
The Genetics of Sudden Infant Death Syndrome—Towards a Gene Reference Resource


 ,
,  and
and 
Abstract
1. Introduction
1.1. Cardiac Defects
1.2. Central Nervous System (CNS) and CNS Pathway Defects
1.3. Immune System Dysfunction
1.4. Metabolism Deficiencies and Other Disorders
2. Materials and Methods
2.1. Collection of Data and Scoring
2.2. Functional Analysis of Genomic Variants
2.3. GO and Pathway Enrichment Analyses
2.4. Network Analysis
2.5. Tissue Gene Expression
3. Results
3.1. Criteria for Scoring Categories
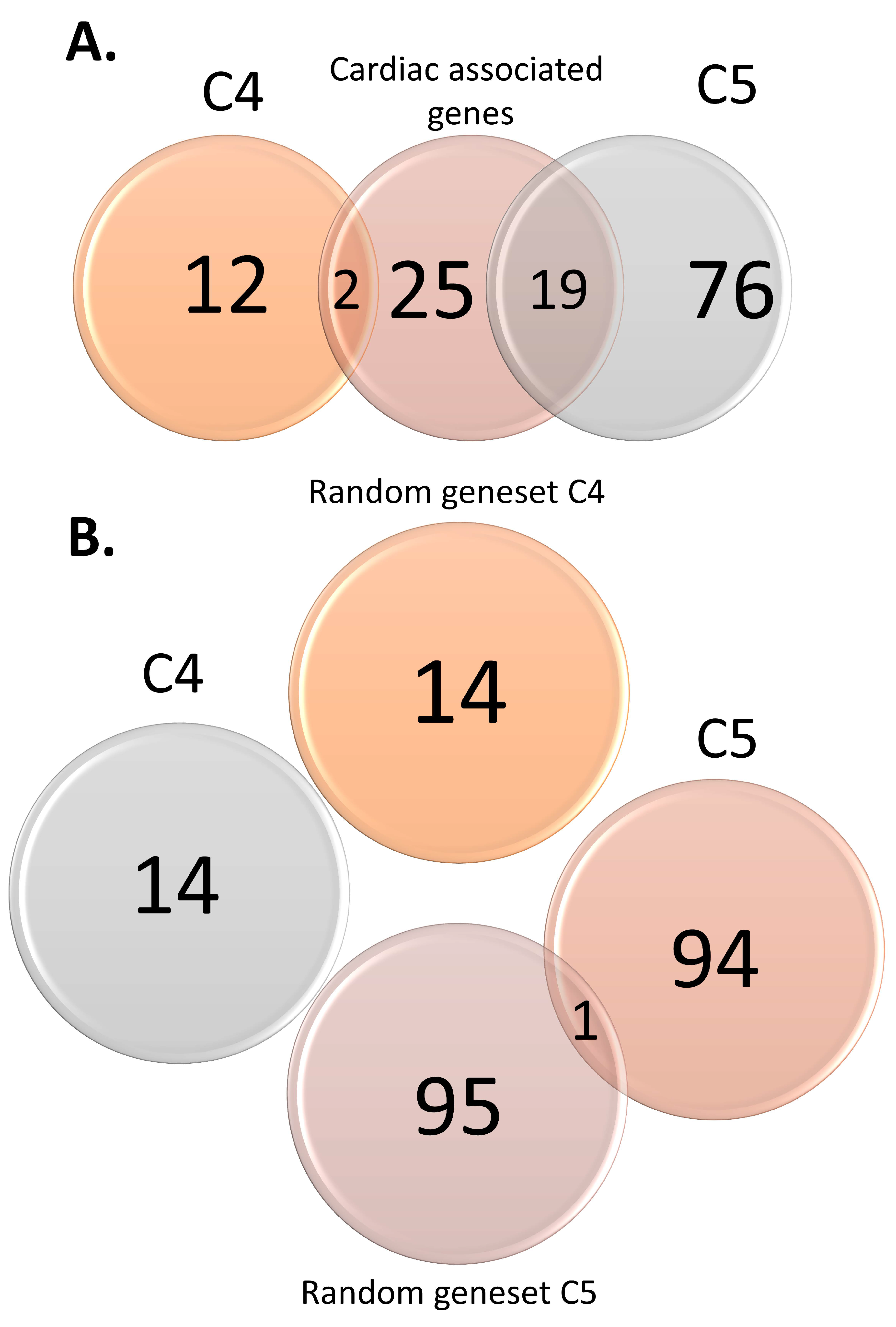
3.2. Functional Analysis of Genomic Variants
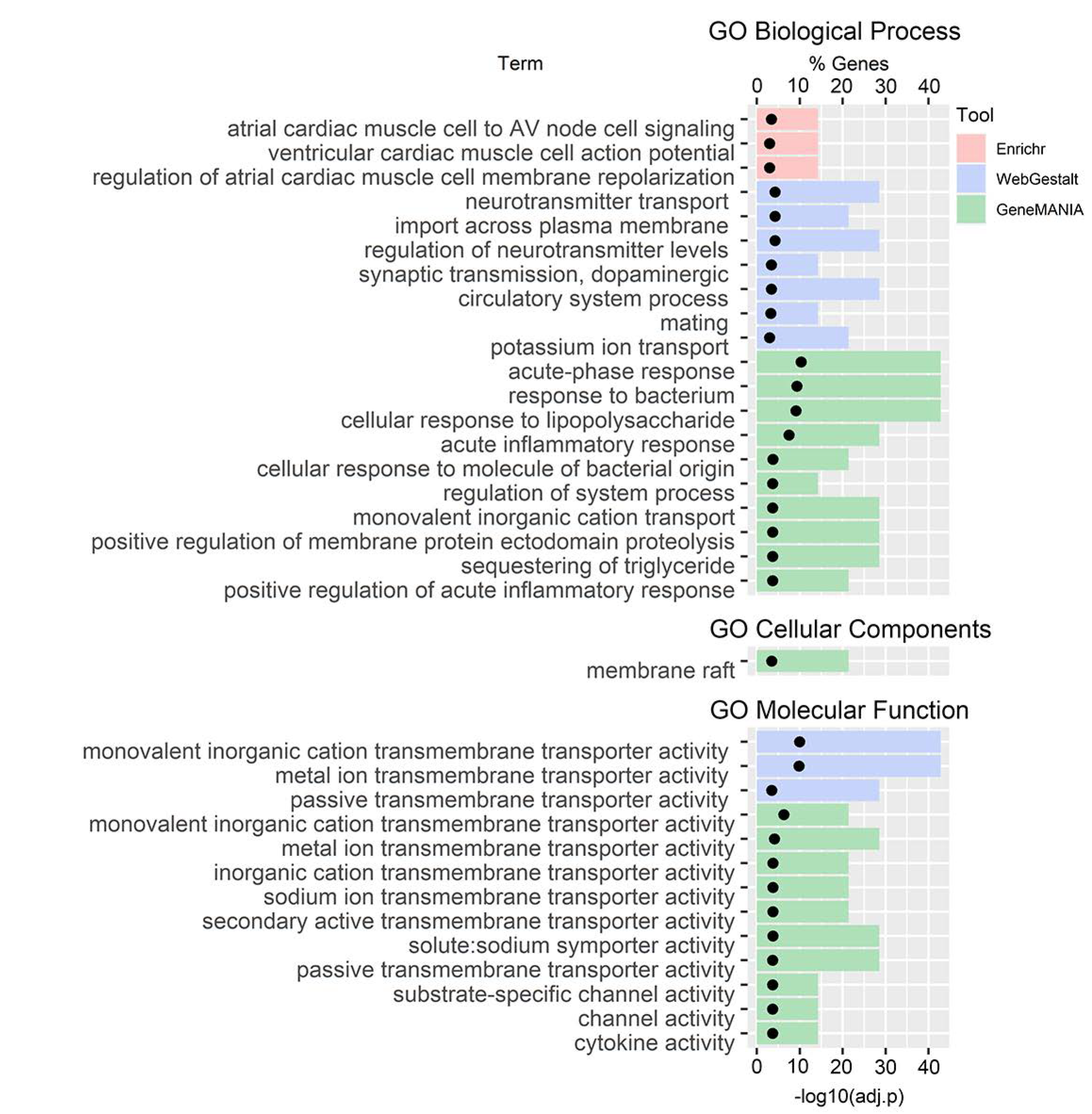
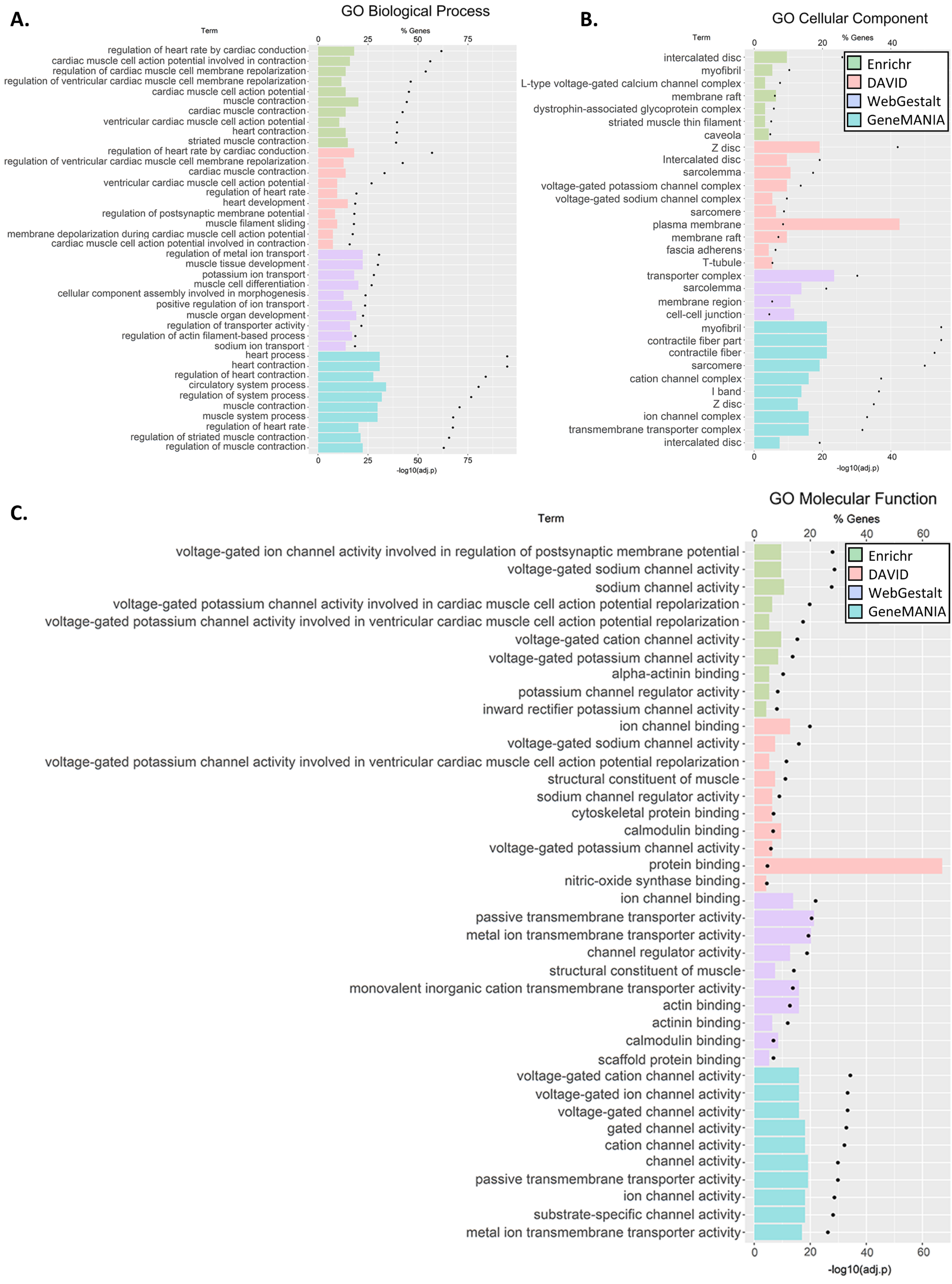
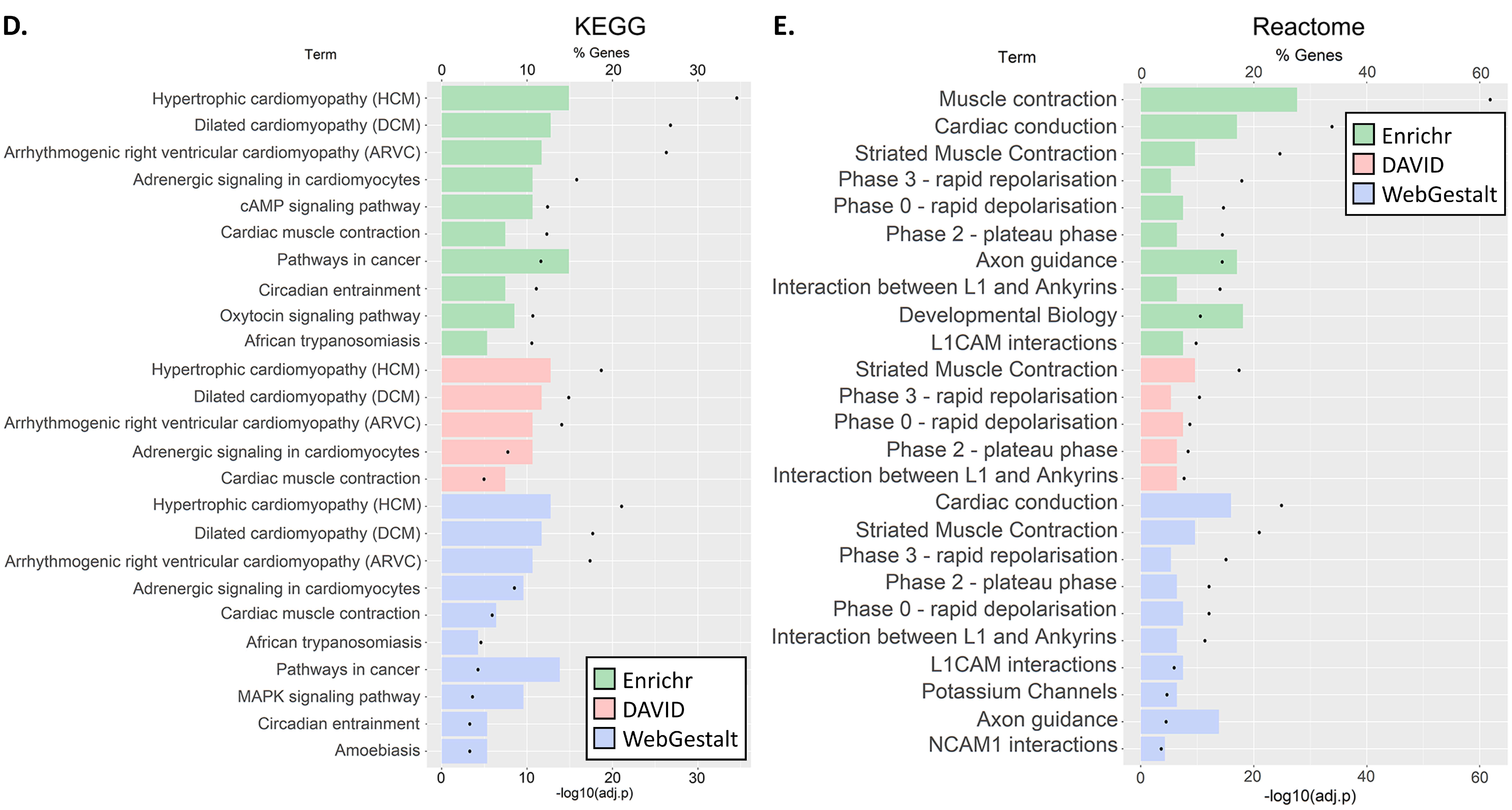
3.3. GO and Pathway Enrichment Analyses
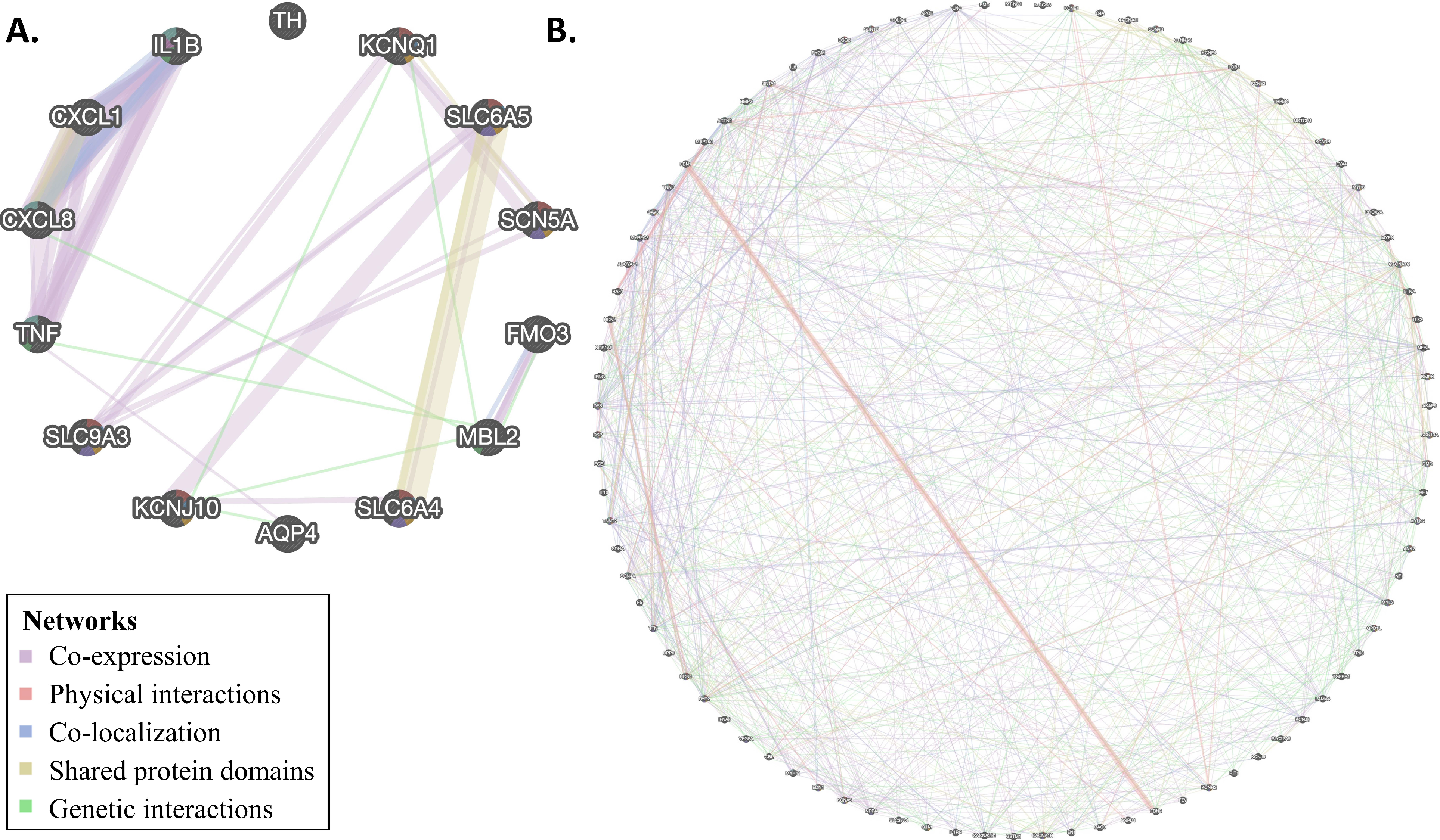
3.4. Network Analysis
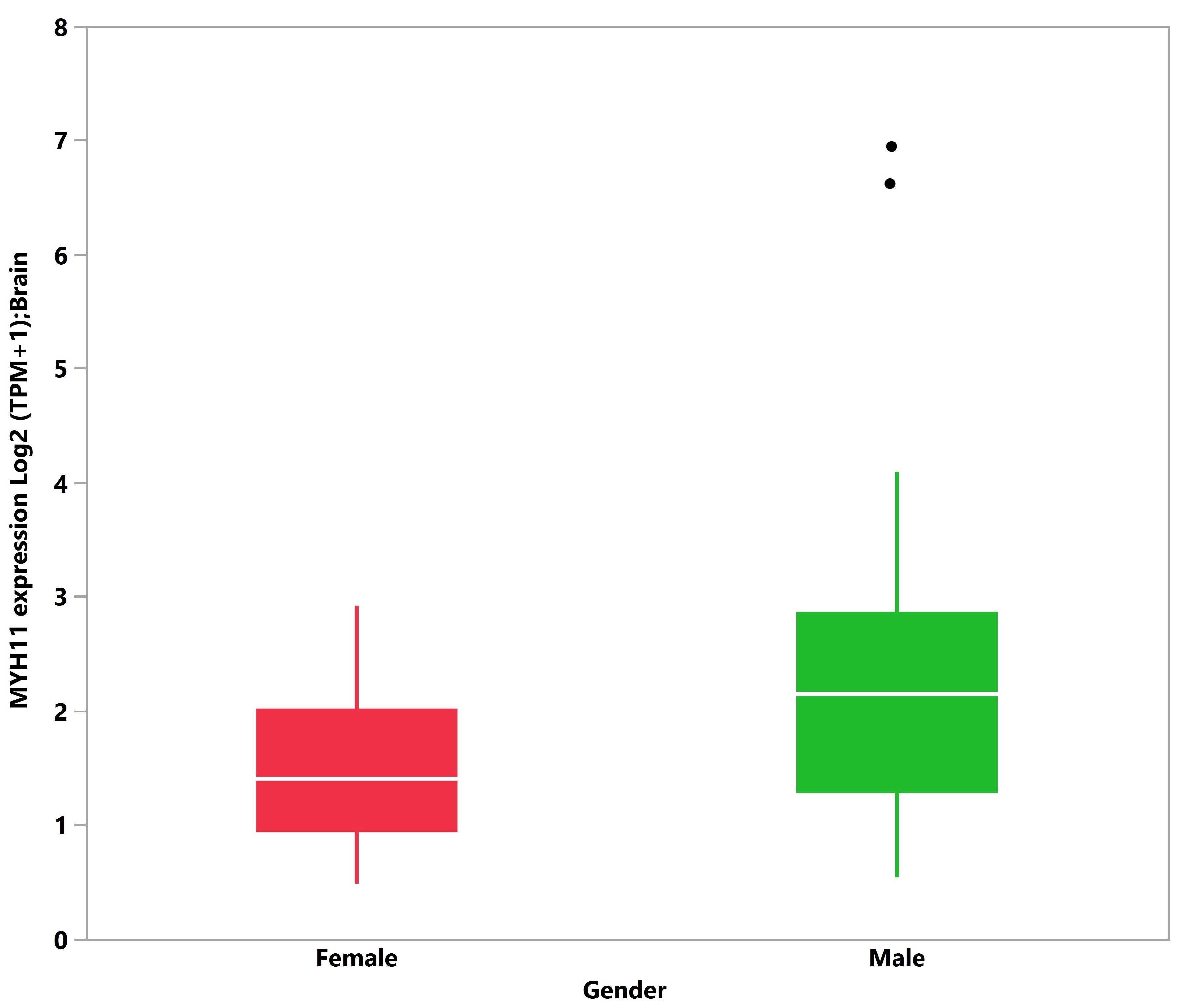
3.5. Tissue Expression Analysis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Werne, J.; Garrow, I. Sudden apparently unexplained death during infancy: I. Pathologic findings in infants found dead. Am. J. Pathol. 1953, 29, 633. [Google Scholar] [PubMed]
- Werne, J.; Garrow, I. Sudden apparently unexplained death during infancy: II. Pathologic findings in infants observed to die suddenly. Am. J. Pathol. 1953, 29, 817. [Google Scholar] [PubMed]
- Garrow, I.; Werne, J. Sudden apparently unexplained death during infancy: III. Pathologic findings in infants dying immediately after violence, contrasted with those after sudden apparently unexplained death. Am. J. Pathol. 1953, 29, 833. [Google Scholar] [PubMed]
- Adelson, L.; Kinney, E.R. Sudden and unexpected death in infancy and childhood. Pediatrics 1956, 17, 663–699. [Google Scholar] [PubMed]
- Beckwith, J.B. The sudden infant death syndrome. Curr. Probl. Pediatr. 1973, 3, 1–36. [Google Scholar] [CrossRef]
- Mitchell, E.A.; Krous, H.F. Sudden unexpected death in infancy: A historical perspective. J. Paediatr. Child Health 2015, 51, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Krous, H.F.; Beckwith, J.B.; Byard, R.W.; Rognum, T.O.; Bajanowski, T.; Corey, T.; Cutz, E.; Hanzlick, R.; Keens, T.G.; Mitchell, E.A. Sudden infant death syndrome and unclassified sudden infant deaths: A definitional and diagnostic approach. Pediatrics 2004, 114, 234–238. [Google Scholar] [CrossRef]
- Fleming, P.J.; Blair, P.S.; Pease, A. Sudden unexpected death in infancy: Aetiology, pathophysiology, epidemiology and prevention in 2015. Arch. Dis. Child. 2015, 100, 984–988. [Google Scholar]
- Tursan d’Espaignet, E.; Bulsara, M.; Wolfenden, L.; Byard, R.W.; Stanley, F.J. Trends in sudden infant death syndrome in Australia from 1980 to 2002. Forensic Sci. Med. Pathol. 2008, 4, 83–90. [Google Scholar] [CrossRef]
- Goldstein, R.D.; Trachtenberg, F.L.; Sens, M.A.; Harty, B.J.; Kinney, H.C. Overall postneonatal mortality and rates of SIDS. Pediatrics 2016, 137, 1–10. [Google Scholar] [CrossRef]
- Moon, R.Y.; Horne, R.S.; Hauck, F.R. Sudden infant death syndrome. Lancet 2007, 370, 1578–1587. [Google Scholar] [CrossRef]
- Centers for Disease Control and Prevention. National Center for Health Statistics CDC Wonder On-Line Database, Compiled from Compressed Mortality File 1999–2016 Series. Available online: https://wonder.cdc.gov/wonder/help/cmf.html (accessed on 8 January 2021).
- Ely, D.M.; Driscoll, A.K. Infant mortality in the united states, 2017: Data from the period linked birth/infant death file. Natl. Vital Stat. Rep. 2019, 68, 1–20. [Google Scholar] [PubMed]
- Filiano, J.; Kinney, H. A perspective on neuropathologic findings in victims of the sudden infant death syndrome: The triple-risk model. Biol. Neonate 1994, 65, 194–197. [Google Scholar] [CrossRef] [PubMed]
- Grether, J.K.; Schulman, J. Sudden infant death syndrome and birth weight. J. Pediatr. 1989, 114, 561–567. [Google Scholar] [CrossRef]
- Mage, D.T.; Donner, M. A unifying theory for SIDS. Int. J. Pediatr. 2009, 2009, 368270. [Google Scholar] [CrossRef]
- Elhaik, E. A “wear and tear” hypothesis to explain sudden infant death syndrome (SIDS). Front. Neurol. 2016, 7. [Google Scholar] [CrossRef]
- Elhaik, E. Neonatal circumcision and prematurity are associated with sudden infant death syndrome (SIDS). J. Clin. Transl. Res. 2018, 4, 136–151. [Google Scholar] [CrossRef]
- Blair, P.S.; Nadin, P.; Cole, T.J.; Fleming, P.J.; Smith, I.J.; Platt, M.W.; Berry, P.J.; Golding, J. Weight gain and sudden infant death syndrome: Changes in weight z scores may identify infants at increased risk. Arch. Dis. Child. 2000, 82, 462–469. [Google Scholar] [CrossRef]
- Taylor, B.J.; Williams, S.M.; Mitchell, E.A.; Ford, R.P. Symptoms, sweating and reactivity of infants who die of SIDS compared with community controls. New Zealand national cot death study group. J. Paediatr. Child Health 1996, 32, 316–322. [Google Scholar] [CrossRef]
- Saetre, E.; Abdelnoor, M. Incidence rate of sudden death in epilepsy: A systematic review and meta-analysis. Epilepsy Behav. 2018, 86, 193–199. [Google Scholar] [CrossRef]
- Gordon, A.E.; Ahmer, O.R.E.; Chan, R.; Madani, O.M.A.; Braun, J.M.; Weir, D.M.; Busuttil, A.; Blackwell, C.C. Why is smoking a risk factor for sudden infant death syndrome? Child Care Health Dev. 2002, 28, 23–25. [Google Scholar] [CrossRef] [PubMed]
- Brownstein, C.A.; Poduri, A.; Goldstein, R.D.; Holm, I.A. The genetics of sudden infant death syndrome. In SIDS Sudden Infant and Early Childhood Death: The Past, The Present and The Future; Duncan, J.R., Byard, R.W., Eds.; University of Adelaide Press: Adelaide, Australia, 2018. [Google Scholar]
- Steer, J.; Annavarapu, S. The genetics of sudden infant death syndrome. In Investigation of Sudden Infant Death Syndrome; Cohen, M.C., Scheimberg, I.B., Beckwith, J.B., Hauck, F., Eds.; Cambridge University Press: Cambridge, UK, 2019. [Google Scholar]
- Weese-Mayer, D.E.; Ackerman, M.J.; Marazita, M.L.; Berry-Kravis, E.M. Sudden infant death syndrome: Review of implicated genetic factors. Am. J. Med. Genet. A 2007, 143A, 771–788. [Google Scholar] [CrossRef] [PubMed]
- Neubauer, J.; Lecca, M.R.; Russo, G.; Bartsch, C.; Medeiros-Domingo, A.; Berger, W.; Haas, C. Post-mortem whole-exome analysis in a large sudden infant death syndrome cohort with a focus on cardiovascular and metabolic genetic diseases. Eur. J. Hum. Genet. 2017, 25, 404–409. [Google Scholar] [CrossRef] [PubMed]
- Gray, B.; Behr, E.R. New insights into the genetic basis of inherited arrhythmia syndromes. Circ. Cardiovasc. Genet. 2016, 9, 569–577. [Google Scholar] [CrossRef]
- Ioakeimidis, N.S.; Papamitsou, T.; Meditskou, S.; Iakovidou-Kritsi, Z. Sudden infant death syndrome due to long QT syndrome: A brief review of the genetic substrate and prevalence. J. Biol. Res. Thessalon. 2017, 24, 6. [Google Scholar] [CrossRef]
- Benito, B.; Brugada, R.; Brugada, J.; Brugada, P. Brugada syndrome. Prog. Cardiovasc. Dis. 2008, 51, 1–22. [Google Scholar] [CrossRef]
- Brugada, R.; Campuzano, O.; Sarquella-Brugada, G.; Brugada, J.; Brugada, P. Brugada syndrome. Methodist Debakey Cardiovasc. J. 2014, 10, 25–28. [Google Scholar] [CrossRef]
- Campuzano, O.; Beltrán-Álvarez, P.; Iglesias, A.; Scornik, F.; Pérez, G.; Brugada, R. Genetics and cardiac channelopathies. Genet. Med. 2010, 12, 260–267. [Google Scholar] [CrossRef]
- Wong, L.C.H.; Behr, E.R. Sudden unexplained death in infants and children: The role of undiagnosed inherited cardiac conditions. Europace 2014, 16, 1706–1713. [Google Scholar] [CrossRef]
- Hof, T.; Liu, H.; Sallé, L.; Schott, J.-J.; Ducreux, C.; Millat, G.; Chevalier, P.; Probst, V.; Guinamard, R.; Bouvagnet, P. Trpm4 non-selective cation channel variants in long QT syndrome. BMC Med. Genet. 2017, 18, 31. [Google Scholar] [CrossRef]
- Osawa, M.; Kimura, R.; Hasegawa, I.; Mukasa, N.; Satoh, F. SNP association and sequence analysis of the nos1ap gene in SIDS. Leg. Med. 2009, 11, S307–S308. [Google Scholar] [CrossRef] [PubMed]
- Van Norstrand, D.W.; Ackerman, M.J. Genomic risk factors in sudden infant death syndrome. Genome Med. 2010, 2, 86. [Google Scholar] [CrossRef] [PubMed]
- Haynes, R.L.; Frelinger, A.L.; Giles, E.K.; Goldstein, R.D.; Tran, H.; Kozakewich, H.P.; Haas, E.A.; Gerrits, A.J.; Mena, O.J.; Trachtenberg, F.L.; et al. High serum serotonin in sudden infant death syndrome. Proc. Natl. Acad. Sci. USA 2017, 114, 7695–7700. [Google Scholar] [CrossRef] [PubMed]
- Salomonis, N. Systems-level perspective of sudden infant death syndrome. Pediatr. Res. 2014, 76, 220–229. [Google Scholar] [CrossRef]
- Narita, N.; Narita, M.; Takashima, S.; Nakayama, M.; Nagai, T.; Okado, N. Serotonin transporter gene variation is a risk factor for sudden infant death syndrome in the Japanese population. Pediatrics 2001, 107, 690–692. [Google Scholar] [CrossRef]
- Pattyn, A.; Goridis, C.; Brunet, J.-F. Specification of the central noradrenergic phenotype by the homeobox gene Phox2b. Mol. Cell. Neurosci. 2000, 15, 235–243. [Google Scholar] [CrossRef]
- Pattyn, A.; Hirsch, M.; Goridis, C.; Brunet, J.F. Control of hindbrain motor neuron differentiation by the homeobox gene Phox2b. Development 2000, 127, 1349–1358. [Google Scholar]
- Liebrechts-Akkerman, G.; Liu, F.; Lao, O.; Ooms, A.H.A.G.; van Duijn, K.; Vermeulen, M.; Jaddoe, V.W.; Hofman, A.; Engelberts, A.C.; Kayser, M. Phox2b polyalanine repeat length is associated with sudden infant death syndrome and unclassified sudden infant death in the Dutch population. Int. J. Leg. Med. 2014, 128, 621–629. [Google Scholar] [CrossRef]
- Wilson, R.J.A.; Cummings, K.J. Pituitary adenylate cyclase-activating polypeptide is vital for neonatal survival and the neuronal control of breathing. Respir. Physiol. Neurobiol. 2008, 164, 168–178. [Google Scholar] [CrossRef]
- Farnham, M.M.J.; Pilowsky, P.M. The role of PACAP in central cardiorespiratory regulation. Respir. Physiol. Neurobiol. 2010, 174, 65–75. [Google Scholar] [CrossRef]
- Courts, C.; Grabmüller, M.; Madea, B. Monoamine oxidase a gene polymorphism and the pathogenesis of sudden infant death syndrome. J. Pediatr. 2013, 163, 89–93. [Google Scholar] [CrossRef] [PubMed]
- Groß, M.; Bajanowski, T.; Vennemann, M.; Poetsch, M. Sudden infant death syndrome (SIDS) and polymorphisms in monoamine oxidase a gene (maoa): A revisit. Int. J. Leg. Med. 2014, 128, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Li, A.; Emond, L.; Nattie, E. Brainstem catecholaminergic neurons modulate both respiratory and cardiovascular function. In Integration in Respiratory Control: From Genes to Systems; Poulin, M.J., Wilson, R.J.A., Eds.; Springer: New York, NY, USA, 2008; pp. 371–376. [Google Scholar]
- Klintschar, M.; Heimbold, C. Association between a functional polymorphism in the MAOA gene and sudden infant death syndrome. Pediatrics 2012, 129, e756–e761. [Google Scholar] [CrossRef] [PubMed]
- Arnestad, M.; Andersen, M.; Vege, A.; Rognum, T.O. Changes in the epidemiological pattern of sudden infant death syndrome in southeast Norway, 1984-1998: Implications for future prevention and research. Arch. Dis. Child. 2001, 85, 108–115. [Google Scholar] [CrossRef][Green Version]
- Summers, A.M.; Summers, C.W.; Drucker, D.B.; Hajeer, A.H.; Barson, A.; Hutchinson, I.V. Association of IL-10 genotype with sudden infant death syndrome. Hum. Immunol. 2000, 61, 1270–1273. [Google Scholar] [CrossRef]
- Rognum, I.J.; Haynes, R.L.; Vege, Ǻ.; Yang, M.; Rognum, T.O.; Kinney, H.C. Interleukin-6 and the serotonergic system of the medulla oblongata in the sudden infant death syndrome. Acta Neuropathol. 2009, 118, 519–530. [Google Scholar] [CrossRef]
- Schneider, P.M.; Wendler, C.; Riepert, T.; Braun, L.; Schacker, U.; Horn, M.; Althoff, H.; Mattern, R.; Rittner, C. Possible association of sudden infant death with partial complement C4 deficiency revealed by post-mortem DNA typing of hla class ii and iii genes. Eur. J. Pediatr. 1989, 149, 170–174. [Google Scholar] [CrossRef]
- Moscovis, S.M.; Gordon, A.E.; Al Madani, O.M.; Gleeson, M.; Scott, R.J.; Hall, S.T.; Burns, C.; Blackwell, C. Genetic and environmental factors affecting TNF-alpha responses in relation to sudden infant death syndrome. Front. Immunol. 2015, 6, 374. [Google Scholar] [CrossRef]
- Moscovis, S.M.; Gordon, A.E.; Al Madani, O.M.; Gleeson, M.; Scott, R.J.; Roberts-Thomson, J.; Hall, S.T.; Weir, D.M.; Busuttil, A.; Blackwell, C.C. Interleukin-10 and sudden infant death syndrome. FEMS Immunol. Med. Microbiol. 2004, 42, 130–138. [Google Scholar] [CrossRef]
- Moscovis, S.M.; Gordon, A.E.; Al Madani, O.M.; Gleeson, M.; Scott, R.J.; Roberts-Thomson, J.; Hall, S.T.; Weir, D.M.; Busuttil, A.; Blackwell, C.C. IL6 G-174C associated with sudden infant death syndrome in a Caucasian Australian cohort. Hum. Immunol. 2006, 67, 819–825. [Google Scholar] [CrossRef]
- Moscovis, S.; Gordon, A.; Almadani, O.; Gleeson, M.; Scott, R.; Hall, S.; Burns, C.; Blackwell, C. Virus infections and sudden death in infancy: The role of interferon-γ. Front. Immunol. 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Dott, M.; Chace, D.; Fierro, M.; Kalas, T.A.; Hannon, W.H.; Williams, J.; Rasmussen, S.A. Metabolic disorders detectable by tandem mass spectrometry and unexpected early childhood mortality: A population-based study. Am. J. Med. Genet. A 2006, 140, 837–842. [Google Scholar] [CrossRef] [PubMed]
- Howat, A.J.; Bennett, M.J.; Variend, S.; Shaw, L.; Engel, P.C. Defects of metabolism of fatty acids in the sudden infant death syndrome. Brit. Med. J. 1985, 290, 1771–1773. [Google Scholar] [CrossRef] [PubMed]
- Lundemose, J.B.; Kølvraa, S.; Gregersen, N.; Christensen, E.; Gregersen, M. Fatty acid oxidation disorders as primary cause of sudden and unexpected death in infants and young children: An investigation performed on cultured fibroblasts from 79 children who died aged between 0–4 years. Mol. Pathol. 1997, 50, 212–217. [Google Scholar] [CrossRef] [PubMed]
- Moczulski, D.; Majak, I.; Mamczur, D. An overview of beta-oxidation disorders. Postepy Hig. Med. Dosw. 2009, 63, 266–277. [Google Scholar]
- Poetsch, M.; Czerwinski, M.; Wingenfeld, L.; Vennemann, M.; Bajanowski, T. A common FMO3 polymorphism may amplify the effect of nicotine exposure in sudden infant death syndrome (SIDS). Int. J. Leg. Med. 2010, 124, 301–306. [Google Scholar] [CrossRef] [PubMed]
- Say, M.; Machaalani, R.; Waters, K.A. Changes in serotoninergic receptors 1A and 2A in the piglet brainstem after intermittent hypercapnic hypoxia (IHH) and nicotine. Brain Res. 2007, 1152, 17–26. [Google Scholar] [CrossRef]
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Hoover, J.; et al. Clinvar: Public archive of interpretations of clinically relevant variants. Nucleic Acids Res. 2016, 44, D862–D868. [Google Scholar] [CrossRef]
- Basu, S.N.; Kollu, R.; Banerjee-Basu, S. AutDB: A gene reference resource for autism research. Nucleic Acids Res. 2009, 37, D832–D836. [Google Scholar] [CrossRef]
- Abrahams, B.S.; Arking, D.E.; Campbell, D.B.; Mefford, H.C.; Morrow, E.M.; Weiss, L.A.; Menashe, I.; Wadkins, T.; Banerjee-Basu, S.; Packer, A. SFARI Gene 2.0: A community-driven knowledgebase for the autism spectrum disorders (ASDs). Mol. Autism 2013, 4, 36. [Google Scholar] [CrossRef]
- Sayers, E.W.; Agarwala, R.; Bolton, E.E.; Brister, J.R.; Canese, K.; Clark, K.; Connor, R.; Fiorini, N.; Funk, K.; Hefferon, T.; et al. Database resources of the national center for biotechnology information. Nucleic Acids Res. 2018, 47, D23–D28. [Google Scholar] [CrossRef] [PubMed]
- Norland, K.; Sveinbjornsson, G.; Thorolfsdottir, R.B.; Davidsson, O.B.; Tragante, V.; Rajamani, S.; Helgadottir, A.; Gretarsdottir, S.; van Setten, J.; Asselbergs, F.W.; et al. Sequence variants with large effects on cardiac electrophysiology and disease. Nat. Commun. 2019, 10, 4803. [Google Scholar] [CrossRef] [PubMed]
- Paludan-Müller, C.; Ghouse, J.; Vad, O.B.; Herfelt, C.B.; Lundegaard, P.; Ahlberg, G.; Schmitt, N.; Svendsen, J.H.; Haunsø, S.; Bundgaard, H.; et al. Reappraisal of variants previously linked with sudden infant death syndrome: Results from three population-based cohorts. Eur. J. Hum. Genet. 2019, 27, 1427–1435. [Google Scholar] [CrossRef] [PubMed]
- Christiansen, S.L.; Hertz, C.L.; Ferrero-Miliani, L.; Dahl, M.; Weeke, P.E.; Ottesen, G.L.; Frank-Hansen, R.; Bundgaard, H.; Morling, N. Genetic investigation of 100 heart genes in sudden unexplained death victims in a forensic setting. Eur. J. Hum. Genet. 2016, 24, 1797–1802. [Google Scholar] [CrossRef] [PubMed]
- Shen, S.; Tao, L.; Wang, X.; Kong, X.; Li, X. Common variants for heart failure. Curr. Genom. 2015, 16, 82–87. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Conrad, D.F.; Jakobsson, M.; Coop, G.; Wen, X.; Wall, J.D.; Rosenberg, N.A.; Pritchard, J.K. A worldwide survey of haplotype variation and linkage disequilibrium in the human genome. Nat. Genet. 2006, 38, 1251–1260. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Boutros, P.C. VennDiagram: A package for the generation of highly-customizable Venn and Euler diagrams in R. BMC Bioinform. 2011, 12, 35. [Google Scholar] [CrossRef]
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.; Thormann, A.; Flicek, P.; Cunningham, F. The Ensembl Variant Effect Predictor. Genome Biol. 2016, 17, 122. [Google Scholar] [CrossRef]
- Wickham, H. Ggplot2: Elegant Graphics for Data Analysis, 2nd ed.; Springer: New York, NY, USA, 2016. [Google Scholar]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The gene ontology consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef]
- Fabregat, A.; Jupe, S.; Matthews, L.; Sidiropoulos, K.; Gillespie, M.; Garapati, P.; Haw, R.; Jassal, B.; Korninger, F.; May, B.; et al. The Reactome pathway Knowledgebase. Nucleic Acids Res. 2017, 46, D649–D655. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. Kegg: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Franz, M.; Rodriguez, H.; Lopes, C.; Zuberi, K.; Montojo, J.; Bader, G.D.; Morris, Q. GeneMANIA update 2018. Nucleic Acids Res. 2018, 46, W60–W64. [Google Scholar] [CrossRef] [PubMed]
- Seifuddin, F.; Singh, K.; Suresh, A.; Judy, J.T.; Chen, Y.C.; Chaitankar, V.; Tunc, I.; Ruan, X.; Li, P.; Chen, Y.; et al. IncRNAKB, a knowledgebase of tissue-specific functional annotation and trait association of long noncoding RNA. Sci. Data 2020, 7, 326. [Google Scholar] [CrossRef] [PubMed]
- Horne, R.S.; Hauck, F.R.; Moon, R.Y. Sudden infant death syndrome and advice for safe sleeping. BMJ 2015, 350, h1989. [Google Scholar] [CrossRef]
- Hunt, C.E.; Hauck, F.R. Sudden infant death syndrome. CMAJ 2006, 174, 1861–1869. [Google Scholar] [CrossRef]
- Goldwater, P.N. Why mainstream SIDS research is not achieving its goal. Am. J. Pediatr. 2018, 4, 104–109. [Google Scholar]
- Phillips, D.P.; Brewer, K.M.; Wadensweiler, P. Alcohol as a risk factor for sudden infant death syndrome (SIDS). Addiction 2011, 106, 516–525. [Google Scholar] [CrossRef]
- Byard, R.W.; Shipstone, R.A.; Young, J. Continuing major inconsistencies in the classification of unexpected infant deaths. J. Forensic Leg. Med. 2019, 64, 20–22. [Google Scholar] [CrossRef]
- Goldstein, R.D.; Blair, P.S.; Sens, M.A.; Shapiro-Mendoza, C.K.; Krous, H.F.; Rognum, T.O.; Moon, R.Y.; Anderson, R.N.; Blair, P.S.; Bundock, E.A.; et al. Inconsistent classification of unexplained sudden deaths in infants and children hinders surveillance, prevention and research: Recommendations from the 3rd international congress on sudden infant and child death. Forensic Sci. Med. Pathol. 2019, 15, 622–628. [Google Scholar] [CrossRef]
- Hefti, M.M.; Kinney, H.C.; Cryan, J.B.; Haas, E.A.; Chadwick, A.E.; Crandall, L.A.; Trachtenberg, F.L.; Armstrong, D.D.; Grafe, M.; Krous, H.F. Sudden unexpected death in early childhood: General observations in a series of 151 cases. Forensic Sci. Med. Pathol. 2016, 12, 4–13. [Google Scholar] [CrossRef]
- Rochtus, A.M.; Goldstein, R.D.; Holm, I.A.; Brownstein, C.A.; Pérez-Palma, E.; Haynes, R.; Lal, D.; Poduri, A.H. The role of sodium channels in sudden unexpected death in pediatrics. Mol. Genet. Genom. Med. 2020, 8, e1309. [Google Scholar] [CrossRef] [PubMed]
- Alhopuro, P.; Phichith, D.; Tuupanen, S.; Sammalkorpi, H.; Nybondas, M.; Saharinen, J.; Robinson, J.P.; Yang, Z.; Chen, L.-Q.; Orntoft, T.; et al. Unregulated smooth-muscle myosin in human intestinal neoplasia. Proc. Natl. Acad. Sci. USA 2008, 105, 5513–5518. [Google Scholar] [CrossRef] [PubMed]
- Castilla, L.H.; Garrett, L.; Adya, N.; Orlic, D.; Dutra, A.; Anderson, S.; Owens, J.; Eckhaus, M.; Bodine, D.; Liu, P.P. The fusion gene Cbfb-MYH11 blocks myeloid differentiation and predisposes mice to acute myelomonocytic leukaemia. Nat. Genet. 1999, 23, 144–146. [Google Scholar] [CrossRef] [PubMed]
- Takeda, N.; Morita, H.; Fujita, D.; Inuzuka, R.; Taniguchi, Y.; Nawata, K.; Komuro, I. A deleterious MYH11 mutation causing familial thoracic aortic dissection. Hum. Genome Var. 2015, 2, 15028. [Google Scholar] [CrossRef]
- Goldwater, P.N. SIDS, prone sleep position and infection: An overlooked epidemiological link in current SIDS research? Key evidence for the “infection hypothesis”. Med. Hypotheses 2020, 144, 110114. [Google Scholar] [CrossRef]
- Middleton, O.; Baxter, S.; Demo, E.; Honeywell, C.; Jentzen, J.; Miller, F.; Pinckard, J.K.; Reichard, R.R.; Rutberg, J.; Stacy, C.; et al. National association of medical examiners position paper: Retaining postmortem samples for genetic testing. Acad. Forensic Pathol. 2013, 3, 191–194. [Google Scholar] [CrossRef]
- Platt, M.W.; Blair, P.S.; Fleming, P.J.; Smith, I.J.; Cole, T.J.; Leach, C.E.; Berry, P.J.; Golding, J. A clinical comparison of SIDS and explained sudden infant deaths: How healthy and how normal? CESDI SUDI research group. Confidential inquiry into stillbirths and deaths in infancy study. Arch. Dis. Child. 2000, 82, 98–106. [Google Scholar] [CrossRef]
- Rossidis, A.C.; Stratigis, J.D.; Chadwick, A.C.; Hartman, H.A.; Ahn, N.J.; Li, H.; Singh, K.; Coons, B.E.; Li, L.; Lv, W.; et al. In utero CRISPR-mediated therapeutic editing of metabolic genes. Nat. Med. 2018, 24, 1513–1518. [Google Scholar] [CrossRef]
- Crotti, L.; Johnson, C.N.; Graf, E.; De Ferrari, G.M.; Cuneo, B.F.; Ovadia, M.; Papagiannis, J.; Feldkamp, M.D.; Rathi, S.G.; Kunic, J.D. Calmodulin mutations associated with recurrent cardiac arrest in infants. Circulation 2013, 127, 1009–1017. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Control Type | List | Size |
|---|---|---|
| Positive | Cardiac-associated genes | 46 |
| Cardiac-associated variants | 122 | |
| Negative | Random gene set C4 | 14 |
| Random SNP set C4 | 65 | |
| Random gene set C5 | 96 | |
| Random SNP set C5 | 188 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Johannsen, E.B.; Baughn, L.B.; Sharma, N.; Zjacic, N.; Pirooznia, M.; Elhaik, E. The Genetics of Sudden Infant Death Syndrome—Towards a Gene Reference Resource. Genes 2021, 12, 216. https://doi.org/10.3390/genes12020216
Johannsen EB, Baughn LB, Sharma N, Zjacic N, Pirooznia M, Elhaik E. The Genetics of Sudden Infant Death Syndrome—Towards a Gene Reference Resource. Genes. 2021; 12(2):216. https://doi.org/10.3390/genes12020216
Chicago/Turabian StyleJohannsen, Emma B., Linda B. Baughn, Neeraj Sharma, Nicolina Zjacic, Mehdi Pirooznia, and Eran Elhaik. 2021. "The Genetics of Sudden Infant Death Syndrome—Towards a Gene Reference Resource" Genes 12, no. 2: 216. https://doi.org/10.3390/genes12020216
APA StyleJohannsen, E. B., Baughn, L. B., Sharma, N., Zjacic, N., Pirooznia, M., & Elhaik, E. (2021). The Genetics of Sudden Infant Death Syndrome—Towards a Gene Reference Resource. Genes, 12(2), 216. https://doi.org/10.3390/genes12020216