
De Novo Sporophyte Transcriptome Assembly and Functional Annotation in the Endangered Fern Species Vandenboschia speciosa (Willd.) G. Kunkel

 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. Next Generation Sequencing and Transcriptome Assembly
2.3. Assembly Filtering
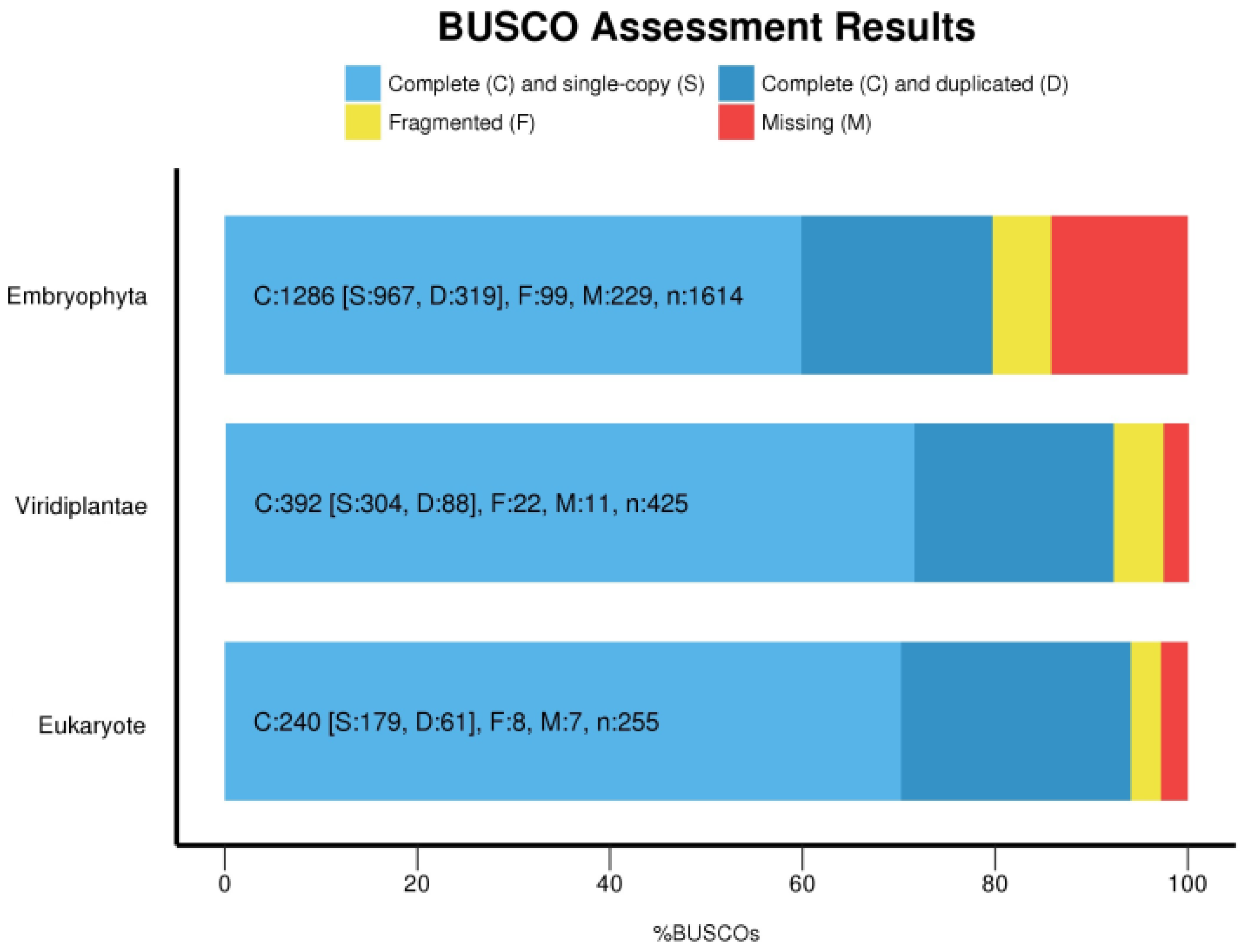
2.4. Assembly Completeness
2.5. Transcript Abundance
2.6. Coding Regions Identification
2.7. Functional Annotation
2.8. Gene Orthology Prediction
3. Results and Discussion
3.1. Sequencing Outputs and De Novo Transcriptome Assembly
3.2. Transcripts Filtering and Assembly Completeness
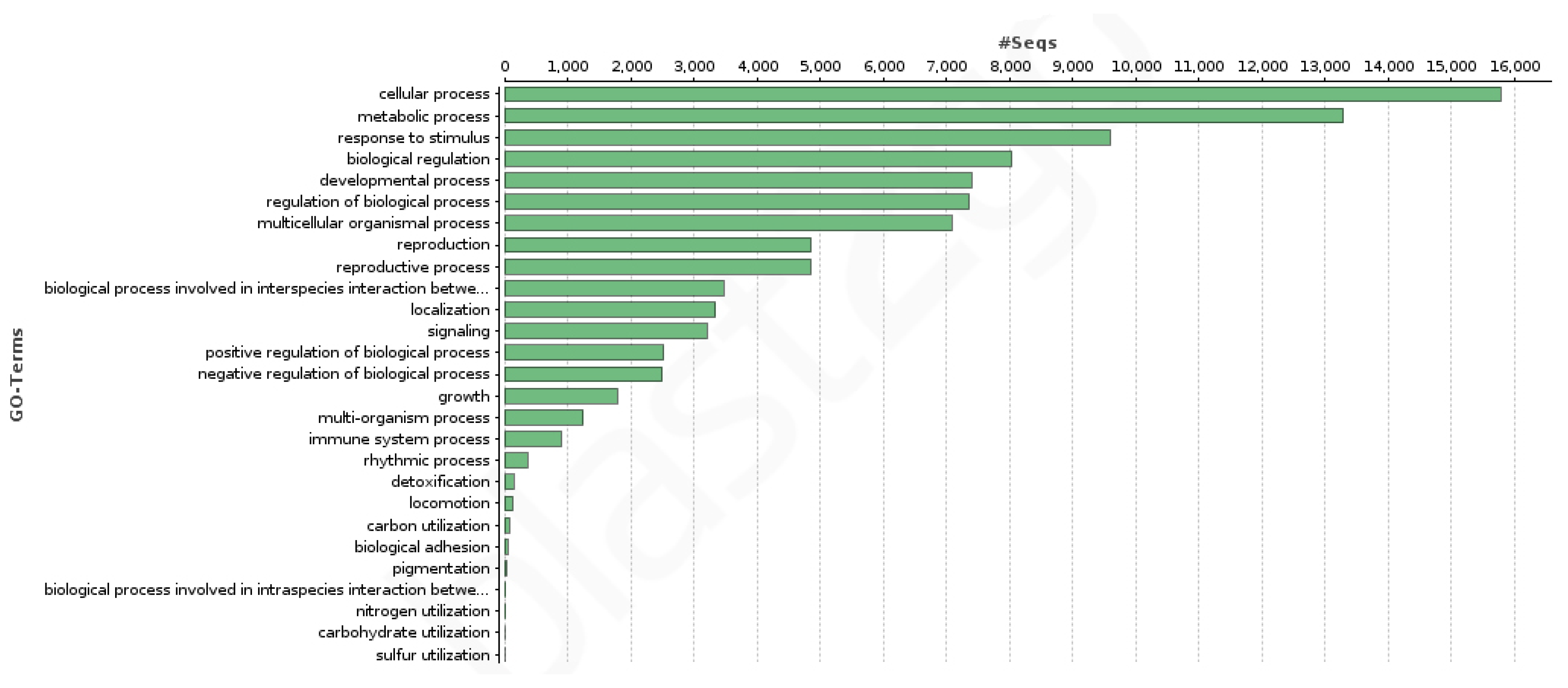
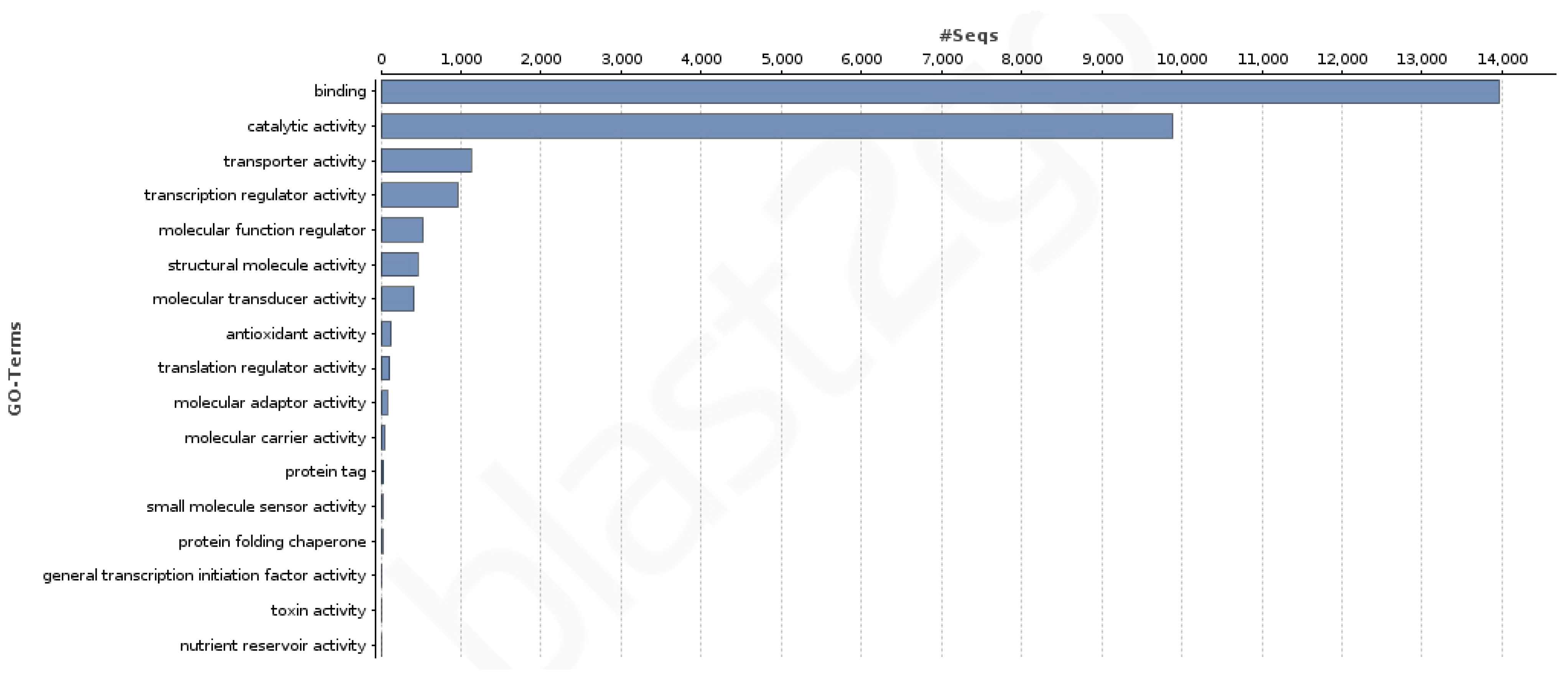
3.3. Coding Regions Identification and Functional Annotation
3.4. Transcript Abundance Analysis and Study of Expressed Genes
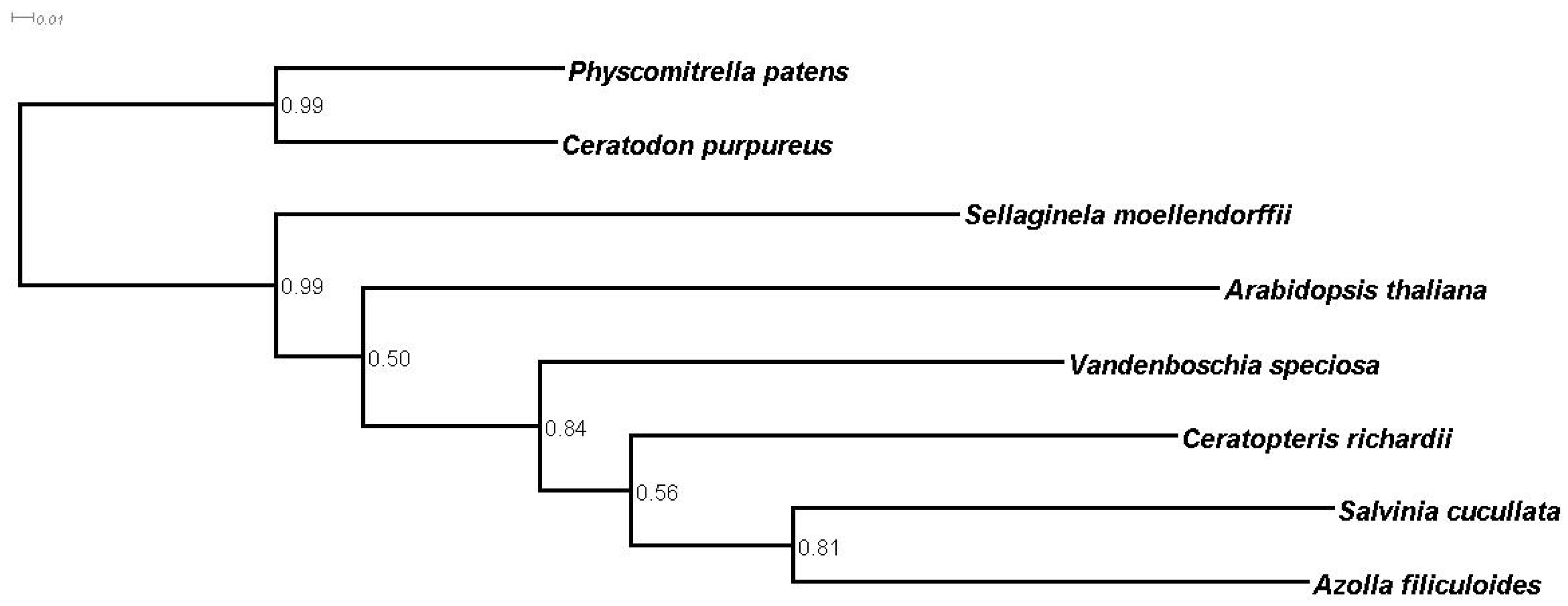
3.5. Homology and Orthology Prediction
3.6. Blastx Comparison between V. speciosa and A. thaliana Transcriptomes
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Pryer, K.M.; Schuettpelz, E.; Wolf, P.G.; Schneider, H.; Smith, A.R.; Cranfill, R. Phylogeny and evolution of ferns (monilophytes) with a focus on the early leptosporangiate divergences. Am. J. Bot. 2004, 91, 1582–1598. [Google Scholar] [CrossRef]
- Smith, A.R.; Pryer, K.M.; Schuettpelz, E.; Korall, P.; Schneider, H.; Wolf, P.G. A classification for extant ferns. Taxon 2006, 55, 705–731. [Google Scholar] [CrossRef]
- Dubuisson, J.; Hennequin, S.; Douzery, E.J.P.; Cranfill, R.B.; Smith, A.R.; Pryer, K.M. rbcL Phylogeny of the Fern Genus Trichomanes (Hymenophyllaceae), with Special Reference to Neotropical Taxa. Int. J. Plant Sci. 2003, 164, 753–761. [Google Scholar] [CrossRef][Green Version]
- Ebihara, A.; Iwatsuki, K.; Ito, M.; Hennequin, S.; Dubuisson, J. A global molecular phylogeny of the fern genus Trichomanes (Hymenophyllaceae) with special reference to stem anatomy. Bot. J. Linn. Soc. 2007, 155, 1–27. [Google Scholar] [CrossRef][Green Version]
- Ebihara, A.; Dubuisson, J.-Y.; Iwatsuki, K.; Ito, M. Systematics of Trichomanes (Hymenophyllaceae: Pteridophyta), progress and future interests. Fern Gazette 2007, 18, 53–58. [Google Scholar]
- Manton, I. Problems of Cytology and Evolution in the Pteridophyta; Smithsonian Institution: Washington, DC, USA, 1950; p. 316. [Google Scholar]
- Manton, I.; Lovis, J.D.; Vida, G.; Gibby, M. Cytology of the fern flora of Madeira. Bulletin of the British Museum (Natural History). Botany 1986, 15, 123–161. [Google Scholar]
- Obermayer, R.; Leitch, I.J.; Hanson, L.; Bennett, M.D. Nuclear DNA C-values in 30 Species Double the Familial Representation in Pteridophytes. Ann. Bot. 2002, 90, 209–217. [Google Scholar] [CrossRef]
- Rumsey, F.J.; Vogel, J.C.; Russell, S.J.; Barrett, J.A.; Gibby, M. Population genetics and conservation biology of the endangered fern Trichomanes speciosum (Hymenophyllaceae) in Scotland. Biol. J. Linn. Soc. 1999, 66, 333–344. [Google Scholar]
- Johnson, G.; Rumsey, F.J.; Headley, A.D.; Sheffield, E. Adaptations to extreme low light in the fern Trichomanes speciosum. New Phytol. 2000, 148, 423–431. [Google Scholar] [CrossRef]
- Makgomol, K.; Sheffield, E. Gametophyte morphology and ultrastructure of the extremely deep shade fern, Trichomanes speciosum. New Phytol. 2001, 151, 243–255. [Google Scholar] [CrossRef]
- Farrar, D.R. Species and Evolution in Asexually Reproducing Independent Fern Gametophytes. Syst. Bot. 1990, 15, 98. [Google Scholar] [CrossRef]
- Farrar, D.R.; Mickel, J.T. Society Vittaria appalachiana: A name for the “Appalachian Gametophyte”. Am. Fern J. 1991, 81, 69–75. [Google Scholar] [CrossRef]
- Farrar, D.R. Trichomanes intricatum: The independent Trichomanes gametophyte in the Eastern United States. Am. Fern J. 1992, 82, 68–74. [Google Scholar] [CrossRef]
- Raine, C.A.; Farrar, D.R.; Sheffield, E. A New Hymenophyllum Species in the Appalachians Represented by Independent Gametophyte Colonies. Am. Fern J. 1991, 81, 109. [Google Scholar] [CrossRef]
- Haas, B.J.; Papanicolaou, A.; Yassour, M.; Grabherr, M.; Blood, P.D.; Bowden, J.; Couger, M.B.; Eccles, D.; Li, B.; Lieber, M.; et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 2013, 8, 1494–1512. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- MacManes, M.D. On the optimal trimming of high-throughput mRNA sequence data. Front. Genet. 2014, 5, 12. [Google Scholar] [CrossRef]
- Smith-Unna, R.; Boursnell, C.; Patro, R.; Hibberd, J.M.; Kelly, S. TransRate: Reference-free quality assessment of de novo transcriptome assemblies. Genome Res. 2016, 26, 1134–1144. [Google Scholar] [CrossRef]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.S.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef]
- Bairoch, A.; Boeckmann, B. The SWISS-PROT protein sequence data bank. Nucleic Acids Res. 1991, 19, 2247–2249. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef] [PubMed]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef]
- Afgan, E.; Baker, D.; Batut, B.; van den Beek, M.; Bouvier, D.; Čech, M.; Chilton, J.; Clements, D.; Coraor, N.; Gruning, B.A.; et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Res. 2018, 46, W537–W544. [Google Scholar] [CrossRef]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016. [Google Scholar]
- R Studio Team. R Studio: Integrated Development for R; R Studio Inc.: Boston, MA, USA, 2015; Available online: https://www.rstudio.com/products/team/ (accessed on 26 June 2021).
- Wagner, G.P.; Kin, K.; Lynch, V.J. Measurement of mRNA abundance using RNA-seq data: RPKM measure is inconsistent among samples. Theory Biosci. 2012, 131, 281–285. [Google Scholar] [CrossRef]
- Conesa, A.; Götz, S.; García-Gómez, J.M.; Terol, J.; Talón, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef]
- Conesa, A.; Götz, S. Blast2GO: A Comprehensive Suite for Functional Analysis in Plant Genomics. Int. J. Plant Genomics 2008, 2008, 619832. [Google Scholar] [CrossRef]
- Myhre, S.; Tveit, H.; Mollestad, T.; Laegreid, A. Additional Gene Ontology structure for improved biological reasoning. Bioinformatics 2006, 22, 2020–2027. [Google Scholar] [CrossRef] [PubMed]
- Emms, D.M.; Kelly, S. OrthoFinder: Solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. Genome Biol. 2015, 16, 157. [Google Scholar] [CrossRef]
- Emms, D.M.; Kelly, S. OrthoFinder: Phylogenetic orthology inference for comparative genomics. Genome Biol. 2019, 20, 1–14. [Google Scholar] [CrossRef]
- Huson, D.H.; Scornavacca, C. Dendroscope 3. An interactive viewer for rooted phylogenetic trees and networks. Syst. Biol. 2012, 61, 1061–1067. [Google Scholar] [CrossRef]
- Ćuković, K.; Dragićević, M.; Bogdanović, M.; Paunović, D.; Giurato, G.; Filipović, B.; Subotić, A.; Todorović, S.; Simonović, A. Plant regeneration in leaf culture of Centaurium erythraea Rafn. Part 3: De novo transcriptome assembly and validation of housekeeping genes for studies of in vitro morphogenesis. Plant Cell Tissue Organ Cult. 2020, 141, 417–433. [Google Scholar] [CrossRef]
- Niu, S.-C.; Xu, Q.; Zhang, G.-Q.; Zhang, Y.-Q.; Tsai, W.-C.; Hsu, J.-L.; Liang, C.-K.; Luo, Y.-B.; Liu, Z.-J. De novo transcriptome assembly databases for the butterfly orchid Phalaenopsis equestris. Sci. Data 2016, 3, 160083. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, X.; Zhou, W.; Li, T.; Tian, C. De novo assembly and transcriptome characterization of spruce dwarf mistletoe Arceuthobium sichuanense uncovers gene expression profiling associated with plant development. BMC Genom. 2016, 17, 771. [Google Scholar] [CrossRef]
- Hu, R.; Yu, C.; Wang, X.; Jia, C.; Pei, S.; He, K.; He, G.; Kong, Y.; Zhou, G. De novo Transcriptome Analysis of Miscanthus lutarioriparius Identifies Candidate Genes in Rhizome Development. Front. Plant Sci. 2017, 8, 492. [Google Scholar] [CrossRef]
- Sigel, E.M.; Schuettpelz, E.; Pryer, K.M.; Der, J.P. Overlapping Patterns of Gene Expression Between Gametophyte and Sporophyte Phases in the Fern Polypodium amorphum (Polypodiales). Front. Plant Sci. 2018, 9, 9. [Google Scholar] [CrossRef]
- Jo, Y.; Lian, S.; Cho, J.K.; Choi, H.; Chu, H.; Cho, W.K. De novo transcriptome assembly of two different Prunus salicina cultivars. Genom. Data 2015, 6, 262–263. [Google Scholar] [CrossRef]
- Geng, Y.; Cai, C.; McAdam, S.A.; Banks, J.A.; Wisecaver, J.H.; Zhou, Y. A De Novo Transcriptome Assembly of Ceratopteris richardii Provides Insights into the Evolutionary Dynamics of Complex Gene Families in Land Plants. Genome Biol. Evol. 2021, 13, evab042. [Google Scholar] [CrossRef] [PubMed]
- Torrens-Spence, M.; Fallon, T.; Weng, J. A Workflow for Studying Specialized Metabolism in Nonmodel Eukaryotic Organisms. Methods Enzymol. 2016, 576, 69–97. [Google Scholar] [CrossRef]
- Waterhouse, R.M.; Seppey, M.; Simão, F.A.; Manni, M.; Ioannidis, P.; Klioutchnikov, G.; Kriventseva, E.V.; Zdobnov, E. BUSCO Applications from Quality Assessments to Gene Prediction and Phylogenomics. Mol. Biol. Evol. 2018, 35, 543–548. [Google Scholar] [CrossRef]
- Aya, K.; Kobayashi, M.; Tanaka, J.; Ohyanagi, H.; Suzuki, T.; Yano, K.; Takano, T.; Yano, K.; Matsuoka, M. De Novo Transcriptome Assembly of a Fern, Lygodium japonicum, and a Web Resource Database, Ljtrans DB. Plant Cell Physiol. 2014, 56, e5. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Estévez, M.; Bakkali, M.; Martín-Blázquez, R.; Garrido-Ramos, M.A. Erratum to “Differential expression patterns of MIKCC-type MADS-box genes in the endangered fern Vandenboschia speciosa” [Plant Gene 12C (2017) 50–56]. Plant Gene 2019, 17, 100125. [Google Scholar] [CrossRef]
- Ruiz-Estévez, M.; Bakkali, M.; Martín-Blázquez, R.; Garrido-Ramos, M.A. Identification and Characterization of TALE Homeobox Genes in the Endangered Fern Vandenboschia speciosa. Genes 2017, 8, 275. [Google Scholar] [CrossRef] [PubMed]
- Silva, G.B.; Ionashiro, M.; Carrara, T.B.; Crivellari, A.C.; Tiné, M.A.; Prado, J.; Carpita, N.C.; Buckeridge, M.S. Cell wall polysaccharides from fern leaves: Evidence for a mannan-rich Type III cell wall in Adiantum raddianum. Phytochemistry 2011, 72, 2352–2360. [Google Scholar] [CrossRef]
- Singh, S.; Singh, G.; Arya, S.K. Mannans: An overview of properties and application in food products. Int. J. Biol. Macromol. 2018, 119, 79–95. [Google Scholar] [CrossRef]
- Li, F.-W.; Brouwer, P.; Carretero-Paulet, L.; Cheng, S.; De Vries, J.; Delaux, P.-M.; Eily, A.; Koppers, N.; Kuo, L.-Y.; Li, Z.; et al. Fern genomes elucidate land plant evolution and cyanobacterial symbioses. Nat. Plants 2018, 4, 460–472. [Google Scholar] [CrossRef]
- Schuettpelz, E.; Pryer, K.M. Fern phylogeny inferred from 400 leptosporangiate species and three plastid genes. Taxon 2007, 56, 1037–1050. [Google Scholar] [CrossRef]
- Rai, H.S.; Graham, S.W. Utility of a large, multigene plastid data set in inferring higher-order relationships in ferns and relatives (monilophytes). Am. J. Bot. 2010, 97, 1444–1456. [Google Scholar] [CrossRef]
- Lehtonen, S. Towards resolving the complete fern tree of life. PLoS ONE 2011, 6, e24851. [Google Scholar] [CrossRef] [PubMed]
- Rothfels, C.J.; Li, F.-W.; Sigel, E.M.; Huiet, L.; Larsson, A.; Burge, D.O.; Ruhsam, M.; Deyholos, M.; Soltis, D.E.; Stewart, C.N., Jr.; et al. The evolutionary history of ferns inferred from 25 low-copy nuclear genes. Am. J. Bot. 2015, 102, 1089–1107. [Google Scholar] [CrossRef] [PubMed]
- Knie, N.; Fischer, S.; Grewe, F.; Polsakiewicz, M.; Knoop, V. Horsetails are the sister group to all other monilophytes and Marattiales are sister to leptosporangiate ferns. Mol. Phylogenet. Evol. 2015, 90, 140–149. [Google Scholar] [CrossRef]
- PPG I. A community-derived classification for extant lycophytes and ferns. J. Syst. Evol. 2016, 54, 563–603. [Google Scholar] [CrossRef]
- Qi, X.; Kuo, L.Y.; Guo, C.; Li, H.; Li, Z.; Qi, J.; Wang, L.; Hu, Y.; Xiang, J.; Zhang, C.; et al. A well-resolved fern nuclear phylogeny reveals the evolution history of numerous transcription factor families. Mol. Phyl. Evol. 2018, 127, 961–977. [Google Scholar] [CrossRef] [PubMed]
- Sessa, E.B.; Der, J.P. Evolutionary genomics of ferns and lycophytes. Adv. Bot. Res. 2016, 78, 215–254. [Google Scholar]
- Marchant, D.B.; Sessa, E.B.; Wolf, P.G.; Heo, K.; Barbazuk, W.B.; Soltis, P.S.; Soltis, D.E. The C-Fern (Ceratopteris richardii) ge-nome: Insights into plant genome evolution with the first partial homosporous fern genome assembly. Sci. Rep. 2019, 9, 18181. [Google Scholar] [CrossRef]
- Dyer, R.J.; Pellicer, J.; Savolainen, V.; Leitch, I.J.; Schneider, H. Genome size expansion and the relationship between nuclear DNA content and spore size in the Asplenium monanthes fern complex (Aspleniaceae). BMC Plant Biol. 2013, 13, 219. [Google Scholar] [CrossRef]
- Wolf, P.G.; Sessa, E.B.; Marchant, D.B.; Li, F.-W.; Rothfels, C.J.; Sigel, E.M.; Gitzendanner, M.; Visger, C.J.; Banks, J.A.; Soltis, D.E.; et al. An Exploration into Fern Genome Space. Genome Biol. Evol. 2015, 7, 2533–2544. [Google Scholar] [CrossRef]
- One Thousand Plant Transcriptomes Initiative. One thousand plant transcriptomes and the phylogenomics of green plants. Nature 2019, 574, 679–685. [Google Scholar] [CrossRef] [PubMed]
- Plackett, A.R.G.; Di Stilio, V.S.; Langdale, J.A. Ferns: The missing link in shoot evolution and development. Front. Plant Sci. 2015, 6, 972. [Google Scholar] [CrossRef] [PubMed]
- Rensing, S.A. Why we need more non-seed plant models. New Phytol. 2017, 216, 355–360. [Google Scholar] [CrossRef]
- Rensing, S.A.; Lang, D.; Zimmer, A.D.; Terry, A.; Salamov, A.; Shapiro, H. The Physcomitrella genome reveals evolu-tionary insights into the conquest of land by plants. Science 2008, 319, 64–69. [Google Scholar] [CrossRef]
- Banks, J.A.; Nishiyama, T.; Hasebe, M.; Bowman, J.L.; Gribskov, M.; Depamphilis, C.; Albert, V.A.; Aono, N.; Aoyama, T.; Ambrose, B.A.; et al. The Selaginella Genome Identifies Genetic Changes Associated with the Evolution of Vascular Plants. Science 2011, 332, 960–963. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, S.; Oldroyd, G.E. GRAS-domain transcription factors that regulate plant development. Plant Signal. Behav. 2009, 4, 698–700. [Google Scholar] [CrossRef]
- Szövényi, P.; Rensing, S.A.; Lang, D.; Wray, G.A.; Shaw, A.J. Generation-biased gene expression in a bryophyte model sys-tem. Mol. Biol. Evol. 2011, 28, 803–812. [Google Scholar] [CrossRef] [PubMed]
- Pires, N.D.; Yi, K.; Breuninger, H.; Catarino, B.; Menand, B.; Dolan, L. Recruitment and remodeling of an ancient gene regu-latory network during land plant evolution. Proc. Natl. Acad. Sci. USA 2013, 110, 9571–9576. [Google Scholar] [CrossRef]
- Ortiz-Ramírez, C.; Hernandez-Coronado, M.; Thamm, A.; Catarino, B.; Wang, M.; Dolan, L.; Feijó, J.; Becker, J.D. A Transcriptome Atlas of Physcomitrella patens Provides Insights into the Evolution and Development of Land Plants. Mol. Plant 2016, 9, 205–220. [Google Scholar] [CrossRef] [PubMed]
- Borges, F.; Gomes, G.; Gardner, R.; Moreno, N.; McCormick, S.; Feijó, J.A.; Becker, J. Comparative Transcriptomics of Arabidopsis Sperm Cells. Plant Physiol. 2008, 148, 1168–1181. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Ruano, F.J.; Navarro-Domínguez, B.; Camacho, J.P.M.; Garrido-Ramos, M.A. Full plastome sequence of the fern Van-denboschia speciosa (Hymenophyllales): Structural singularities and evolutionary insights. J. Plant Res. 2019, 132, 3–17. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Raw Data | after Quality Trimming | |
|---|---|---|
| Number of paired-end reads | 66.3 million | 65.2 million |
| Number of bases | 6700 million | 6590 million |
| Before Filtering | After Filtering | |
|---|---|---|
| Total transcripts | 84,759 | 36,430 |
| Percent GC | 45.18 | 45.18 |
| Contig N50 (bp) | 1955 | 2085 |
| Contig N70 (bp) | 1332 | 1511 |
| Contig N90 (bp) | 479 | 729 |
| Ex90N50 (bp) | 2039 | 2299 |
| Number transcripts corresponding to the Ex90 peak | 14,645 | 21,543 |
| Size of the smallest contig (bp) | 201 | 201 |
| Size of the largest contig (bp) | 13,225 | 13,224 |
| Number of contigs greater than 1 Kb long | 35,801 | 20,532 |
| Number of contigs greater than 10 Kb long | 18 | 12 |
| Median contig length (bp) | 722 | 1197 |
| Average contig (bp) | 1,144.86 | 1,437.37 |
| Total number of assembled bases | 97,037,551 | 52,363,571 |
| Before Filtering | After Filtering | |||
|---|---|---|---|---|
| Percentage Intervals | Number of Proteins * | Accumulated Number of Proteins ** | Number of Proteins * | Accumulated Number of Proteins ** |
| 91–100 | 3301 | 3301 (>90%) | 3196 | 3196 (>90%) |
| 81–90 | 1330 | 4631 (>80%) | 1352 | 4548 (>80%) |
| 71–80 | 916 | 5547 (>70%) | 878 | 5426 (>70%) |
| 61–70 | 646 | 6193 (>60%) | 569 | 5995 (>60%) |
| 51–60 | 551 | 6744 (>50%) | 449 | 6444 (>50%) |
| 41–50 | 586 | 7330 (>40%) | 448 | 6892 (>40%) |
| 31–40 | 538 | 7868 (>30%) | 381 | 7273 (>30%) |
| 21–30 | 506 | 8374 (>20%) | 302 | 7575 (>20%) |
| 11–20 | 411 | 8785 (>10%) | 225 | 7800 (>10%) |
| 1–10 | 90 | 8875 (>1%) | 51 | 7851 (>1%) |
| TOTAL | 8875 | 8875 | 7851 | 7851 |
| Mosses | Lycophyte | Seed Plant | Leptosporangiate Ferns | |||||
|---|---|---|---|---|---|---|---|---|
| Statistics | Cp | Pp | Sm | At | Vs | Cr | Af | Sc |
| Number of proteins | 40,806 | 38,354 | 22,285 | 48,359 | 29,220 | 44,668 | 20,203 | 19,779 |
| Number of proteins in orthogroups | 31,747 | 26,769 | 20,136 | 44,174 | 23,964 | 35,963 | 17,948 | 16,818 |
| Number of unassigned proteins | 9059 | 11,585 | 2149 | 4185 | 5256 | 8705 | 2255 | 2961 |
| Percentage of proteins in orthogroups | 77.8 | 69.8 | 90.4 | 91.3 | 82.0 | 80.5 | 88.8 | 85.0 |
| Percentage of unassigned proteins | 22.2 | 30.2 | 9.6 | 8.7 | 18.0 | 19.5 | 11.2 | 15.0 |
| Number of orthogroups containing species | 12,351 | 11,499 | 9586 | 12,003 | 11,195 | 12,715 | 9915 | 9882 |
| Percentage of orthogroups containing species | 46.0 | 42.8 | 35.7 | 44.7 | 41.7 | 47.3 | 36.9 | 36.8 |
| Number of species-specific orthogroups | 1654 | 813 | 1461 | 4018 | 1384 | 2207 | 344 | 282 |
| Nº of proteins in species-specific orthogroups | 6849 | 2540 | 6545 | 18,005 | 3696 | 8205 | 1142 | 921 |
| % of proteins in species-specific orthogroups | 16.8 | 6.6 | 29.4 | 37.2 | 12.6 | 18.4 | 5.7 | 4.7 |
| A. filiculoides | C. purpureus | C. richardii | P. patens | S. cucullata | S. moellendorffii | V. speciosa | |
|---|---|---|---|---|---|---|---|
| A. thaliana | 6905 | 6921 | 7275 | 6954 | 6882 | 6753 | 7243 |
| A. filiculoides | 7169 | 8675 | 7188 | 8448 | 6901 | 8028 | |
| C. purpureus | 7576 | 10339 | 7145 | 7072 | 7433 | ||
| C. richardii | 7576 | 8665 | 7309 | 8916 | |||
| P. patens | 7167 | 7070 | 7443 | ||||
| S. cucullata | 6901 | 8052 | |||||
| S. moellendorffii | 7198 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bakkali, M.; Martín-Blázquez, R.; Ruiz-Estévez, M.; Garrido-Ramos, M.A. De Novo Sporophyte Transcriptome Assembly and Functional Annotation in the Endangered Fern Species Vandenboschia speciosa (Willd.) G. Kunkel. Genes 2021, 12, 1017. https://doi.org/10.3390/genes12071017
Bakkali M, Martín-Blázquez R, Ruiz-Estévez M, Garrido-Ramos MA. De Novo Sporophyte Transcriptome Assembly and Functional Annotation in the Endangered Fern Species Vandenboschia speciosa (Willd.) G. Kunkel. Genes. 2021; 12(7):1017. https://doi.org/10.3390/genes12071017
Chicago/Turabian StyleBakkali, Mohammed, Rubén Martín-Blázquez, Mercedes Ruiz-Estévez, and Manuel A. Garrido-Ramos. 2021. "De Novo Sporophyte Transcriptome Assembly and Functional Annotation in the Endangered Fern Species Vandenboschia speciosa (Willd.) G. Kunkel" Genes 12, no. 7: 1017. https://doi.org/10.3390/genes12071017
APA StyleBakkali, M., Martín-Blázquez, R., Ruiz-Estévez, M., & Garrido-Ramos, M. A. (2021). De Novo Sporophyte Transcriptome Assembly and Functional Annotation in the Endangered Fern Species Vandenboschia speciosa (Willd.) G. Kunkel. Genes, 12(7), 1017. https://doi.org/10.3390/genes12071017