
Investigating Cellular Trajectories in the Severity of COVID-19 and Their Transcriptional Programs Using Machine Learning Approaches


Abstract
1. Introduction
2. Materials and Methods
2.1. COVID-19 BALF scRNA-Seq Data
2.2. Trajectory Inference Using Slingshot
2.3. Transcriptional Program Identification
3. Results
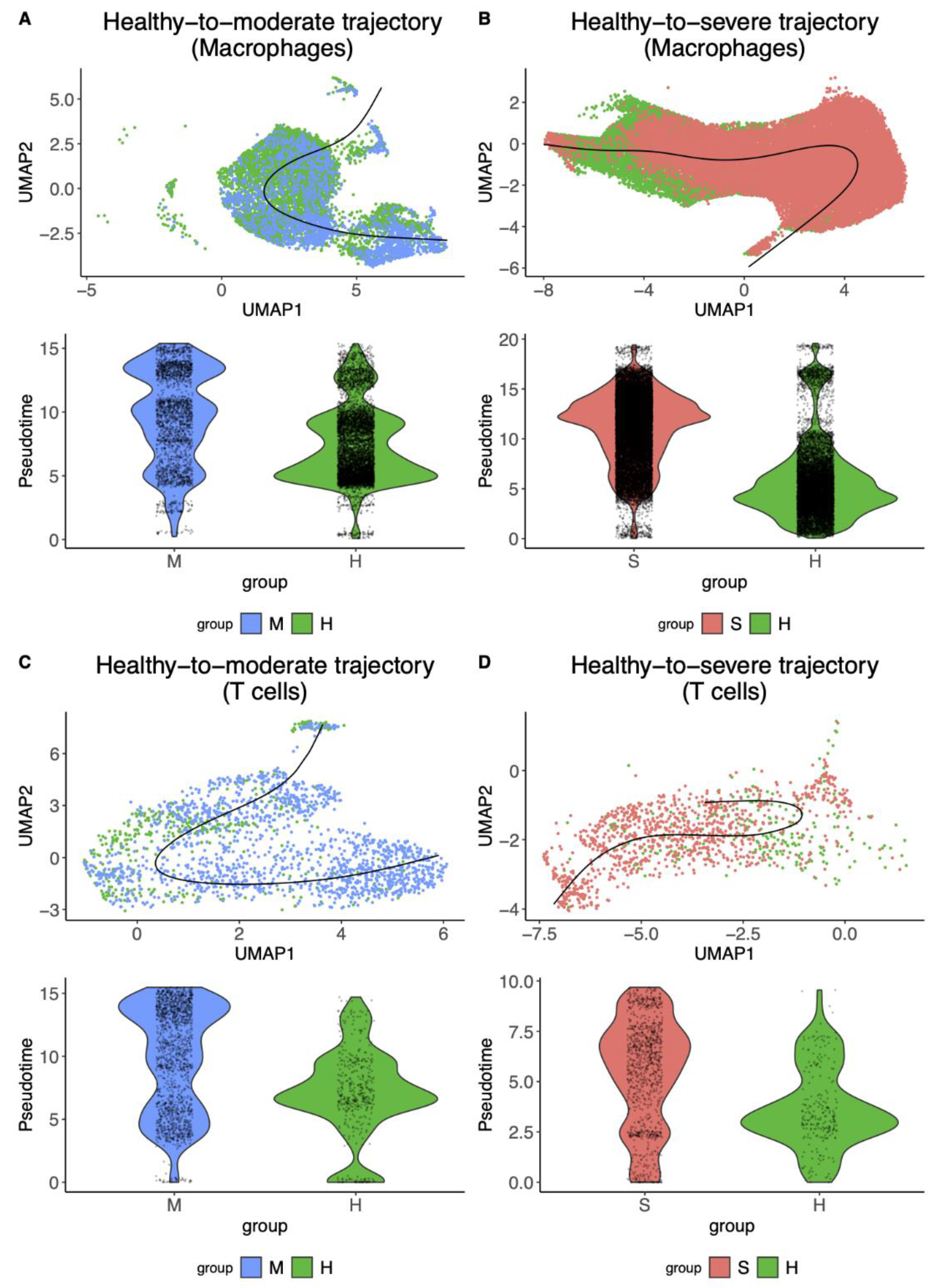
3.1. Macrophages and T Cells Exhibited More Diverse Cellular Trajectories from Healthy Controls to COVID-19 Patients
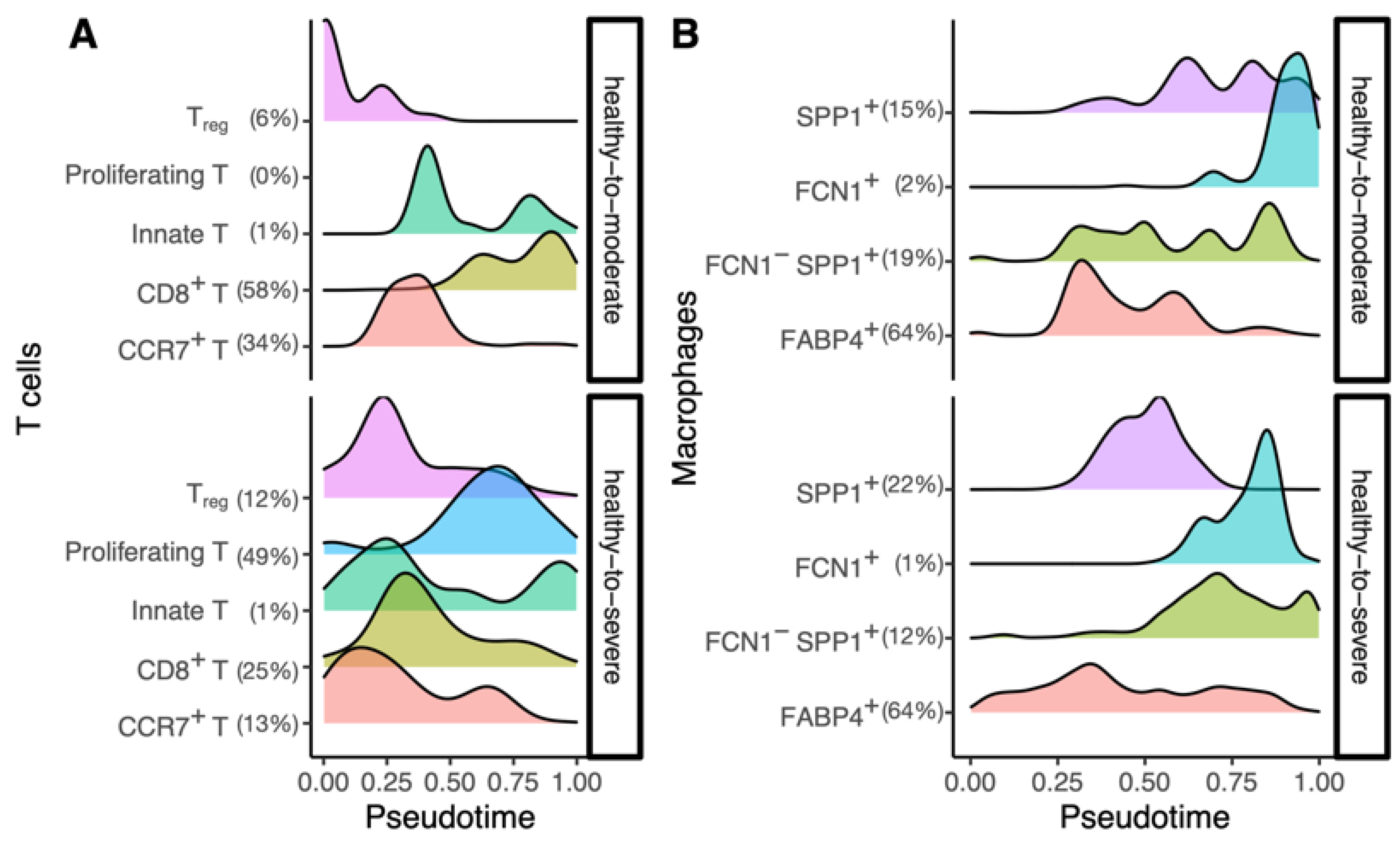
3.2. SARS-CoV-2 Gene-Expression Pattern Could Dissect the Healthy-to-Severe Trajectories into Three Different Stages of COVID-19
3.3. The Inferred Cellular-Trajectories Model the Cell Type-Specific Immune Response
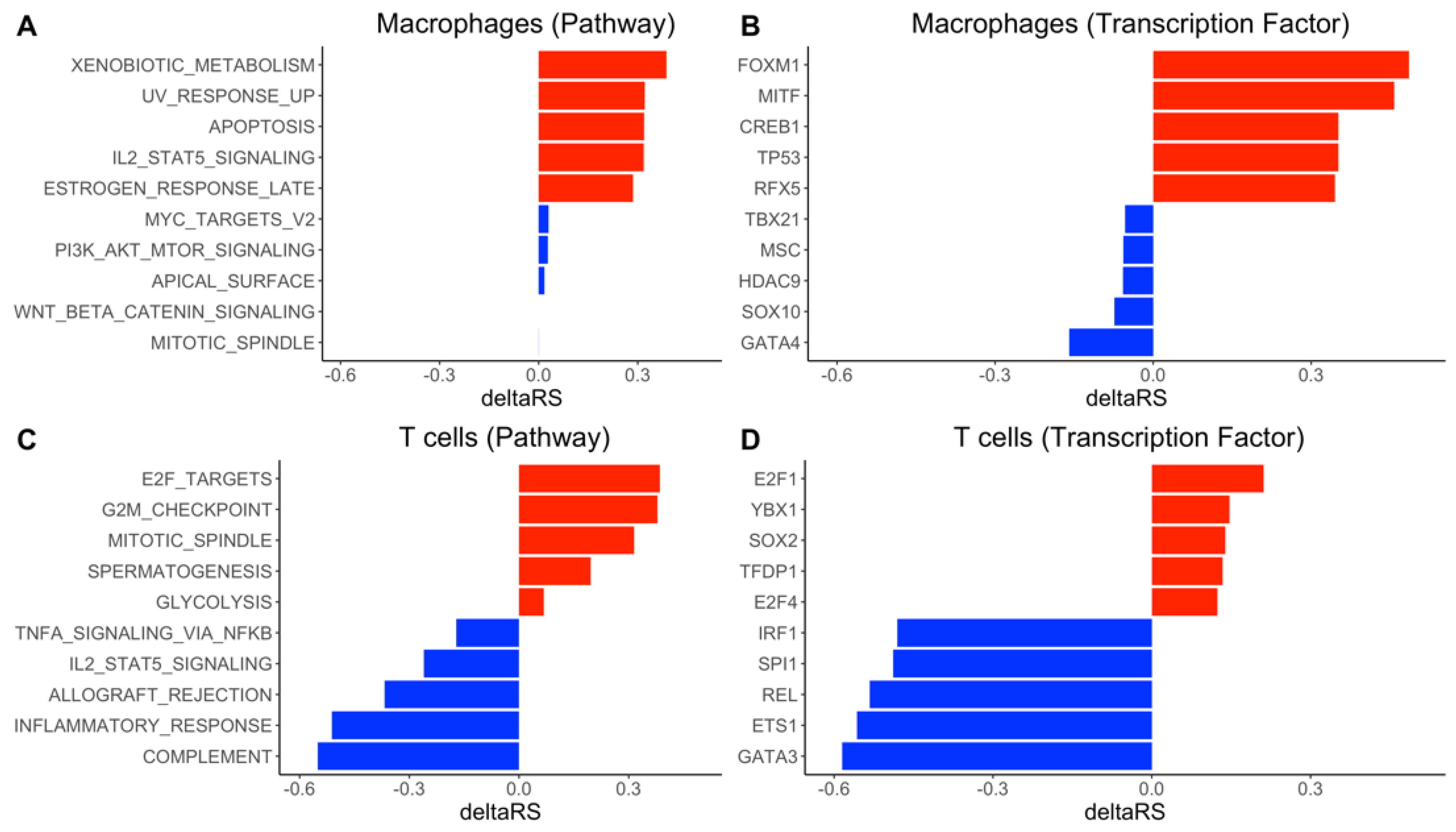
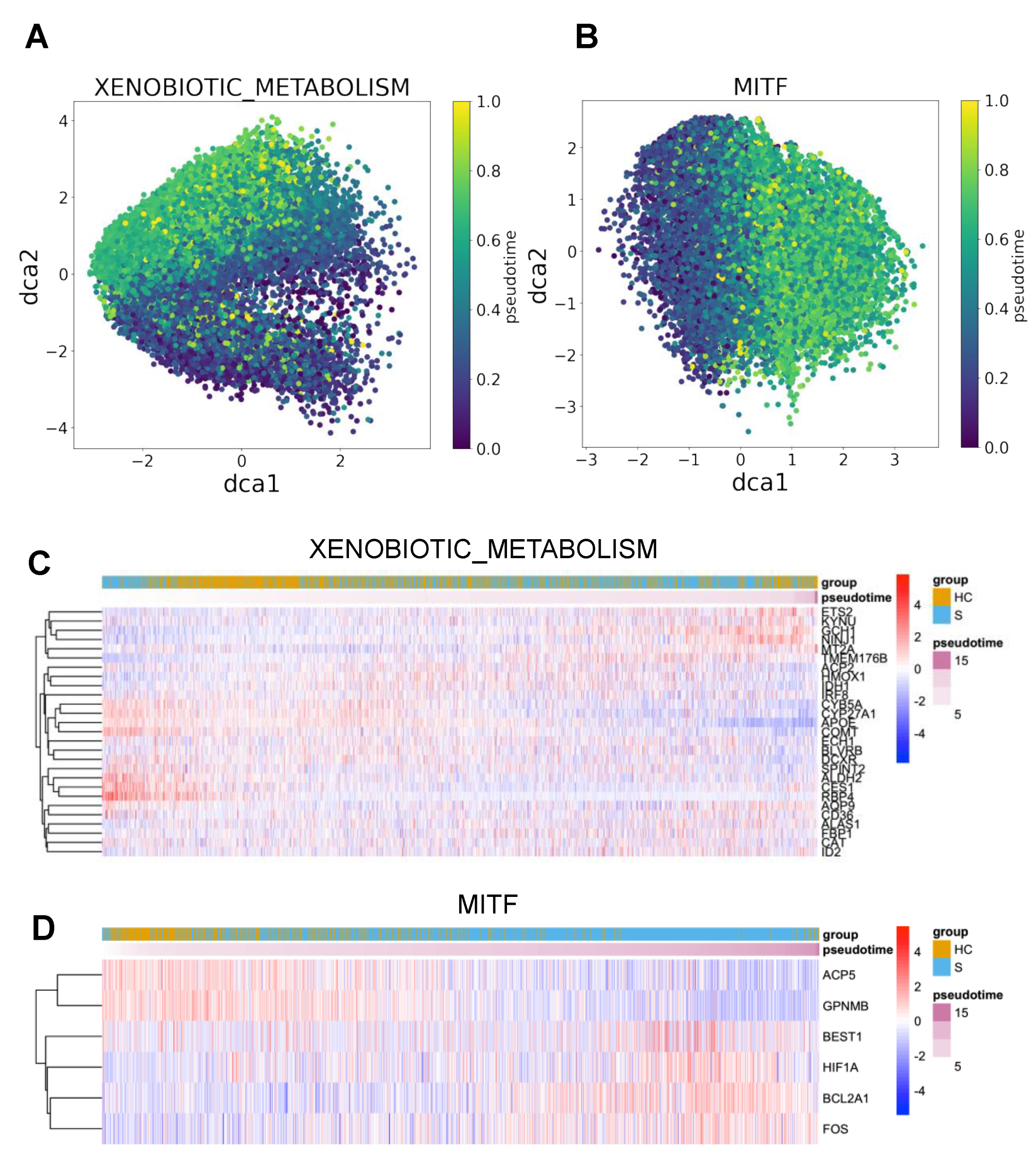
3.4. DrivAER Identified Potential Transcriptional Programs That Differentiate The Severity of COVID-19
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- CDC COVID Data Tracker. Available online: https://covid.cdc.gov/covid-data-tracker/ (accessed on 3 February 2021).
- Ahmed, A.; Ali, A.; Hasan, S. Comparison of Epidemiological Variations in COVID-19 Patients Inside and Outside of China-A Meta-Analysis. Front. Public Health 2020, 8, 193. [Google Scholar] [CrossRef] [PubMed]
- Goodman, K.E.; Magder, L.S.; Baghdadi, J.D.; Pineles, L.; Levine, A.R.; Perencevich, E.N.; Harris, A.D. Impact of Sex and Metabolic Comorbidities on COVID-19 Mortality Risk Across Age Groups: 66,646 Inpatients Across 613 U.S. Hospitals. Clin. Infect. Dis. 2020. [Google Scholar] [CrossRef]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef]
- Xiong, J.; Lipsitz, O.; Nasri, F.; Lui, L.M.W.; Gill, H.; Phan, L.; Chen-Li, D.; Iacobucci, M.; Ho, R.; Majeed, A.; et al. Impact of COVID-19 pandemic on mental health in the general population: A systematic review. J. Affect. Disord. 2020, 277, 55–64. [Google Scholar] [CrossRef]
- Gustine, J.N.; Jones, D. Immunopathology of Hyperinflammation in COVID-19. Am. J. Pathol. 2021, 191, 4–17. [Google Scholar] [CrossRef] [PubMed]
- Ye, Q.; Wang, B.; Mao, J. The pathogenesis and treatment of the `Cytokine Storm’ in COVID-19. J. Infect. 2020, 80, 607–613. [Google Scholar] [CrossRef]
- Dai, Y.; Hu, R.; Manuel, A.M.; Liu, A.; Jia, P.; Zhao, Z. CSEA-DB: An omnibus for human complex trait and cell type associations. Nucleic Acids Res. 2021, 49, 862–870. [Google Scholar] [CrossRef] [PubMed]
- Liao, M.; Liu, Y.; Yuan, J.; Wen, Y.; Xu, G.; Zhao, J.; Cheng, L.; Li, J.; Wang, X.; Wang, F.; et al. Single-cell landscape of bronchoalveolar immune cells in patients with COVID-19. Nat. Med. 2020, 26, 842–844. [Google Scholar] [CrossRef]
- Liu, T.; Jia, P.; Fang, B.; Zhao, Z. Differential Expression of Viral Transcripts From Single-Cell RNA Sequencing of Moderate and Severe COVID-19 Patients and Its Implications for Case Severity. Front. Microbiol. 2020, 11, 603509. [Google Scholar] [CrossRef]
- Xu, G.; Qi, F.; Li, H.; Yang, Q.; Wang, H.; Wang, X.; Liu, X.; Zhao, J.; Liao, X.; Liu, Y.; et al. The differential immune responses to COVID-19 in peripheral and lung revealed by single-cell RNA sequencing. Cell Discov. 2020, 6, 73. [Google Scholar] [CrossRef]
- Zhao, Z.; Li, H.; Wu, X.; Zhong, Y.; Zhang, K.; Zhang, Y.-P.; Boerwinkle, E.; Fu, Y.-X. Moderate mutation rate in the SARS coronavirus genome and its implications. BMC Evol. Biol. 2004, 4, 21. [Google Scholar] [CrossRef]
- Liu, S.; Shen, J.; Fang, S.; Li, K.; Liu, J.; Yang, L.; Hu, C.-D.; Wan, J. Genetic spectrum and distinct evolution patterns of SARS-CoV-2. Front. Microbiol. 2020, 11, 593548. [Google Scholar] [CrossRef] [PubMed]
- Street, K.; Risso, D.; Fletcher, R.B.; Das, D.; Ngai, J.; Yosef, N.; Purdom, E.; Dudoit, S. Slingshot: Cell lineage and pseudotime inference for single-cell transcriptomics. BMC Genom. 2018, 19, 477. [Google Scholar] [CrossRef] [PubMed]
- Simon, L.M.; Yan, F.; Zhao, Z. DrivAER: Identification of driving transcriptional programs in single-cell RNA sequencing data. Gigascience 2020, 9. [Google Scholar] [CrossRef]
- UCSC Cell Browser. Available online: https://cells.ucsc.edu/?ds=covid19-balf (accessed on 5 February 2021).
- McInnes, L.; Healy, J.; Melville, J. UMAP: Uniform Manifold Approximation and Projection for Dimension Reduction. arXiv 2018, arXiv:1802.03426. [Google Scholar]
- Stuart, T.; Butler, A.; Hoffman, P.; Hafemeister, C.; Papalexi, E.; Mauck, W.M., 3rd; Hao, Y.; Stoeckius, M.; Smibert, P.; Satija, R. Comprehensive Integration of Single-Cell Data. Cell 2019, 177, 1888–1902. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Akat, K.M.; Sun, Z.; Zhang, W.; Schlondorff, D.; Liu, Z.; Tuschl, T.; Lee, K.; He, J.C. Single-Cell RNA Profiling of Glomerular Cells Shows Dynamic Changes in Experimental Diabetic Kidney Disease. J. Am. Soc. Nephrol. 2019, 30, 533–545. [Google Scholar] [CrossRef]
- Saelens, W.; Cannoodt, R.; Todorov, H.; Saeys, Y. A comparison of single-cell trajectory inference methods. Nat. Biotechnol. 2019, 37, 547–554. [Google Scholar] [CrossRef] [PubMed]
- Heimberg, G.; Bhatnagar, R.; El-Samad, H.; Thomson, M. Low Dimensionality in Gene Expression Data Enables the Accurate Extraction of Transcriptional Programs from Shallow Sequencing. Cell Syst. 2016, 2, 239–250. [Google Scholar] [CrossRef]
- Eraslan, G.; Simon, L.M.; Mircea, M.; Mueller, N.S.; Theis, F.J. Single-cell RNA-seq denoising using a deep count autoencoder. Nat. Commun. 2019, 10, 390. [Google Scholar] [CrossRef] [PubMed]
- Liberzon, A.; Birger, C.; Thorvaldsdóttir, H.; Ghandi, M.; Mesirov, J.P.; Tamayo, P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 2015, 1, 417–425. [Google Scholar] [CrossRef]
- Han, H.; Cho, J.-W.; Lee, S.; Yun, A.; Kim, H.; Bae, D.; Yang, S.; Kim, C.Y.; Lee, M.; Kim, E.; et al. TRRUST v2: An expanded reference database of human and mouse transcriptional regulatory interactions. Nucleic Acids Res. 2018, 46, 380–386. [Google Scholar] [CrossRef] [PubMed]
- Wickham, H. ggplot2. WIRes Comput. Stat. 2011, 3, 180–185. [Google Scholar] [CrossRef]
- Patterson, A.D.; Gonzalez, F.J.; Idle, J.R. Xenobiotic metabolism: A view through the metabolometer. Chem. Res. Toxicol. 2010, 23, 851–860. [Google Scholar] [CrossRef] [PubMed]
- El-Ghiaty, M.A.; Shoieb, S.M.; El-Kadi, A.O.S. Cytochrome P450-mediated drug interactions in COVID-19 patients: Current findings and possible mechanisms. Med. Hypotheses 2020, 144, 110033. [Google Scholar] [CrossRef] [PubMed]
- Hukkanen, J.; Pelkonen, O.; Raunio, H. Expression of xenobiotic-metabolizing enzymes in human pulmonary tissue: Possible role in susceptibility for ILD. Eur. Respir. J. Suppl. 2001, 32, 122–126. [Google Scholar]
- Tian, L.-X.; Tang, X.; Zhu, J.-Y.; Luo, L.; Ma, X.-Y.; Cheng, S.-W.; Zhang, W.; Tang, W.-Q.; Ma, W.; Yang, X.; et al. Cytochrome P450 1A1 enhances inflammatory responses and impedes phagocytosis of bacteria in macrophages during sepsis. Cell Commun. Signal. 2020, 18, 70. [Google Scholar] [CrossRef]
- Cecchini, R.; Cecchini, A.L. SARS-CoV-2 infection pathogenesis is related to oxidative stress as a response to aggression. Med. Hypotheses 2020, 143, 110102. [Google Scholar] [CrossRef]
- Otsuka, R.; Seino, K.-I. Macrophage activation syndrome and COVID-19. Inflamm. Regen. 2020, 40, 19. [Google Scholar] [CrossRef]
- Kawakami, A.; Fisher, D.E. The master role of microphthalmia-associated transcription factor in melanocyte and melanoma biology. Lab. Investig. 2017, 97, 649–656. [Google Scholar] [CrossRef]
- Garraway, L.A.; Sellers, W.R. Lineage dependency and lineage-survival oncogenes in human cancer. Nat. Rev. Cancer 2006, 6, 593–602. [Google Scholar] [CrossRef]
- Bost, P.; Giladi, A.; Liu, Y.; Bendjelal, Y.; Xu, G.; David, E.; Blecher-Gonen, R.; Cohen, M.; Medaglia, C.; Li, H.; et al. Host-Viral Infection Maps Reveal Signatures of Severe COVID-19 Patients. Cell 2020, 181, 1475–1488. [Google Scholar] [CrossRef]
- Harris, M.L.; Fufa, T.D.; Palmer, J.W.; Joshi, S.S.; Larson, D.M.; Incao, A.; Gildea, D.E.; Trivedi, N.S.; Lee, A.N.; Day, C.-P.; et al. A direct link between MITF, innate immunity, and hair graying. PLoS Biol. 2018, 16, 2003648. [Google Scholar] [CrossRef]
- Douglass, T.G.; Driggers, L.; Zhang, J.G.; Hoa, N.; Delgado, C.; Williams, C.C.; Dan, Q.; Sanchez, R.; Jeffes, E.W.B.; Wepsic, H.T.; et al. Macrophage colony stimulating factor: Not just for macrophages anymore! A gateway into complex biologies. Int. Immunopharmacol. 2008, 8, 1354–1376. [Google Scholar] [CrossRef] [PubMed]
- Mehta, P.; Porter, J.C.; Manson, J.J.; Isaacs, J.D.; Openshaw, P.J.M.; McInnes, I.B.; Summers, C.; Chambers, R.C. Therapeutic blockade of granulocyte macrophage colony-stimulating factor in COVID-19-associated hyperinflammation: Challenges and opportunities. Lancet Respir. Med. 2020, 8, 822–830. [Google Scholar] [CrossRef]
- Cheng, L.-L.; Guan, W.-J.; Duan, C.-Y.; Zhang, N.-F.; Lei, C.-L.; Hu, Y.; Chen, A.-L.; Li, S.-Y.; Zhuo, C.; Deng, X.-L.; et al. Effect of Recombinant Human Granulocyte Colony-Stimulating Factor for Patients With Coronavirus Disease 2019 (COVID-19) and Lymphopenia: A Randomized Clinical Trial. JAMA Intern. Med. 2021, 181, 71–78. [Google Scholar] [CrossRef] [PubMed]
- de Candia, P.; Prattichizzo, F.; Garavelli, S.; Matarese, G. T Cells: Warriors of SARS-CoV-2 Infection. Trends Immunol. 2021, 42, 18–30. [Google Scholar] [CrossRef] [PubMed]
- Qin, C.; Zhou, L.; Hu, Z.; Zhang, S.; Yang, S.; Tao, Y.; Xie, C.; Ma, K.; Shang, K.; Wang, W.; et al. Dysregulation of Immune Response in Patients With Coronavirus 2019 (COVID-19) in Wuhan, China. Clin. Infect. Dis. 2020, 71, 762–768. [Google Scholar] [CrossRef] [PubMed]
- Dai, Y.; Wang, J.; Jeong, H.-H.; Chen, W.; Jia, P.; Zhao, Z. Association of CXCR6 with COVID-19 severity: Delineating the host genetic factors in transcriptomic regulation. bioRxiv 2021. [Google Scholar] [CrossRef]
- Liu, L.; Xu, L.; Lin, C. T cell response in patients with COVID-19. Blood Sci. 2020, 2, 76. [Google Scholar] [CrossRef]
- Stark, G.R.; Taylor, W.R. Analyzing the G2/M checkpoint. Methods Mol. Biol. 2004, 280, 51–82. [Google Scholar] [CrossRef]
- Kirkham, P.A.; Lam, E.W.; Takamatsu, H.H.; Parkhouse, R.M. Transcription factor E2F controls the reversible gamma delta T cell growth arrest mediated through WC1. J. Immunol. 1998, 161, 1630–1636. [Google Scholar] [PubMed]
- Inoué, S. Cell division and the mitotic spindle. J. Cell Biol. 1981, 91, 131–147. [Google Scholar] [CrossRef] [PubMed]
- Janeway, C.A., Jr.; Travers, P.; Walport, M.; Shlomchik, M.J. The Complement System and Innate Immunity; Garland Science: New York, NY, USA, 2001. [Google Scholar]
- Dunkelberger, J.R.; Song, W.-C. Complement and its role in innate and adaptive immune responses. Cell Res. 2010, 20, 34–50. [Google Scholar] [CrossRef] [PubMed]
- Holter, J.C.; Pischke, S.E.; de Boer, E.; Lind, A.; Jenum, S.; Holten, A.R.; Tonby, K.; Barratt-Due, A.; Sokolova, M.; Schjalm, C.; et al. Systemic complement activation is associated with respiratory failure in COVID-19 hospitalized patients. Proc. Natl. Acad. Sci. USA 2020, 117, 25018–25025. [Google Scholar] [CrossRef]
- Java, A.; Apicelli, A.J.; Liszewski, M.K.; Coler-Reilly, A.; Atkinson, J.P.; Kim, A.H.; Kulkarni, H.S. The complement system in COVID-19: Friend and foe? JCI Insight 2020, 5. [Google Scholar] [CrossRef] [PubMed]
- Defendi, F.; Thielens, N.M.; Clavarino, G.; Cesbron, J.-Y.; Dumestre-Pérard, C. The Immunopathology of Complement Proteins and Innate Immunity in Autoimmune Disease. Clin. Rev. Allergy Immunol. 2020, 58, 229–251. [Google Scholar] [CrossRef] [PubMed]
- Markiewski, M.M.; Lambris, J.D. The role of complement in inflammatory diseases from behind the scenes into the spotlight. Am. J. Pathol. 2007, 171, 715–727. [Google Scholar] [CrossRef]
- Kwan, W.-H.; van der Touw, W.; Heeger, P.S. Complement regulation of T cell immunity. Immunol. Res. 2012, 54, 247–253. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Peng, W.; McKenzie, J.A.; Hwu, P. Complementing T-cell Function: An Inhibitory Role of the Complement System in T-cell-Mediated Antitumor Immunity. Cancer Discov. 2016, 6, 953–955. [Google Scholar] [CrossRef]
- Lee, S.H.; Kwon, J.Y.; Kim, S.-Y.; Jung, K.; Cho, M.-L. Interferon-gamma regulates inflammatory cell death by targeting necroptosis in experimental autoimmune arthritis. Sci. Rep. 2017, 7, 10133. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, T.; Onodera, A.; Hosokawa, H.; Watanabe, Y.; Horiuchi, S.; Yamashita, J.; Tanaka, H.; Ogawa, Y.; Suzuki, Y.; Nakayama, T. Genome-Wide Gene Expression Profiling Revealed a Critical Role for GATA3 in the Maintenance of the Th2 Cell Identity. PLoS ONE 2013, 8, 66468. [Google Scholar] [CrossRef] [PubMed]
- Tindemans, I.; Serafini, N.; Di Santo, J.P.; Hendriks, R.W. GATA-3 function in innate and adaptive immunity. Immunity 2014, 41, 191–206. [Google Scholar] [CrossRef] [PubMed]
- Koyasu, S.; Moro, K. Type 2 innate immune responses and the natural helper cell. Immunology 2011, 132, 475–481. [Google Scholar] [CrossRef]
- Roncati, L.; Nasillo, V.; Lusenti, B.; Riva, G. Signals of Th2 immune response from COVID-19 patients requiring intensive care. Ann. Hematol. 2020, 99, 1419–1420. [Google Scholar] [CrossRef] [PubMed]
- Li, C.K.-F.; Wu, H.; Yan, H.; Ma, S.; Wang, L.; Zhang, M.; Tang, X.; Temperton, N.J.; Weiss, R.A.; Brenchley, J.M.; et al. T cell responses to whole SARS coronavirus in humans. J. Immunol. 2008, 181, 5490–5500. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Type | Trajectory | Number of Healthy Cells | Number of Moderate/Severe Cells | Fold-Change | p-Value |
|---|---|---|---|---|---|
| Macrophage | 6370 | 2168 | 0.37 | ||
| 10,492 | 19,798 | 0.94 | |||
| T cell | 414 | 1524 | 0.47 | ||
| 237 | 975 | 0.56 |
| Cell Type | Trajectory | Gene | Number of Infected Cells | Average (Pseudotime) |
|---|---|---|---|---|
| Macrophage | S | 104 | 0.46 0.19 | |
| ORF8 | 41 | 0.46 0.20 | ||
| N | 258 | 0.48 0.21 | ||
| ORF10 | 67 | 0.49 0.20 | ||
| ORF3a | 24 | 0.49 0.21 | ||
| M | 34 | 0.50 0.22 | ||
| ORF1ab | 467 | 0.52 0.21 | ||
| ORF7a | 403 | 0.57 0.18 | ||
| T cell | ORF1ab | 38 | 0.61 0.27 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jeong, H.-H.; Jia, J.; Dai, Y.; Simon, L.M.; Zhao, Z. Investigating Cellular Trajectories in the Severity of COVID-19 and Their Transcriptional Programs Using Machine Learning Approaches. Genes 2021, 12, 635. https://doi.org/10.3390/genes12050635
Jeong H-H, Jia J, Dai Y, Simon LM, Zhao Z. Investigating Cellular Trajectories in the Severity of COVID-19 and Their Transcriptional Programs Using Machine Learning Approaches. Genes. 2021; 12(5):635. https://doi.org/10.3390/genes12050635
Chicago/Turabian StyleJeong, Hyun-Hwan, Johnathan Jia, Yulin Dai, Lukas M. Simon, and Zhongming Zhao. 2021. "Investigating Cellular Trajectories in the Severity of COVID-19 and Their Transcriptional Programs Using Machine Learning Approaches" Genes 12, no. 5: 635. https://doi.org/10.3390/genes12050635
APA StyleJeong, H.-H., Jia, J., Dai, Y., Simon, L. M., & Zhao, Z. (2021). Investigating Cellular Trajectories in the Severity of COVID-19 and Their Transcriptional Programs Using Machine Learning Approaches. Genes, 12(5), 635. https://doi.org/10.3390/genes12050635