
The Trifecta of Single-Cell, Systems-Biology, and Machine-Learning Approaches

 ,
, 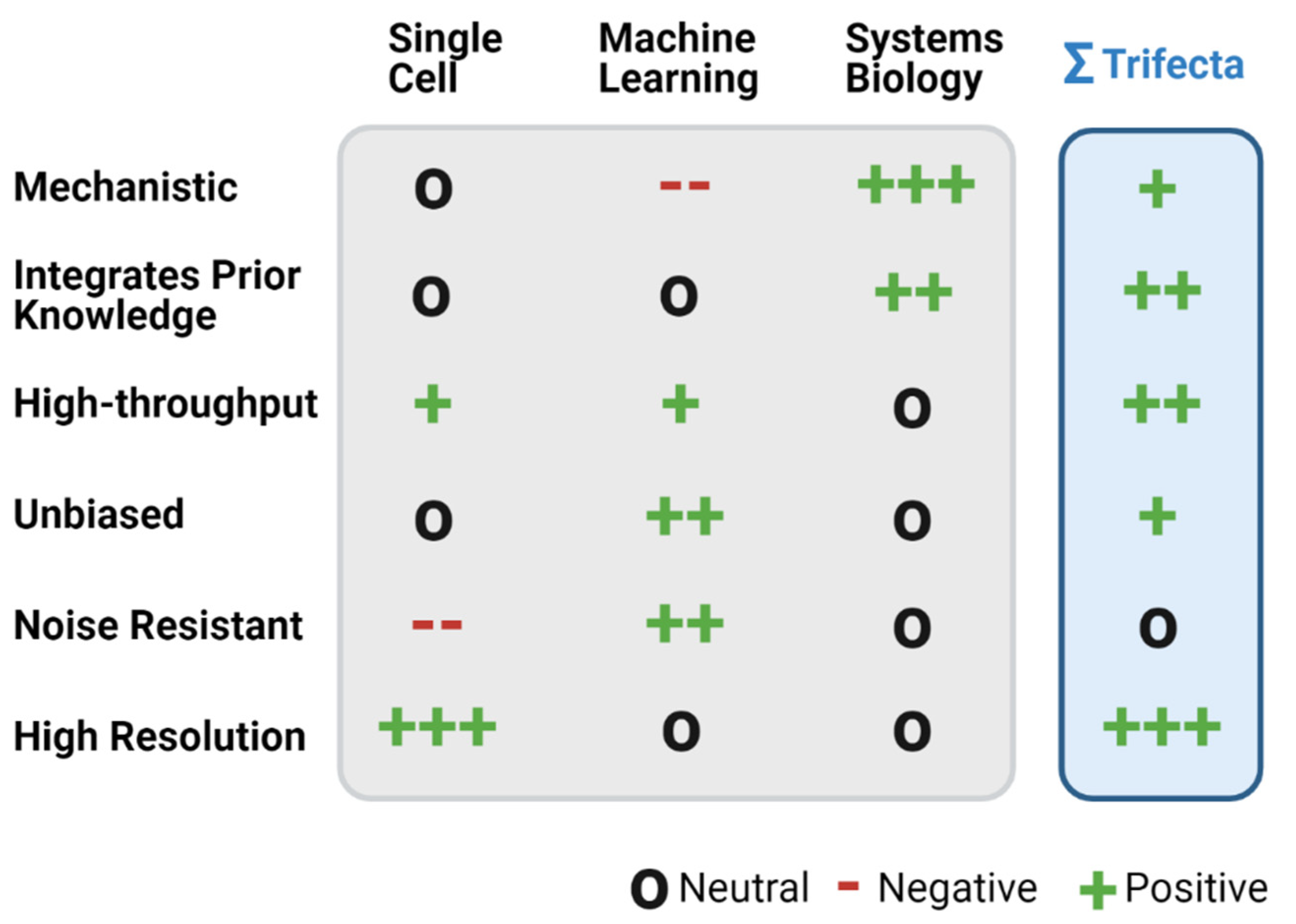{kind=link}
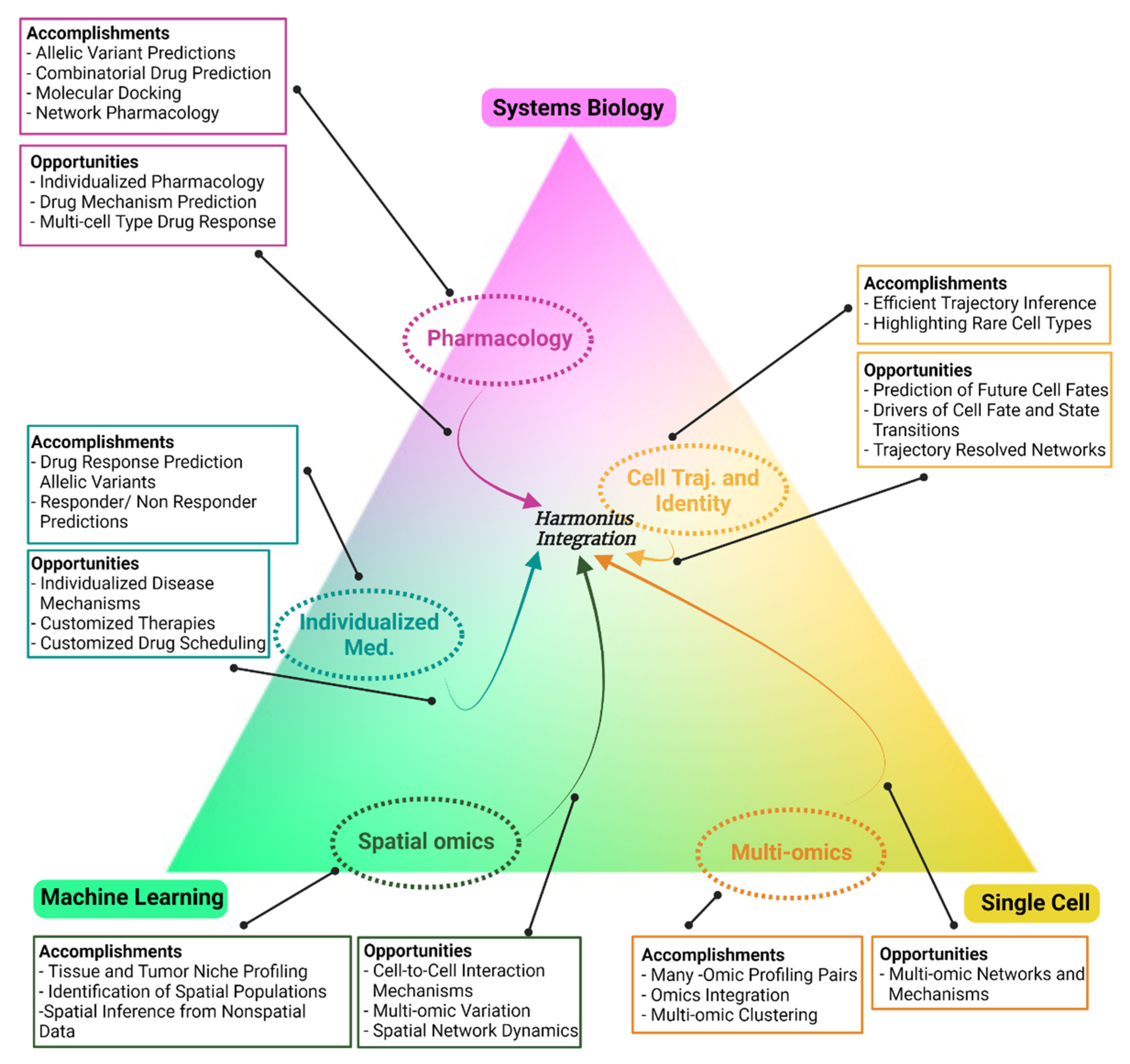{kind=link}
Abstract
1. Introduction
2. Cell Identities and Trajectories
3. Pharmacology
4. Spatial Omics
5. Multi-Omic Characterizations
6. Individualized Medicine
7. The Future of the Trifecta
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Yuan, G.C.; Cai, L.; Elowitz, M.; Enver, T.; Fan, G.; Guo, G.; Irizarry, R.; Kharchenko, P.; Kim, J.; Orkin, S.; et al. Challenges and emerging directions in single-cell analysis. Genome Biol. 2017, 18, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Svensson, V. Droplet scRNA-seq is not zero-inflated. Nat. Biotechnol. 2020, 38, 147–150. [Google Scholar] [CrossRef] [PubMed]
- Argelaguet, R.; Cuomo, A.S.E.; Stegle, O.; Marioni, J.C. Computational principles and challenges in single-cell data integration. Nat. Biotechnol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Zhao, Y.; Liao, X.; Shi, W.; Li, K.; Zou, Q.; Peng, S. Deep learning in omics: A survey and guideline. Brief. Funct. Genomics 2019, 18, 41–57. [Google Scholar] [CrossRef] [PubMed]
- Emmert-Streib, F.; Yang, Z.; Feng, H.; Tripathi, S.; Dehmer, M. An Introductory Review of Deep Learning for Prediction Models With Big Data. Front. Artif. Intell. 2020, 3, 1–23. [Google Scholar] [CrossRef]
- Nicora, G.; Vitali, F.; Dagliati, A.; Geifman, N.; Bellazzi, R. Integrated Multi-Omics Analyses in Oncology: A Review of Machine Learning Methods and Tools. Front. Oncol. 2020, 10. [Google Scholar] [CrossRef] [PubMed]
- Martorell-Marugán, J.; Tabik, S.; Benhammou, Y.; del Val, C.; Zwir, I.; Herrera, F.; Carmona-Sáez, P. Deep Learning in Omics Data Analysis and Precision Medicine. Comput. Biol. 2019, 37–53. [Google Scholar] [CrossRef]
- Nagarajan, R.; Scutari, M.; Lèbre, S. Bayesian Networks in R with Applications in Systems Biology; Springer: New York, NY, USA, 2013. [Google Scholar]
- Russell, S.; Norvig, P. Artificial Intelligence: A Modern Approach; Pearson: London, UK, 2015. [Google Scholar]
- Fortelny, N.; Bock, C. Knowledge-primed neural networks enable biologically interpretable deep learning on single-cell sequencing data. Genome Biol. 2020, 21, 1–36. [Google Scholar] [CrossRef]
- Angelov, P.; Soares, E. Towards explainable deep neural networks (xDNN). Neural Netw. 2020, 130, 185–194. [Google Scholar] [CrossRef]
- Chen, G.; Ning, B.; Shi, T. Single-cell RNA-seq technologies and related computational data analysis. Front. Genet. 2019, 10, 1–13. [Google Scholar] [CrossRef]
- Wang, D.; Bodovitz, S. Single cell analysis: The new frontier in “omics”. Trends Biotechnol. 2010, 28, 281–290. [Google Scholar] [CrossRef]
- Xing, Q.R.; El Farran, C.A.; Zeng, Y.Y.; Yi, Y.; Warrier, T.; Gautam, P.; Collins, J.J.; Xu, J.; Dröge, P.; Koh, C.G.; et al. Parallel bimodal single-cell sequencing of transcriptome and chromatin accessibility. Genome Res. 2020, 30, 1027–1039. [Google Scholar] [CrossRef]
- Wang, X.; Allen, W.E.; Wright, M.A.; Sylwestrak, E.L.; Samusik, N.; Vesuna, S.; Evans, K.; Liu, C.; Ramakrishnan, C.; Liu, J.; et al. Three-dimensional intact-tissue sequencing of single-cell transcriptional states. Science 2018, 361. [Google Scholar] [CrossRef]
- Cannoodt, R.; Saelens, W.; Saeys, Y. Computational methods for trajectory inference from single-cell transcriptomics. Eur. J. Immunol. 2016, 46, 2496–2506. [Google Scholar] [CrossRef] [PubMed]
- Saelens, W.; Cannoodt, R.; Todorov, H.; Saeys, Y. A comparison of single-cell trajectory inference methods. Nat. Biotechnol. 2019, 37, 547–554. [Google Scholar] [CrossRef]
- Li, X.; Guo, X.; Zhu, Y.; Wei, G.; Zhang, Y.; Li, X.; Xu, H.; Cui, J.; Wu, W.; He, J.; et al. Single-Cell Transcriptomic Analysis Reveals BCMA CAR-T Cell Dynamics in a Patient with Refractory Primary Plasma Cell Leukemia. Mol. Ther. 2021, 29, 645–657. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; MacLean, A.; Nie, Q. Low-Rank Similarity Matrix Optimization Identifies Subpopulation Structure and Orders Single Cells in Pseudotime. bioRxiv 2017, 168922. [Google Scholar] [CrossRef]
- Chickarmane, V.; Enver, T.; Peterson, C. Computational modeling of the hematopoietic erythroid-myeloid switch reveals insights into cooperativity, priming, and irreversibility. PLoS Comput. Biol. 2009, 5. [Google Scholar] [CrossRef] [PubMed]
- Marr, C.; Strasser, M.; Schwarzfischer, M.; Schroeder, T.; Theis, F.J. Multi-scale modeling of GMP differentiation based on single-cell genealogies. FEBS J. 2012, 279, 3488–3500. [Google Scholar] [CrossRef]
- Wagner, A.; Regev, A.; Yosef, N. Revealing the vectors of cellular identity with single-cell genomics. Nat. Biotechnol. 2016, 34, 1145–1160. [Google Scholar] [CrossRef] [PubMed]
- Woo, J.H.; Shimoni, Y.; Yang, W.S.; Subramaniam, P.; Iyer, A.; Nicoletti, P.; Rodríguez Martínez, M.; López, G.; Mattioli, M.; Realubit, R.; et al. Elucidating Compound Mechanism of Action by Network Perturbation Analysis. Cell 2015, 162, 441–451. [Google Scholar] [CrossRef]
- Ghanat Bari, M.; Ung, C.Y.; Zhang, C.; Zhu, S.; Li, H. Machine Learning-Assisted Network Inference Approach to Identify a New Class of Genes that Coordinate the Functionality of Cancer Networks. Sci. Rep. 2017, 7, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Ung, C.Y.; Ghanat Bari, M.; Zhang, C.; Liang, J.; Correia, C.; Li, H. Regulostat Inferelator: A novel network biology platform to uncover molecular devices that predetermine cellular response phenotypes. Nucleic Acids Res. 2019, 47, e82. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Narayan, R.; Corsello, S.M.; Peck, D.D.; Natoli, T.E.; Lu, X.; Gould, J.; Davis, J.F.; Tubelli, A.A.; Asiedu, J.K.; et al. A Next Generation Connectivity Map: L1000 Platform and the First 1,000,000 Profiles. Cell 2017, 171, 1437–1452.e17. [Google Scholar] [CrossRef]
- Tsherniak, A.; Vazquez, F.; Montgomery, P.G.; Weir, B.A.; Kryukov, G.; Cowley, G.S.; Gill, S.; Harrington, W.F.; Krill-burger, J.M.; Meyers, R.M.; et al. HHS Public Access Defining a Cancer Dependency Map. Natl. Lab. Med. 2018, 170, 564–576. [Google Scholar] [CrossRef]
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z.; et al. DrugBank 5.0: A major update to the DrugBank database for 2018. Nucleic Acids Res. 2018, 46, D1074–D1082. [Google Scholar] [CrossRef]
- Zhao, S.; Iyengar, R. Systems Pharmacology: Network Analysis to Identify Multiscale Mechanisms of Drug Action. Annu. Rev. Pharmacol. Toxicol. 2012, 52, 505–512. [Google Scholar] [CrossRef] [PubMed]
- Sharifi-Noghabi, H.; Zolotareva, O.; Collins, C.C.; Ester, M. MOLI: Multi-omics late integration with deep neural networks for drug response prediction. Bioinformatics 2019, 35, i501–i509. [Google Scholar] [CrossRef]
- Kandaswamy, C.; Silva, L.M.; Alexandre, L.A.; Santos, J.M. High-Content Analysis of Breast Cancer Using Single-Cell Deep Transfer Learning. J. Biomol. Screen. 2016, 21, 252–259. [Google Scholar] [CrossRef][Green Version]
- Gautam, P.; Jaiswal, A.; Aittokallio, T.; Al-Ali, H.; Wennerberg, K. Phenotypic Screening Combined with Machine Learning for Efficient Identification of Breast Cancer-Selective Therapeutic Targets. Cell Chem. Biol. 2019, 26, 970–979.e4. [Google Scholar] [CrossRef]
- O’Duibhir, E.; Paris, J.; Lawson, H.; Sepulveda, C.; Shenton, D.D.; Carragher, N.O.; Kranc, K.R. Machine Learning Enables Live Label-Free Phenotypic Screening in Three Dimensions. Assay Drug Dev. Technol. 2018, 16, 51–63. [Google Scholar] [CrossRef] [PubMed]
- Waylen, L.N.; Nim, H.T.; Martelotto, L.G.; Ramialison, M. From whole-mount to single-cell spatial assessment of gene expression in 3D. Commun. Biol. 2020, 3, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Satija, R.; Farrell, J.A.; Gennert, D.; Schier, A.F.; Regev, A. Spatial reconstruction of single-cell gene expression data. Nat. Biotechnol. 2015, 33, 495–502. [Google Scholar] [CrossRef] [PubMed]
- Ren, X.; Zhong, G.; Zhang, Q.; Zhang, L.; Sun, Y.; Zhang, Z. Reconstruction of cell spatial organization from single-cell RNA sequencing data based on ligand-receptor mediated self-assembly. Cell Res. 2020, 30, 763–778. [Google Scholar] [CrossRef]
- Dries, R.; Zhu, Q.; Dong, R.; Eng, C.H.L.; Li, H.; Liu, K.; Fu, Y.; Zhao, T.; Sarkar, A.; Bao, F.; et al. Giotto: A toolbox for integrative analysis and visualization of spatial expression data. Genome Biol. 2021, 22, 1–31. [Google Scholar] [CrossRef] [PubMed]
- Cang, Z.; Nie, Q. Inferring spatial and signaling relationships between cells from single cell transcriptomic data. Nat. Commun. 2020, 11, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Hyeon, D.Y.; Hwang, D. Single-cell multiomics: Technologies and data analysis methods. Exp. Mol. Med. 2020, 52, 1428–1442. [Google Scholar] [CrossRef]
- Argelaguet, R.; Arnol, D.; Bredikhin, D.; Deloro, Y.; Velten, B.; Marioni, J.C.; Stegle, O. MOFA+: A statistical framework for comprehensive integration of multi-modal single-cell data. Genome Biol. 2020, 21, 1–17. [Google Scholar] [CrossRef]
- Welch, J.D.; Kozareva, V.; Ferreira, A.; Vanderburg, C.; Martin, C.; Macosko, E.Z. Single-Cell Multi-omic Integration Compares and Contrasts Features of Brain Cell Identity. Cell 2019, 177, 1873–1887.e17. [Google Scholar] [CrossRef]
- Pinu, F.R.; Beale, D.J.; Paten, A.M.; Kouremenos, K.; Swarup, S.; Schirra, H.J.; Wishart, D. Systems biology and multi-omics integration: Viewpoints from the metabolomics research community. Metabolites 2019, 9, 76. [Google Scholar] [CrossRef]
- Ma, T.; Zhang, A. Integrate multi-omics data with biological interaction networks using Multi-view Factorization AutoEncoder (MAE). BMC Genom. 2019, 20, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Ramazzotti, D.; Lal, A.; Wang, B.; Batzoglou, S.; Sidow, A. Multi-omic tumor data reveal diversity of molecular mechanisms that correlate with survival. Nat. Commun. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Poplin, R.; Chang, P.C.; Alexander, D.; Schwartz, S.; Colthurst, T.; Ku, A.; Newburger, D.; Dijamco, J.; Nguyen, N.; Afshar, P.T.; et al. A universal snp and small-indel variant caller using deep neural networks. Nat. Biotechnol. 2018, 36, 983. [Google Scholar] [CrossRef]
- Xiong, H.Y.; Alipanahi, B.; Lee, L.J.; Bretschneider, H.; Yuen, R.K.C.; Hua, Y.; Gueroussov, S.; Hamed, S.; Hughes, T.R.; Morris, Q.; et al. The human splicing code reveals new insights into the genetic determinants of disease. Science 2015, 347. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Theesfeld, C.L.; Yao, K.; Chen, K.M.; Wong, A.K.; Troyanskaya, O.G. Deep learning sequence-based ab initio prediction of variant effects on expression and disease risk. Nat. Genet. 2018, 50, 1171–1179. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Wu, P.; Chen, Y.; Meng, Q.; Dawood, H.; Dawood, H. A hierarchical integration deep flexible neural forest framework for cancer subtype classification by integrating multi-omics data. BMC Bioinform. 2019, 20, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Azuaje, F. Artificial intelligence for precision oncology: Beyond patient stratification. NPJ Precis. Oncol. 2019, 3, 1–5. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Weiskittel, T.M.; Correia, C.; Yu, G.T.; Ung, C.Y.; Kaufmann, S.H.; Billadeau, D.D.; Li, H. The Trifecta of Single-Cell, Systems-Biology, and Machine-Learning Approaches. Genes 2021, 12, 1098. https://doi.org/10.3390/genes12071098
Weiskittel TM, Correia C, Yu GT, Ung CY, Kaufmann SH, Billadeau DD, Li H. The Trifecta of Single-Cell, Systems-Biology, and Machine-Learning Approaches. Genes. 2021; 12(7):1098. https://doi.org/10.3390/genes12071098
Chicago/Turabian StyleWeiskittel, Taylor M., Cristina Correia, Grace T. Yu, Choong Yong Ung, Scott H. Kaufmann, Daniel D. Billadeau, and Hu Li. 2021. "The Trifecta of Single-Cell, Systems-Biology, and Machine-Learning Approaches" Genes 12, no. 7: 1098. https://doi.org/10.3390/genes12071098
APA StyleWeiskittel, T. M., Correia, C., Yu, G. T., Ung, C. Y., Kaufmann, S. H., Billadeau, D. D., & Li, H. (2021). The Trifecta of Single-Cell, Systems-Biology, and Machine-Learning Approaches. Genes, 12(7), 1098. https://doi.org/10.3390/genes12071098