
Understanding the Adaptive Evolutionary Histories of South American Ancient and Present-Day Populations via Genomics


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
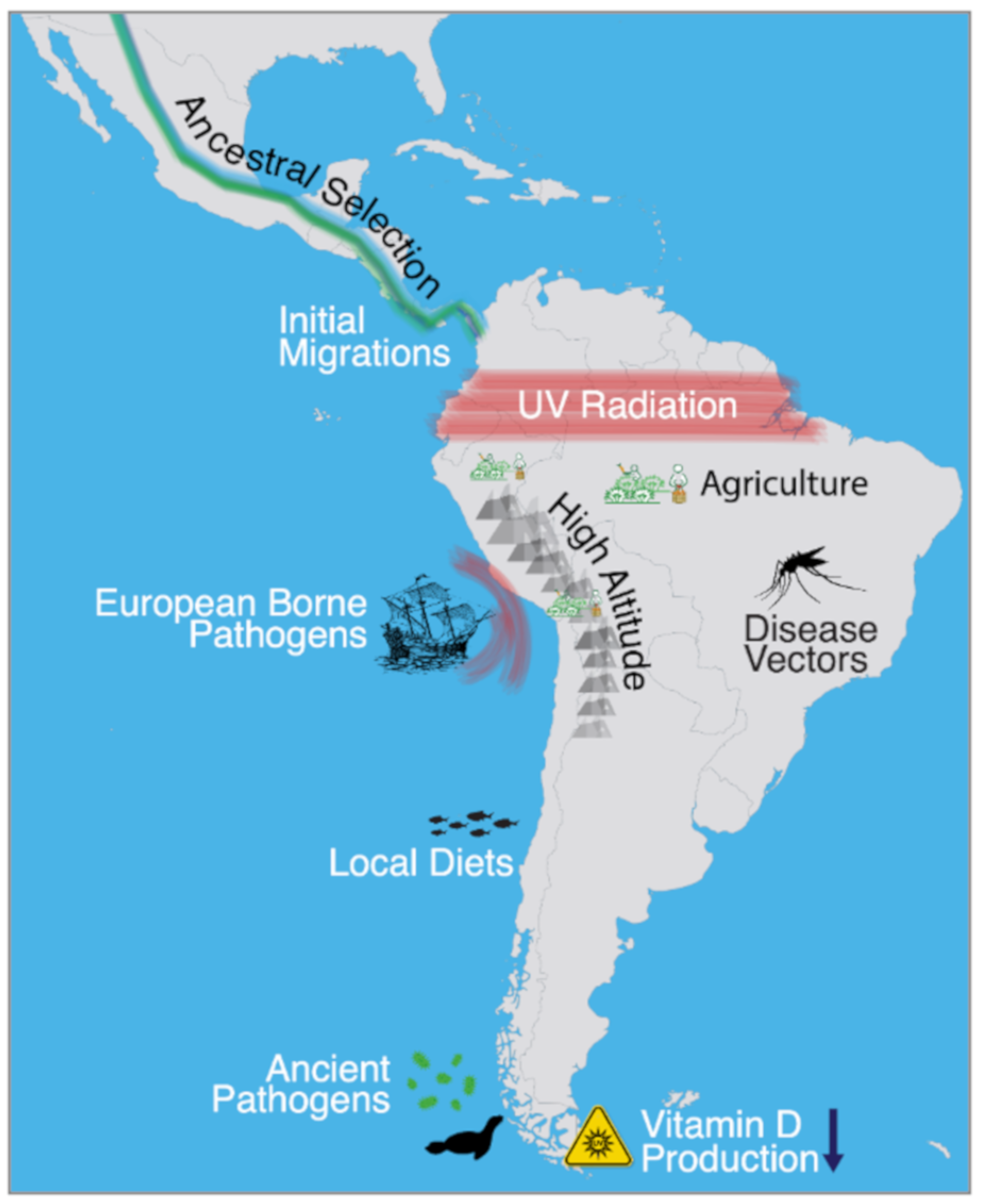
1. Introduction
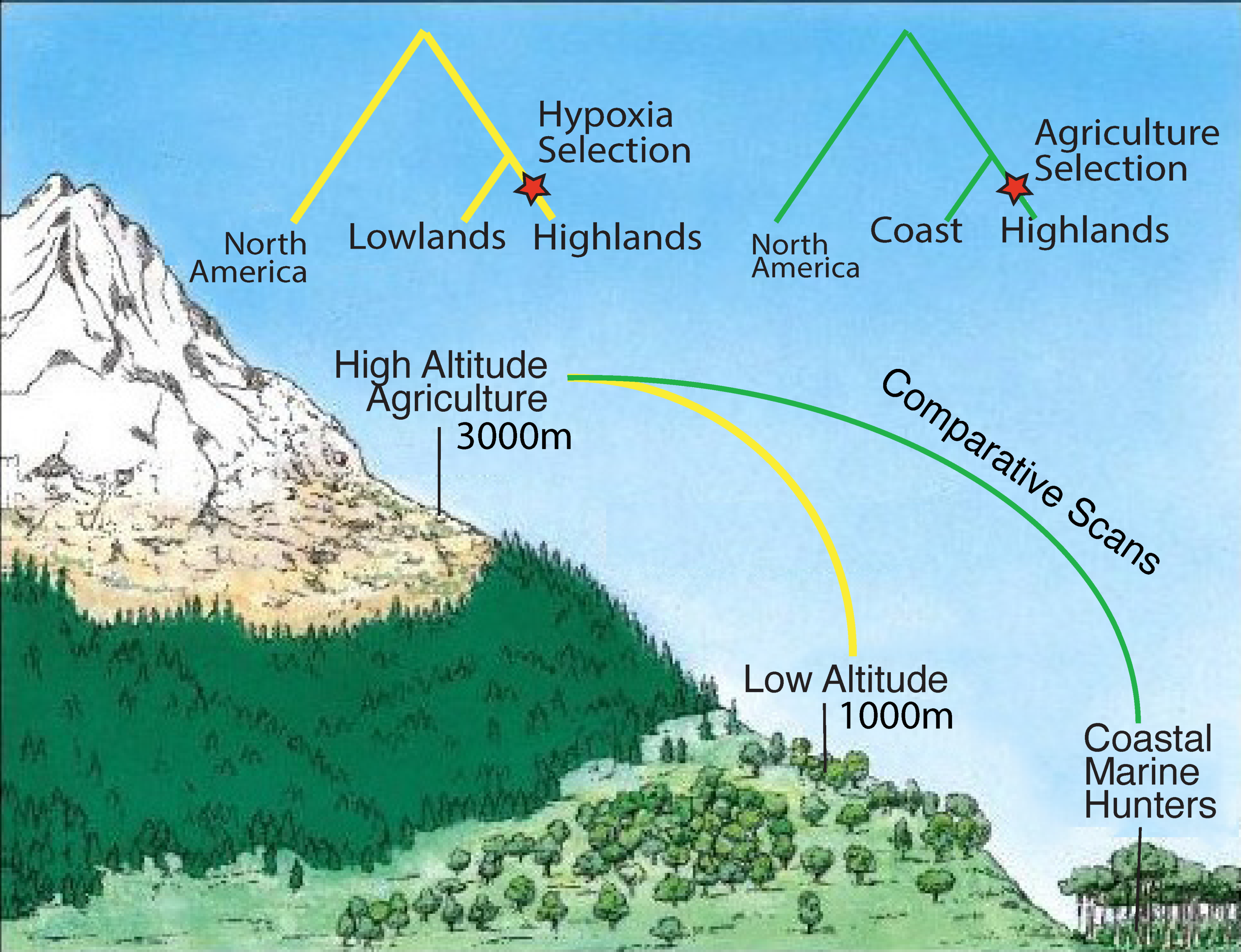
2. High-Altitude Adaptation
3. Ultraviolet Radiation
4. Adaptive and Plastic Responses to Culture
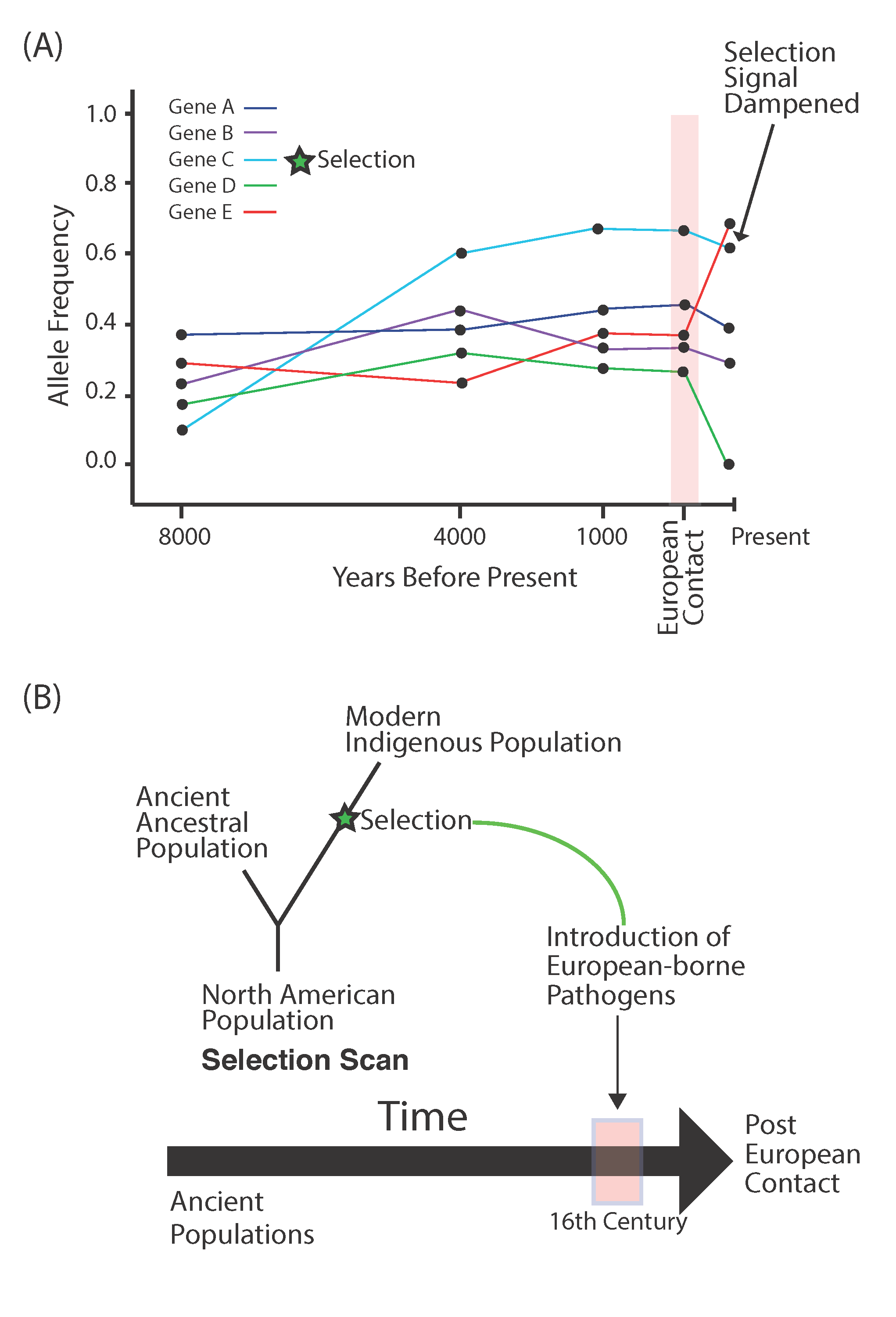
5. Ancient Pathogens and European-Borne Disease
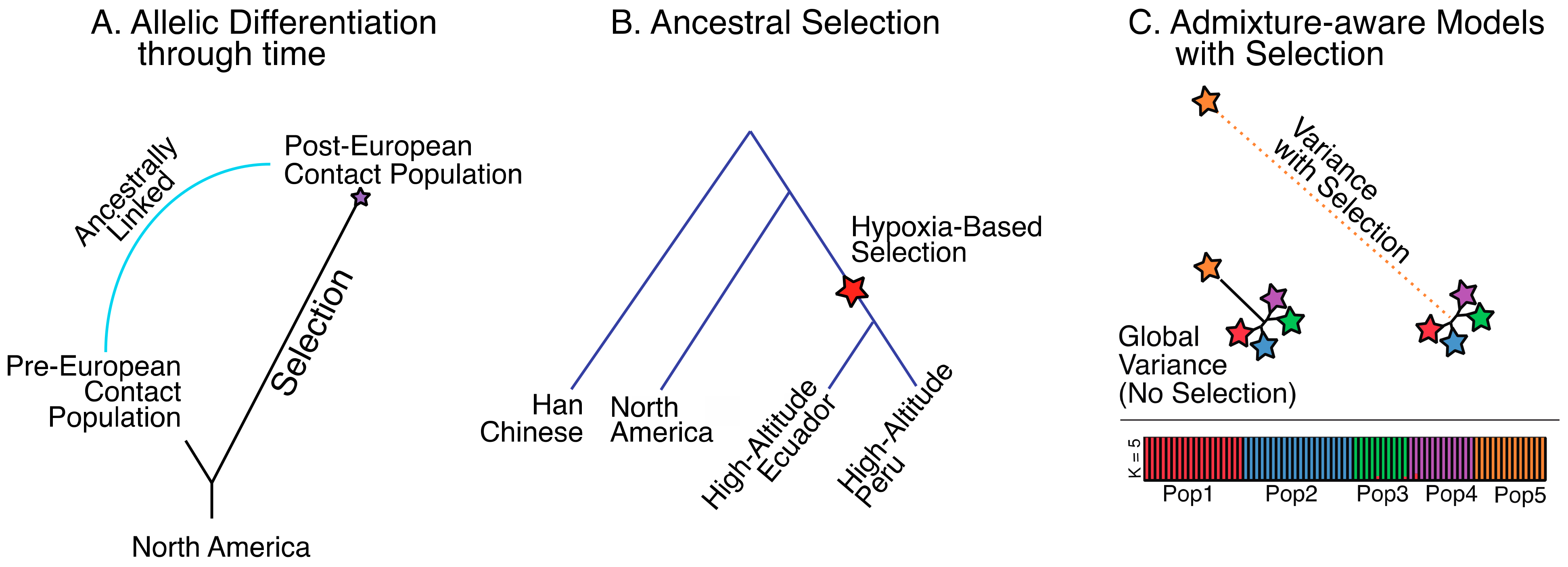
6. Methods for Detecting Positive Selection
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Dillehay, T.D.; Ramírez, C.; Pino, M.; Collins, M.B.; Rossen, J.; Pino-Navarro, J.D. Monte Verde: Seaweed, food, medicine, and the peopling of South America. Science 2008, 320, 784–786. [Google Scholar] [CrossRef]
- Moreno-Mayar, J.V.; Vinner, L.; de Barros Damgaard, P.; de la Fuente, C.; Chan, J.; Spence, J.P.; Allentoft, M.E.; Vimala, T.; Racimo, F.; Pinotti, T.; et al. Early human dispersals within the Americas. Science 2018, 525, eaav2621. [Google Scholar] [CrossRef]
- Scheib, C.L.; Li, H.; Desai, T.; Link, V.; Kendall, C.; Dewar, G.; Griffith, P.W.; Mörseburg, A.; Johnson, J.R.; Potter, A.; et al. Ancient human parallel lineages within North America contributed to a coastal expansion. Science 2018, 360, 1024–1027. [Google Scholar] [CrossRef]
- Rademaker, K.; Hodgins, G.; Moore, K.; Zarrillo, S.; Miller, C.; Bromley, G.R.M.; Leach, P.; Reid, D.A.; Álvarez, W.Y.; Sandweiss, D.H. Paleoindian settlement of the high-altitude Peruvian Andes. Science 2014, 346, 466–469. [Google Scholar] [CrossRef] [PubMed]
- Da-Gloria, P.; Hubbe, M.; Neves, W.A. Lagoa Santa’s contribution to the origins and life of early Americans. Evol. Anthropol. 2018, 27, 121–133. [Google Scholar] [CrossRef] [PubMed]
- Dillehay, T.D. The late Pleistocene cultures of South America. Evol. Anthropol. 1999, 7, 206–216. [Google Scholar] [CrossRef]
- Lindo, J.; Haas, R.; Hofman, C.; Apata, M.; Moraga, M.; Verdugo, R.A.; Watson, J.T.; Viviano Llave, C.; Witonsky, D.; Beall, C.; et al. The genetic prehistory of the Andean highlands 7000 years BP though European contact. Sci. Adv. 2018, 4, eaau4921. [Google Scholar] [CrossRef] [PubMed]
- Crawford, J.E.; Amaru, R.; Song, J.; Julian, C.G.; Racimo, F.; Cheng, J.Y.; Guo, X.; Yao, J.; Ambale-Venkatesh, B.; Lima, J.A.; et al. Natural Selection on Genes Related to Cardiovascular Health in High-Altitude Adapted Andeans. Am. J. Hum. Genet. 2017, 101, 752–767. [Google Scholar] [CrossRef] [PubMed]
- Beall, C.M. Adaptation to High Altitude: Phenotypes and Genotypes. Annu. Rev. Anthropol. 2014, 43, 251–272. [Google Scholar] [CrossRef]
- Bigham, A.W.; Mao, X.; Mei, R.; Brutsaert, T.; Wilson, M.J.; Julian, C.G.; Parra, E.J.; Akey, J.M.; Moore, L.G.; Shriver, M.D. Identifying positive selection candidate loci for high-altitude adaptation in Andean populations. Hum. Genom. 2009, 4, 79–90. [Google Scholar] [CrossRef]
- Yi, X.; Liang, Y.; Huerta-Sánchez, E.; Jin, X.; Cuo, Z.X.P.; Pool, J.E.; Xu, X.; Jiang, H.; Vinckenbosch, N.; Korneliussen, T.S.; et al. Sequencing of 50 Human Exomes Reveals Adaptation to High Altitude. Science 2010, 329, 75–78. [Google Scholar] [CrossRef]
- Huerta-Sánchez, E.; DeGiorgio, M.; Pagani, L.; Tarekegn, A.; Ekong, R.; Antão, T.; Cardona, A.; Montgomery, H.E.; Cavalleri, G.L.; Robbins, P.A.; et al. Genetic signatures reveal high-altitude adaptation in a set of ethiopian populations. Mol. Biol. Evol. 2013, 30, 1877–1888. [Google Scholar] [CrossRef]
- Cook, N.D. Demographic Collapse; Cambridge University Press: Cambridge, UK, 2004. [Google Scholar]
- Bélisle, V.; Quispe-Bustamante, H.; Hardy, T.J.; Davis, A.R.; Condori, E.A.; González, C.D.; Avendaño, J.V.G.; Reid, D.A.; Williams, P.R. Wari impact on regional trade networks_Patterns of obsidian exchange in Cusco, Peru. J. Archaeol. Sci. Rep. 2020, 32, 102439. [Google Scholar]
- Cheng, J.Y.; Racimo, F.; Nielsen, R. Ohana: Detecting selection in multiple populations by modelling ancestral admixture components. bioRxiv 2019. [Google Scholar] [CrossRef]
- Herman, J.R.; Krotkov, N.; Celarier, E.; Larko, D.; Labow, G. Distribution of UV radiation at the Earth’s surface from TOMS-measured UV-backscattered radiances. J. Geophys. Res. Atmos. 1999, 104, 12059–12076. [Google Scholar] [CrossRef]
- Jablonski, N.G.; Chaplin, G. Colloquium paper: Human skin pigmentation as an adaptation to UV radiation. Proc. Natl. Acad. Sci. USA 2010, 107 (Suppl. 2), 8962–8968. [Google Scholar] [CrossRef]
- Jablonski, N.G. The Anthropology of Skin Colors: An Examination of the Evolution of Skin Pigmentation and the Concepts of Race and Skin of Color. In Dermatoanthropology of Ethnic Skin and Hair; Vashi, N.A., Maibach, H.I., Eds.; Springer: Cham, Switzerland, 2017; Volume 33, pp. 1–11. [Google Scholar]
- Adhikari, K.; Mendoza-Revilla, J.; Sohail, A.; Fuentes-Guajardo, M.; Lampert, J.; Chacón-Duque, J.C.; Hurtado, M.; Villegas, V.; Granja, V.; Acuña-Alonzo, V.; et al. A GWAS in Latin Americans highlights the convergent evolution of lighter skin pigmentation in Eurasia. Nat. Commun. 2019, 10, 358. [Google Scholar] [CrossRef]
- Fu, Q.; Posth, C.; Hajdinjak, M.; Petr, M.; Mallick, S.; Fernandes, D.; Furtwängler, A.; Haak, W.; Meyer, M.; Mittnik, A.; et al. The genetic history of Ice Age Europe. Nature 2016, 534, 200–205. [Google Scholar] [CrossRef]
- Meyer-Lindenberg, A.; Domes, G.; Kirsch, P.; Heinrichs, M. Oxytocin and vasopressin in the human brain: Social neuropeptides for translational medicine. Nat. Rev. Neurosci. 2011, 1–15. [Google Scholar] [CrossRef]
- Gokhman, D.; Meshorer, E.; Carmel, L. Epigenetics: It’s Getting Old. Past Meets Future in Paleoepigenetics. Trends Ecol. Evol. 2016, 31, 290–300. [Google Scholar] [CrossRef]
- Nakatsuka, N.; Lazaridis, I.; Barbieri, C.; Skoglund, P.; Rohland, N.; Mallick, S.; Posth, C.; Harkins-Kinkaid, K.; Ferry, M.; Harney, E.; et al. A Paleogenomic Reconstruction of the Deep Population History of the Andes. Cell 2020, 181, 1131–1145.e21. [Google Scholar] [CrossRef]
- Posth, C.; Nakatsuka, N.; Lazaridis, I.; Skoglund, P.; Mallick, S.; Lamnidis, T.C.; Rohland, N.; Nägele, K.; Adamski, N.; Bertolini, E.; et al. Reconstructing the Deep Population History of Central and South America. Cell 2018, 175, 1185–1197.e22. [Google Scholar] [CrossRef]
- Miller, M.J.; Agarwal, S.C.; Aristizabal, L.; Langebaek, C. The daily grind: Sex- and age-related activity patterns inferred from cross-sectional geometry of long bones in a pre-Columbian muisca population from Tibanica, Colombia. Am. J. Phys. Anthropol. 2018, 167, 311–326. [Google Scholar] [CrossRef] [PubMed]
- Tishkoff, S.A.; Reed, F.A.; Ranciaro, A.; Voight, B.F.; Babbitt, C.C.; Silverman, J.S.; Powell, K.; Mortensen, H.M.; Hirbo, J.B.; Osman, M.; et al. Convergent adaptation of human lactase persistence in Africa and Europe. Nat. Genet. 2007, 39, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Rumold, C.U.; Aldenderfer, M.S. Late Archaic-Early Formative period microbotanical evidence for potato at Jiskairumoko in the Titicaca Basin of southern Peru. Proc. Natl. Acad. Sci. USA 2016, 113, 13672–13677. [Google Scholar] [CrossRef]
- Perry, G.H.; Dominy, N.J.; Claw, K.G.; Lee, A.S.; Fiegler, H.; Redon, R.; Werner, J.; Villanea, F.A.; Mountain, J.L.; Misra, R.; et al. Diet and the evolution of human amylase gene copy number variation. Nat. Genet. 2007, 39, 1256–1260. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, D.; Mitchell, L.M.; Armour, J.A.L. Copy number variation of human AMY1 is a minor contributor to variation in salivary amylase expression and activity. Hum. Genom. 2017, 11, 2. [Google Scholar] [CrossRef]
- Fernández, C.I.; Wiley, A.S. Rethinking the starch digestion hypothesis for AMY1copy number variation in humans. Am. J. Phys. Anthropol. 2017, 163, 645–657. [Google Scholar] [CrossRef]
- Lindo, J.; Huerta-Sánchez, E.; Nakagome, S.; Rasmussen, M.; Petzelt, B.; Mitchell, J.; Cybulski, J.S.; Willerslev, E.; DeGiorgio, M.; Malhi, R.S. A time transect of exomes from a Native American population before and after European contact. Nat. Commun. 2016, 7, 13175. [Google Scholar] [CrossRef]
- Dobyns, H. Disease transfer at contact. Annu. Rev. Anthropol. 1993, 22, 273–291. [Google Scholar] [CrossRef]
- Norris, E.T.; Rishishwar, L.; Chande, A.T.; Conley, A.B.; Ye, K.; Valderrama-Aguirre, A.; Jordan, I.K. Admixture-enabled selection for rapid adaptive evolution in the Americas. Genome Biol. 2020, 21, 29. [Google Scholar] [CrossRef] [PubMed]
- Vicuña, L.; Klimenkova, O.; Norambuena, T.; Martinez, F.I.; Fernandez, M.I.; Shchur, V.; Eyheramendy, S. Postadmixture Selection on Chileans Targets Haplotype Involved in Pigmentation, Thermogenesis and Immune Defense against Pathogens. Genome Biol. Evol. 2020, 12, 1459–1470. [Google Scholar] [CrossRef]
- Ramenofsky, A. Native American disease history: Past, present and future directions. World Archaeol. 2010, 35, 241–257. [Google Scholar] [CrossRef]
- Parker, M. Panama Fever; Anchor: New York, NY, USA, 2009. [Google Scholar]
- Bos, K.I.; Harkins, K.M.; Herbig, A.; Coscolla, M.; Weber, N.; Comas, I.; Forrest, S.A.; Bryant, J.M.; Harris, S.R.; Schuenemann, V.J.; et al. Pre-Columbian mycobacterial genomes reveal seals as a source of New World human tuberculosis. Nature 2014, 514, 494–497. [Google Scholar] [CrossRef]
- Shriver, M.D.; Kennedy, G.C.; Parra, E.J.; Lawson, H.A.; Sonpar, V.; Huang, J.; Akey, J.M.; Jones, K.W. The genomic distribution of population substructure in four populations using 8525 autosomal SNPs. Hum. Genom. 2004, 1, 274–286. [Google Scholar] [CrossRef]
- Cheng, J.Y.; Mailund, T.; Nielsen, R. Fast admixture analysis and population tree estimation for SNP and NGS data. Bioinformatics 2017, 33, 2148–2155. [Google Scholar] [CrossRef]
- Librado, P.; Gamba, C.; Gaunitz, C.; Der Sarkissian, C.; Pruvost, M.; Albrechtsen, A.; Fages, A.; Khan, N.; Schubert, M.; Jagannathan, V.; et al. Ancient genomic changes associated with domestication of the horse. Science 2017, 356, 442–445. [Google Scholar] [CrossRef] [PubMed]
- Librado, P.; Orlando, L. Detecting Signatures of Positive Selection along Defined Branches of a Population Tree Using LSD. Mol. Biol. Evol. 2018, 35, 1520–1535. [Google Scholar] [CrossRef] [PubMed]
- Racimo, F. Testing for Ancient Selection Using Cross-population Allele Frequency Differentiation. Genetics 2016, 202, 733–750. [Google Scholar] [CrossRef]
- Bonhomme, M.; Chevalet, C.; Servin, B.; Boitard, S.; Abdallah, J.; Blott, S.; Sancristobal, M. Detecting selection in population trees: The Lewontin and Krakauer test extended. Genetics 2010, 186, 241–262. [Google Scholar] [CrossRef]
- Fariello, M.I.; Boitard, S.; Naya, H.; Sancristobal, M.; Servin, B. Detecting Signatures of Selection Through Haplotype Differentiation Among Hierarchically Structured Populations. Genetics 2013, 193, 929–941. [Google Scholar] [CrossRef]
- Peyrégne, S.; Boyle, M.J.; Dannemann, M.; Prüfer, K. Detecting ancient positive selection in humans using extended lineage sorting. Genome Res. 2017, 27, 1563–1572. [Google Scholar] [CrossRef] [PubMed]
- Johnson, K.E.; Voight, B.F. Patterns of shared signatures of recent positive selection across human populations. Nat. Ecol. Evol. 2018, 2, 713–720. [Google Scholar] [CrossRef] [PubMed]
- Harris, A.M.; DeGiorgio, M. Identifying and Classifying Shared Selective Sweeps from Multilocus Data. Genetics 2020, 215, 143–171. [Google Scholar] [CrossRef]
- Waters, M.R. Late Pleistocene exploration and settlement of the Americas by modern humans. Science 2019, 365, eaat5447. [Google Scholar] [PubMed]
- Scheinsohn, V. Hunter-gatherer archaeology in South America. Annu. Rev. Anthropol. 2003, 32, 339–361. [Google Scholar] [CrossRef]
- Ilardo, M.A.; Moltke, I.; Korneliussen, T.S.; Cheng, J.; Stern, A.J.; Racimo, F.; de Barros Damgaard, P.; Sikora, M.; Seguin-Orlando, A.; Rasmussen, S.; et al. Physiological and Genetic Adaptations to Diving in Sea Nomads. Cell 2018, 173, 569–573.e15. [Google Scholar] [CrossRef]
- Schrider, D.R.; Kern, A.D. S/HIC: Robust Identification of Soft and Hard Sweeps Using Machine Learning. PLoS Genet. 2016, 12, e1005928. [Google Scholar] [CrossRef]
- Sheehan, S.; Song, Y.S. Deep Learning for Population Genetic Inference. PLoS Comput. Biol. 2016, 12, e1004845. [Google Scholar] [CrossRef]
- Schrider, D.R.; Ayroles, J.; Matute, D.R.; Kern, A.D. Supervised machine learning reveals introgressed loci in the genomes of Drosophila simulans and D. sechellia. PLoS Genet. 2018, 14, e1007341. [Google Scholar] [CrossRef]
- Sugden, L.A.; Atkinson, E.G.; Fischer, A.P.; Rong, S.; Henn, B.M.; Ramachandran, S. Localization of adaptive variants in human genomes using averaged one-dependence estimation. Nat. Commun. 2018, 9, 703–714. [Google Scholar] [CrossRef] [PubMed]
- Lin, K.; Li, H.; Schlötterer, C.; Futschik, A. Distinguishing positive selection from neutral evolution: Boosting the performance of summary statistics. Genetics 2011, 187, 229–244. [Google Scholar] [CrossRef] [PubMed]
- Flagel, L.; Brandvain, Y.; Schrider, D.R. The Unreasonable Effectiveness of Convolutional Neural Networks in Population Genetic Inference. Mol. Biol. Evol. 2019, 36, 220–238. [Google Scholar] [CrossRef]
- Adrion, J.R.; Galloway, J.G.; Kern, A.D. Predicting the Landscape of Recombination Using Deep Learning. Mol. Biol. Evol. 2020, 37, 1790–1808. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Wang, J.; Kourakos, M.; Hoang, N. Automatic Inference of Demographic Parameters Using Generative Adversarial Networks. bioRxiv 2020. [Google Scholar] [CrossRef]
- Mughal, M.R.; DeGiorgio, M. Localizing and Classifying Adaptive Targets with Trend Filtered Regression. Mol. Biol. Evol. 2019, 36, 252–270. [Google Scholar] [CrossRef]
- Mughal, M.R.; Koch, H.; Huang, J.; Chiaromonte, F.; DeGiorgio, M. Learning the properties of adaptive regions with functional data analysis. PLoS Genet. 2020, 16, e1008896. [Google Scholar] [CrossRef]
- Torada, L.; Lorenzon, L.; Beddis, A.; Isildak, U.; Pattini, L.; Mathieson, S.; Fumagalli, M. ImaGene: A convolutional neural network to quantify natural selection from genomic data. BMC Bioinform. 2019, 20, 337. [Google Scholar] [CrossRef]
- Isildak, U.; Stella, A.; Fumagalli, M. Distinguishing between recent balancing selection and incomplete sweep using deep neural networks. bioRxiv 2020. [Google Scholar] [CrossRef]
- Berg, J.J.; Coop, G. A population genetic signal of polygenic adaptation. PLoS Genet. 2014, 10, e1004412. [Google Scholar] [CrossRef]
- Racimo, F.; Berg, J.J.; Pickrell, J.K. Detecting Polygenic Adaptation in Admixture Graphs. Genetics 2018, 208, 1565–1584. [Google Scholar] [CrossRef] [PubMed]
- Edge, M.D.; Coop, G. Reconstructing the History of Polygenic Scores Using Coalescent Trees. Genetics 2019, 211, 235–262. [Google Scholar] [CrossRef] [PubMed]
- Speidel, L.; Forest, M.; Shi, S.; Myers, S.R. A method for genome-wide genealogy estimation for thousands of samples. Nat. Genet. 2019, 51, 1321–1329. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lindo, J.; DeGiorgio, M. Understanding the Adaptive Evolutionary Histories of South American Ancient and Present-Day Populations via Genomics. Genes 2021, 12, 360. https://doi.org/10.3390/genes12030360
Lindo J, DeGiorgio M. Understanding the Adaptive Evolutionary Histories of South American Ancient and Present-Day Populations via Genomics. Genes. 2021; 12(3):360. https://doi.org/10.3390/genes12030360
Chicago/Turabian StyleLindo, John, and Michael DeGiorgio. 2021. "Understanding the Adaptive Evolutionary Histories of South American Ancient and Present-Day Populations via Genomics" Genes 12, no. 3: 360. https://doi.org/10.3390/genes12030360
APA StyleLindo, J., & DeGiorgio, M. (2021). Understanding the Adaptive Evolutionary Histories of South American Ancient and Present-Day Populations via Genomics. Genes, 12(3), 360. https://doi.org/10.3390/genes12030360