRef]
- Connors, L.H.; Théberge, R.; Skare, R.; Costello, C.E.; Falk, R.H.; Skinner, M. A new transthyretin variant (Ser23Asn) associated with familial amyloidosis in a Portuguese patient. Amyloid 1999, 6, 114–118. [Google Scholar] [CrossRef]
- Long, D.; Zeng, I.; Wu, L.Q.; Tang, L.S.; Wang, H.L.; Wang, H. Vitreous amyloidosis in two large mainland Chinese kindreds resulting from transthyretin variant Lys35Thr and Leu55Arg. Ophthalmic Genet. 2012, 33, 28–33. [Google Scholar] [CrossRef]
- Ishida, K.; Nishida, T.; Niimi, T.; Yoshinaga, T.; Hashimoto, R.; Sato, T.; Sekijima, Y.; Tamaoka, A. Elderly onset vitreous opacities as the initial manifestation in hereditary transthyretin (ATTR Val30Met) carriers. Ophthalmic Genet. 2017, 38, 387–391. [Google Scholar] [CrossRef]
- Jacobson, D.; McFarlin, D.; Kane, I.; Buxbaum, J. Transthyretin Pro55, a variant associated with early-onset, aggressive, diffuse amyloidosis with cardiac and neurologic involvement. Qual. Life Res. 1992, 89, 353–356. [Google Scholar] [CrossRef]
- Martin, S.E.; Benson, M.D.; Hattab, E.M. The pathologic spectrum of oculoleptomeningeal amyloidosis with Val30Gly transthyretin gene mutation in a postmortem case. Hum. Pathol. 2014, 45, 1105–1108. [Google Scholar] [CrossRef]
- Saeki, Y.; Ueno, S.; Yorifuji, S.; Sugiyama, Y.; Ide, Y.; Matsuzawa, Y. New mutant gene (transthyretin Arg 58) in cases with hereditary polyneuropathy detected by non-isotope method of single-strand conformation polymorphism analysis. Biochem. Biophys. Res. Commun. 1991, 180, 380–385. [Google Scholar] [CrossRef]
- Lim, A.; Prokaeva, T.; McComb, M.E.; Connors, L.H.; Skinner, M.; Costello, C.E. Identification ofS-sulfonation andS-thiolation of a novel transthyretin Phe33Cys variant from a patient diagnosed with familial transthyretin amyloidosis. Protein Sci. 2003, 12, 1775–1785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uemichi, T.; Uitti, R.J.; Koeppen, A.H.; Donat, J.R.; Benson, M.D. Oculoleptomeningeal Amyloidosis Associated With a New Transthyretin Variant Ser64. Arch. Neurol. 1999, 56, 1152–1155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacobson, D.R.; Santiago-Schwartz, F.; Buxbaum, J.N.; Santiago-Schwarz, F. Restriction fragment analysis confirms the position 33 mutation in transthyretin from an Israeli patient (SKO) with familial amyloidotic polyneuropathy. Biochem. Biophys. Res. Commun. 1988, 153, 198–202. [Google Scholar] [CrossRef]
- Schweitzer, K.; Ehmann, D.; Garcia, R.; Alport, E. Oculoleptomeningeal amyloidosis in 3 individuals with the transthyretin variant Tyr69His. Can. J. Ophthalmol. 2009, 44, 317–319. [Google Scholar] [CrossRef]
- Hawkins, P.N.; Rowczenio, D.; Godfrey, T.; Godfrey, T.; Stawell, R.; Zamir, E. Familial amyloid polyneuropathy associated with the novel transthyretin variant Arg34Gly. Amyloid 2012, 19, 201–203. [Google Scholar]
- Izumoto, S.; Younger, D.; Hays, A.P.; Martone, R.L.; Smith, R.T.; Herbert, J. Familial amyloidotic polyneuropathy presenting with carpal tunnel syndrome and a new transthyretin mutation, asparagine 70. Neurology 1992, 42, 2094–2102. [Google Scholar] [CrossRef]
- Almeida, M.R.; Lopez-Andreu, F.; Munar-Qués, M.; Costa, P.P.; Saraiva, M.J. Transthyretin ALA 71: A new transthyretin variant in a Spanish family with familial amyloidotic polyneuropathy. Hum. Mutat. 1993, 2, 420–421. [Google Scholar] [CrossRef]
- Jones, L.; Skare, J.C.; Harding, J.; Cohen, A.S.; Milunsky, A.; Skinner, M. Proline at position 36: A new transthyretin mutation associated with familial amyloidotic polyneuropathy. Am. J. Hum. Genet. 1991, 48, 979–982. [Google Scholar]
- Xie, Y.; Zhao, Y.; Zhou, J.J.; Wang, X. Identification of a TTR gene mutation in a family with hereditary vitreous amyloidosis. Zhonghua Yi Xue Yi Chuan Xue Za Zhi 2012, 29, 13–15. [Google Scholar]
- Yazaki, M.; Connors, L.H.; Eagle, R.C.; Leff, S.R.; Skinner, M.; Benson, M.D. Transthyretin amyloidosis associated with a novel variant (Trp41Leu) presenting with vitreous opacities. Amyloid 2002, 9, 263–267. [Google Scholar] [CrossRef]
- Skinner, M.; Harding, J.; Skare, I.; Jones, L.A.; Cohen, A.S.; Milunsky, A.; Skare, J. A new transthyretin mutation associated with amyloidotic vitreous opacities. Asparagine for isoleucine at position 84. Ophthalmology 1992, 99, 503–508. [Google Scholar] [CrossRef]
- Mazzeo, A.; Russo, M.; Di Bella, G.; Minutoli, F.; Stancanelli, C.; Gentile, L.; Baldari, S.; Carerj, S.; Toscano, A.; Vitaa, G. Transthyretin-Related Familial Amyloid Polyneuropathy (TTR-FAP): A Single-Center Experience in Sicily, an Italian Endemic Area. J. Neuromuscul. Dis. 2015, 2, S39–S48. [Google Scholar] [CrossRef] [Green Version]
- Dwulet, F.; Benson, M.D. Characterization of a transthyretin (prealbumin) variant associated with familial amyloidotic polyneuropathy type II (Indiana/Swiss). J. Clin. Investig. 1986, 78, 880–886. [Google Scholar] [CrossRef] [Green Version]
- Douglass, C.; Suvarna, K.; Reilly, M.M.; Hawkins, P.N.; Hadjivassiliou, M. A novel amyloidogenic transthyretin variant, Gly53Ala, associated with intermittent headaches and ataxia. J. Neurol. Neurosurg. Psychiatry 2007, 78, 193–195. [Google Scholar] [CrossRef]
- Tachibana, N.; Tokuda, T.; Yoshida, K.; Taketomi, T.; Nakazato, M.; Li, Y.F.; Masuda, Y.; Ikeda, S. Usefulness of MALDI/TOF mass spectrometry of immunoprecipitated serum variant transthyretin in the diagnosis of familial amyloid polyneuropathy. Amyloid 1999, 6, 282–288. [Google Scholar] [CrossRef]
- Reilly, M.M.; Adams, D.; Booth, D.R.; Davis, M.B.; Said, G.; Laubriat-Bianchin, M.; Pepys, M.B.; Thomas, P.K.; Harding, A.E. Transthyretin gene analysis in European patients with suspected familial amyloid polyneuropathy. Brain 1995, 118, 849–856. [Google Scholar] [CrossRef]
- Ueno, S.; Uemichi, T.; Yorifuji, S.; Tarui, S. A novel variant of transthyretin (Tyr114 to Cys) deduced from the nucleotide sequences of gene fragments from familial amyloidotic polyneuropathy in Japanese sibling cases. Biochem. Biophys. Res. Commun. 1990, 169, 143–147. [Google Scholar] [CrossRef]
- Togashi, S.; Watanabe, H.; Nagasaka, T.; Shindo, K.; Shiozawa, Z.; Maeda, S.; Tawata, M.; Onaya, T. An aggressive familial amyloidotic polyneuropathy caused by a new variant transthyretin Lys 54. Neurology 1999, 53, 637–639. [Google Scholar] [CrossRef]
- Théberge, R.; Connors, L.; Skare, J.; Skinner, M.; Falk, R.H.; Costello, C. A new amyloidogenic transthyretin variant (Val122Ala) found in a compound heterozygous patient. Amyloid 1999, 6, 54–58. [Google Scholar] [CrossRef]
- Kakihara, S.; Hirano, T.; Imai, A.; Miyahara, T.; Murata, T. Small gauge vitrectomy for vitreous amyloidosis and subsequent management of secondary glaucoma in patients with hereditary transthyretin amyloidosis. Sci. Rep. 2020, 10, 1–8. [Google Scholar] [CrossRef]
- Beirão, N.M.; Matos, E.; Beirão, I.; Costa, P.P.; Torres, P. Recurrence of vitreous amyloidosis and need of surgical reintervention in Portuguese patients with familial amyloidosis ATTR V30M. Retina 2011, 31, 1373–1377. [Google Scholar] [CrossRef]
- Ferreira, N.N.; Dias, D.A.C.; Carvalho, R.P.A.; Coelho, M.T.P.M. Re-Intervention in De Novo Vitreous Opacities After Pars Plana Vitrectomy in Familial Amyloidotic Polyneuropathy TTR Val30Met Portuguese Patients. Retin Cases Brief Rep. 2017, 1–6. [Google Scholar] [CrossRef]
- Ando, T.; Oshitari, T.; Saito, M.; Tawada, A.; Baba, T.; Yotsukura, J.; Yamamoto, S. A Case of Conjunctival Amyloidosis with Repeated Subconjunctival Hemorrhage. Case Rep. Ophthalmol. Med. 2017, 2017, 5423027. [Google Scholar] [CrossRef]
- Zloto, O.; Rosner, M.; Vishnevskia-Dai, V. Yellow subconjunctival deposits. BMJ Case Rep. 2019, 12, e230370. [Google Scholar] [CrossRef]
- Hayek, S.; Adam, C.; Adams, D.; Cauquil, C.; Barreau, E.; Guiochon-Mantel, A.; Labetoulle, M.; Rousseau, A. Conjunctival lymphangiectasia: A novel ocular manifestation of hereditary transthyretin amyloidosis. Amyloid 2019, 26, 94–95. [Google Scholar] [CrossRef]
- Bunod, R.; Adams, D.; Cauquil, C.; Francou, B.; Labeyrie, C.; Bourenane, H.; Adam, C.; Algalarrondo, V.; Slama, M.; Darce-Bello, M.; et al. Conjunctival lymphangiectasia as a biomarker of severe systemic disease in Ser77Tyr hereditary transthyretin amyloidosis. Br. J. Ophthalmol. 2020, 104, 1363–1367. [Google Scholar] [CrossRef]
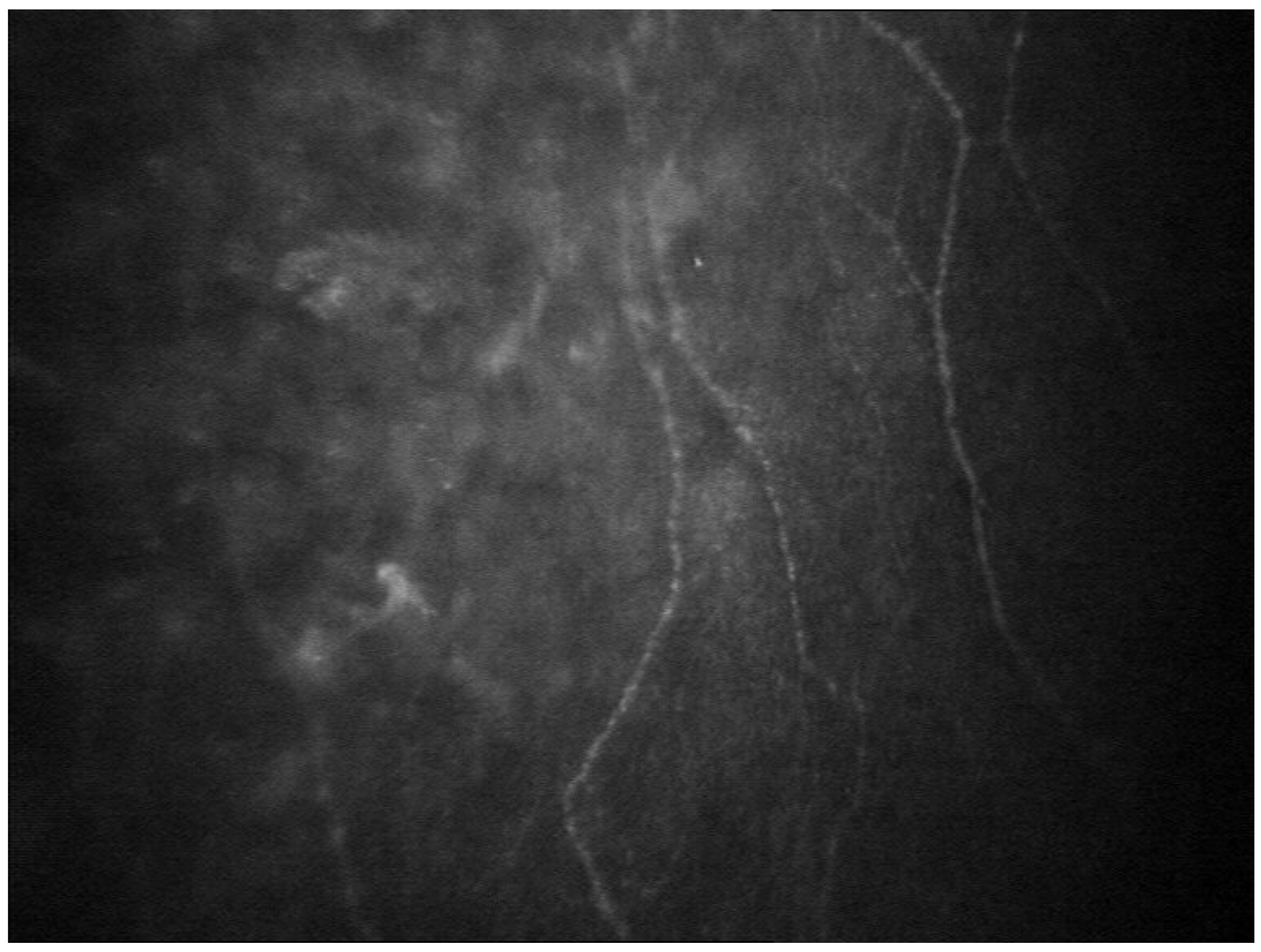
- Rousseau, A.; Cauquil, C.; Dupas, B.; Labbé, A.; Baudouin, C.; Barreau, E.; Théaudin, M.; Lacroix, C.; Guiochon-Mantel, A.; Benmalek, A.; et al. Potential Role of In Vivo Confocal Microscopy for Imaging Corneal Nerves in Transthyretin Familial Amyloid Polyneuropathy. JAMA Ophthalmol. 2016, 134, 983–989. [Google Scholar] [CrossRef]
- Chen, X.; Graham, J.; Dabbah, M.; Petropoulos, I.N.; Ponirakis, G.; Asghar, O.; Alam, U.; Marshall, A.; Fadavi, H.; Ferdousi, M.; et al. Small Nerve Fiber Quantification in the Diagnosis of Diabetic Sensorimotor Polyneuropathy: Comparing Corneal Confocal Microscopy With Intraepidermal Nerve Fiber Density. Diabetes Care 2015, 38, 1138–1144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tavakoli, M.; Marshall, A.; Thompson, L.; Kenny, M.; Waldek, S.; Efron, N.; Malik, R.A. Corneal confocal microscopy: A novel noninvasive means to diagnose neuropathy in patients with fabry disease. Muscle Nerve 2009, 40, 976–984. [Google Scholar] [CrossRef]
- Tavakoli, M.; Marshall, A.; Banka, S.; Msc, I.N.P.; Fadavi, H.; Kingston, H.; Malik, R.A. Corneal confocal microscopy detects small-fiber neuropathy in Charcot-Marie-Tooth disease type 1A patients. Muscle Nerve 2012, 46, 698–704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beirão, M.; Matos, E.; Beirão, I.; Costa, P.P.E.; Torres, P. Anticipation of presbyopia in Portuguese familial amyloidosis ATTR V30M. Amyloid 2011, 18, 92–97. [Google Scholar] [CrossRef] [PubMed]
- Kakihara, S.; Hirano, T.; Matsuda, Y.; Takano, D.; Imai, A.; Miyahara, T.; Murata, T. Deposits on Retinal Surface Seen on OCT in Ocular Amyloidosis. Ophthalmol. Retin. 2021, 2468. [Google Scholar] [CrossRef]
- Marques, J.H.; Coelho, J.; Malheiro, J.; Pessoa, B.; Beirão, J.M. Subclinical retinal angiopathy associated with hereditary transthyretin amyloidosis–Assessed with optical coherence tomography angiography. Amyloid 2021, 28, 66–71. [Google Scholar] [CrossRef] [PubMed]
- Mano, F.; Dispenzieri, A.; Kusaka, S.; Pavesio, C.; Khalid, H.; Keane, P.A.; Pulido, J.S. Association between Choroidal Characteristics and Systemic Severity in Amyloidosis. Retina 2021, 41, 1037–1046. [Google Scholar] [CrossRef]
- Marques, J.H.; Malheiro, L.; Malheiro, J.; Oliveira, L.; Menéres, M.J.; Beirão, J.M. Pupillometry: An objective test to assess endocular hereditary transthyretin amyloidosis. Eur. J. Ophthalmol. 2021, 18. [Google Scholar] [CrossRef]
- Sandgren, O. Ocular amyloidosis, with special reference to thehereditary forms with vitreous involvement. Surv. Ophthalmol. 1995, 40, 173–196. [Google Scholar] [CrossRef]
- Dermarkarian, C.R.; Bhatt, A.; Chévez-Barrios, P.; Allen, R.C. Bilateral acquired nasolacrimal duct obstruction secondary to amyloidosis in a 15-year-old. J. Am. Assoc. Pediatr. Ophthalmol. Strabismus 2021, 1091. [Google Scholar] [CrossRef]
- Chean, C.S.; Sovani, V.; Boden, A.; Knapp, C. Lacrimal gland extranodal marginal zone B-cell lymphoma in the presence of amyloidosis. Orbit 2020. [Google Scholar] [CrossRef]
- Blandford, A.D.; Yordi, S.; Kapoor, S.; Yeaney, G.; Cotta, C.V.; Valent, J.; Perry, J.D.; Singh, A.D. Ocular Adnexal Amyloidosis: A Mass Spectrometric Analysis. Am. J. Ophthalmol. 2018, 193, 28–32. [Google Scholar] [CrossRef]
- Jiménez, R.M.; España, J.C.S.; Vásquez, L.M.; Bahamondes, A.T.; Rondón, M.; Francesc, T.; Barroso, E.A. Orbital and peri-orbital amyloidosis: A report of four cases. Orbit 2018, 38, 148–153. [Google Scholar] [CrossRef]
- Dodd, M.-M.U.; Wolkow, N.; Cunnane, M.E.; Ma, L.; Dryja, T.P.; Hunter, D. Isolated orbital amyloidosis causing internal and external ophthalmoplegia. J. Am. Assoc. Pediatr. Ophthalmol. Strabismus 2020, 24, 48–51.e1. [Google Scholar] [CrossRef]
- Kang, S.; Dehabadi, M.H.; Rose, G.E.; Verity, D.H.; Amin, S.; Das-Bhaumik, R. Ocular amyloid: Adnexal and systemic involvement. Orbit 2019, 39, 13–17. [Google Scholar] [CrossRef]
- Chan, T.M.; Ponce, C.M.P.; Allen, R.C.; Bell, D.; Lee, A.G. Orbital AL amyloid. Orbit 2019, 39, 68–70. [Google Scholar] [CrossRef]
- Plante-Bordeneuve, V. Transthyretin familial amyloid polyneuropathy: An update. J. Neurol. 2018, 265, 976–983. [Google Scholar] [CrossRef]
- Ackermann, E.J.; Guo, S.; Benson, M.D.; Booten, S.; Freier, S.; Hughes, S.G.; Kim, T.-W.; Kwoh, T.J.; Matson, J.; Norris, D.; et al. Suppressing transthyretin production in mice, monkeys and humans using 2nd-Generation antisense oligonucleotides. Amyloid 2016, 23, 148–157. [Google Scholar] [CrossRef]
- Love, K.T.; Mahon, K.P.; Levins, C.G.; Whitehead, K.A.; Querbes, W.; Dorkin, J.R.; Qin, J.; Cantley, W.; Qin, L.L.; Racie, T.; et al. Lipid-like materials for low-dose, in vivo gene silencing. Proc. Natl. Acad. Sci. USA 2010, 107, 1864–1869, Erratum in 2010, 107, 9915. [Google Scholar] [CrossRef] [Green Version]
- Coelho, T.; Adams, D.; Silva, A.; Lozeron, P.; Hawkins, P.N.; Mant, T.; Perez, J.; Chiesa, J.; Warrington, S.; Tranter, E.; et al. Safety and Efficacy of RNAi Therapy for Transthyretin Amyloidosis. N. Engl. J. Med. 2013, 369, 819–829. [Google Scholar] [CrossRef]
- Maurer, M.S.; Schwartz, J.H.; Gundapaneni, B.; Elliott, P.M.; Merlini, G.; Waddington-Cruz, M.; Kristen, A.V.; Grogan, M.; Witteles, R.; Damy, T.; et al. Tafamidis Treatment for Patients with Transthyretin Amyloid Cardiomyopathy. N. Engl. J. Med. 2018, 379, 1007–1016. [Google Scholar] [CrossRef]
- Berk, J.L.; Suhr, O.B.; Obici, L.; Sekijima, Y.; Zeldenrust, S.R.; Yamashita, T.; Heneghan, M.A.; Gorevic, P.D.; Litchy, W.J.; Wiesman, J.F.; et al. Diflunisal Trial Consortium. Repurposing diflunisal for familial amyloid polyneuropathy: A randomized clinical trial. JAMA 2013, 310, 2658–2667. [Google Scholar] [CrossRef] [Green Version]
- Kiuru, S. Gelsolin-related familial amyloidosis, Finnish type (FAF), and its variants found worldwide. Amyloid 1998, 5, 55–66. [Google Scholar] [CrossRef]
- Solomon, J.P.; Page, L.J.; Balch, W.E.; Kelly, J.W. Gelsolin amyloidosis: Genetics, biochemistry, pathology and possible strategies for therapeutic intervention. Crit. Rev. Biochem. Mol. Biol. 2012, 47, 282–296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nikoskinen, T.; Schmidt, E.-K.; Strbian, D.; Kiuru-Enari, S.; Atula, S. Natural course of Finnish gelsolin amyloidosis. Ann. Med. 2015, 47, 506–511. [Google Scholar] [CrossRef] [PubMed]
- Potrč, M.; Volk, M.; de Rosa, M.; Pižem, J.; Teran, N.; Jaklič, H.; Maver, A.; Drnovšek-Olup, B.; Bollati, M.; Vogelnik, K.; et al. Clinical and Histopathological Features of Gelsolin Amyloidosis Associated with a Novel GSN Variant p.Glu580Lys. Int. J. Mol. Sci. 2021, 22, 1084. [Google Scholar] [CrossRef] [PubMed]
- Kiuru-Enari, S.; Haltia, M. Chapter 39–Hereditary Gelsolin Amyloidosis. In Peripheral Nerve Disorders; Said, G., Krarup CBT-H of CN, Eds.; Elsevier: Amsterdam, The Netherlands, 2013; Volume 115, pp. 659–681. [Google Scholar] [CrossRef]
- Maury, C.; Kere, J.; Tolvanen, R.; De La Chapelle, A. Homozygosity for the Asn187 gelsolin mutation in Finnish-type familial amyloidosis is associated with severe renal disease. Genomics 1992, 13, 902–903. [Google Scholar] [CrossRef]
- Sethi, S.; Theis, J.D.; Quint, P.; Maierhofer, W.; Kurtin, P.J.; Dogan, A.; Highsmith, E.W. Renal Amyloidosis Associated With a Novel Sequence Variant of Gelsolin. Am. J. Kidney Dis. 2013, 61, 161–166. [Google Scholar] [CrossRef]
- Efebera, Y.A.; Sturm, A.; Baack, E.C.; Hofmeister, C.C.; Satoskar, A.; Nadasdy, T.; Nadasdy, G.; Benson, D.M.; Gillmore, J.D.; Hawkins, P.N.; et al. Novel gelsolin variant as the cause of nephrotic syndrome and renal amyloidosis in a large kindred. Amyloid 2014, 21, 110–112. [Google Scholar] [CrossRef]
- Carrwik, C.; Stenevi, U. Lattice corneal dystrophy, gelsolin type (Meretoja’s syndrome). Acta Ophthalmol. 2009, 87, 813–819. [Google Scholar] [CrossRef]
- Yuan, C.; Berscheit, H.L.; Huang, A.J. Identification of an amyloidogenic region on keratoepithelin via synthetic peptides. FEBS Lett. 2006, 581, 241–247. [Google Scholar] [CrossRef] [Green Version]
- Suesskind, D.; Auw-Haedrich, C.; Schorderet, D.F.; Munier, F.L.; Loeffler, K.U. Keratoepithelin in secondary corneal amyloidosis. Graefe’s Arch. Clin. Exp. Ophthalmol. 2005, 244, 725–731. [Google Scholar] [CrossRef]
- Dammacco, R.; Merlini, G.; Lisch, W.; Kivelä, T.T.; Giancipoli, E.; Vacca, A.; Dammacco, F. Amyloidosis and Ocular Involvement: An Overview. Semin. Ophthalmol. 2020, 35, 7–26. [Google Scholar] [CrossRef]
- Klintworth, G.K.; Sommer, J.R.; Han, L.; Ahmed, M.N.; Qumsiyeh, M.B.; Lin, P.Y.; Basti, S.; Reddy, M.K.; Kanai, A.; Hotta, Y.; et al. Familial subepithelial corneal amyloidosis (gelatinous drop-like corneal dystrophy): Exclusion of linkage to lactoferrin gene. Mol. Vis. 1998, 4, 31. [Google Scholar]
- Kawasaki, S.; Kinoshita, S. Clinical and basic aspects of gelatinous drop-like corneal dystrophy. Dev. Ophthalmol. 2011, 48, 97–115. [Google Scholar] [CrossRef]
- Tasaki, M.; Ueda, M.; Matsumoto, K.; Kawaji, T.; Misumi, Y.; Eiki, D.; Suenaga, G.; Obayashi, K.; Yamashita, T.; Tanihara, H.; et al. Clinico-histopathological and biochemical analyses of corneal amyloidosis in gelatinous drop-like corneal dystrophy. Amyloid 2014, 22, 67–69. [Google Scholar] [CrossRef]
- Klintworth, G.K.; Valnickova, Z.; Kielar, R.; Baratz, K.H.; Campbell, R.J.; Enghild, J.J. Familial subepithelial corneal amyloidosis--a lactoferrin-related amyloidosis. Investig. Ophthalmol. Vis. Sci. 1997, 38, 2756–2763. [Google Scholar]
- Vincent, A.L. Corneal dystrophies and genetics in the International Committee for Classification of Corneal Dystrophies era: A review. Clin. Exp. Ophthalmol. 2014, 42, 4–12. [Google Scholar] [CrossRef] [Green Version]
- Araki-Sasaki, K.; Hirano, K.; Osakabe, Y.; Kuroda, M.; Kitagawa, K.; Mishima, H.; Obata, H.; Yamada, M.; Maeda, N.; Nishida, K.; et al. Classification of Secondary Corneal Amyloidosis and Involvement of Lactoferrin. Ophthalmology 2013, 120, 1166–1172. [Google Scholar] [CrossRef]
- Araki-Sasaki, K.; Osakabe, Y.; Ideta, R.; Hirano, K.; Fukuoka, H. Findings of secondary corneal amyloidosis with ultrahigh-resolution optical coherence tomography. Clin. Ophthalmol. 2014, 8, 2115–2119. [Google Scholar] [CrossRef] [Green Version]



| Fibril Protein | Precursor Protein | Systemic and/or Localized | Acquired or Hereditary | Target Organs |
|---|---|---|---|---|
| AL | Immunoglobulin light chain | S,L | A,H | All organs, usually except CNS |
| AH | Immunoglobulin heavy chain | S,L | A | All organs except CNS |
| AA | (Apo) Serum amyloid A | S | A | All organs except CNS |
| ATTR | Transthyretin, wild type | S | A | Heart mainly in males, Lung, Ligaments, Tenosynovium |
| Transthyretin, variants | S | H | PNS, ANS, heart, eye, leptomeninges | |
| AGel | Gelsolin, variants | S | H | PNS, cornea |
| AKer | Keratoepithelin | L | A,H | Cornea |
| ALac | Lactoferrin | L | A | Cornea |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Minnella, A.M.; Rissotto, R.; Antoniazzi, E.; Di Girolamo, M.; Luigetti, M.; Maceroni, M.; Bacherini, D.; Falsini, B.; Rizzo, S.; Obici, L. Ocular Involvement in Hereditary Amyloidosis. Genes 2021, 12, 955. https://doi.org/10.3390/genes12070955
Minnella AM, Rissotto R, Antoniazzi E, Di Girolamo M, Luigetti M, Maceroni M, Bacherini D, Falsini B, Rizzo S, Obici L. Ocular Involvement in Hereditary Amyloidosis. Genes. 2021; 12(7):955. https://doi.org/10.3390/genes12070955
Chicago/Turabian StyleMinnella, Angelo Maria, Roberta Rissotto, Elena Antoniazzi, Marco Di Girolamo, Marco Luigetti, Martina Maceroni, Daniela Bacherini, Benedetto Falsini, Stanislao Rizzo, and Laura Obici. 2021. "Ocular Involvement in Hereditary Amyloidosis" Genes 12, no. 7: 955. https://doi.org/10.3390/genes12070955
APA StyleMinnella, A. M., Rissotto, R., Antoniazzi, E., Di Girolamo, M., Luigetti, M., Maceroni, M., Bacherini, D., Falsini, B., Rizzo, S., & Obici, L. (2021). Ocular Involvement in Hereditary Amyloidosis. Genes, 12(7), 955. https://doi.org/10.3390/genes12070955