
Effect of An 84-bp Deletion of the Receptor-Binding Domain on the ACE2 Binding Affinity of the SARS-CoV-2 Spike Protein: An In Silico Analysis

 , ,
, , 
 ,
, 
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection, Andsequencing
2.2. Clinical History and Other Details
2.3. In Vitro Isolation
2.4. In Silico Protein Analysis
3. Results
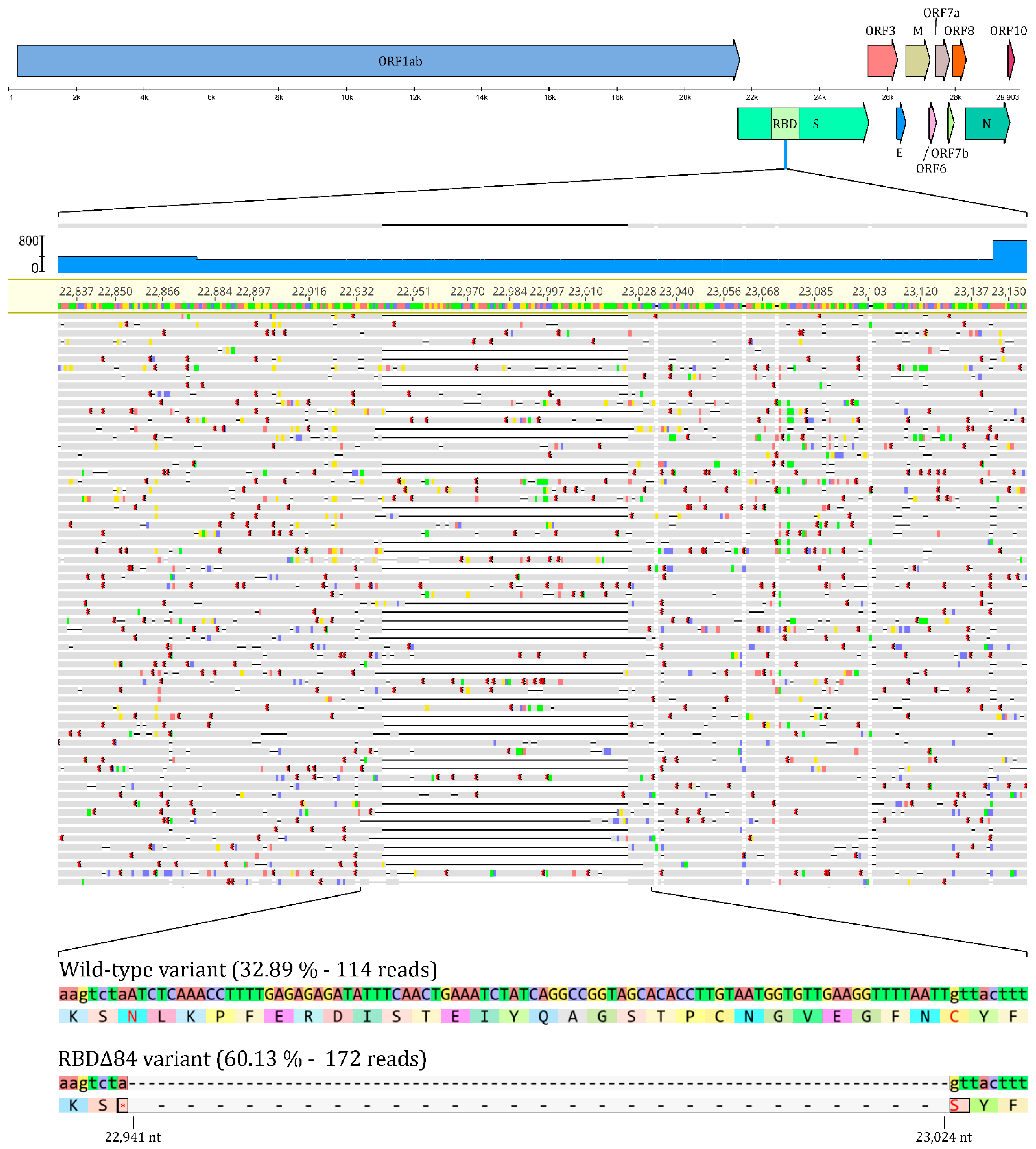
3.1. Sequence Analysis
3.2. In Vitro Isolation
3.3. Protein Modelling Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dong, G.-M.; Xu, G.-L.; Xiao, Q.-Y.; Wang, D.-M.; Hu, Y.-M.; Zhou, D.-J.; Wang, P.; Zhang, Y.-Z.; Yang, X.-M.; Zhu, F.-C.; et al. An epidemiological study of rabies virus in domestic dogs, cats and wildlife and the immunogenicity study for rabies vaccines derived from different cell cultured virus strains. Bing du xue bao = Chin. J. Virol. 2007, 23, 417–423. [Google Scholar]
- Amendola, A.; Bianchi, S.; Gori, M.; Colzani, D.; Canuti, M.; Borghi, E.; Raviglione, M.C.; Zuccotti, G.V.; Tanzi, E. Evidence of SARS-CoV-2 RNA in an Oropharyngeal Swab Specimen, Milan, Italy, Early December 2019. Emerg. Infect. Dis. 2021, 27. [Google Scholar] [CrossRef] [PubMed]
- Dong, E.; Du, H.; Gardner, L. An interactive web-based dashboard to track COVID-19 in real time. Lancet Infect. Dis. 2020, 20, 533–534. [Google Scholar] [CrossRef]
- Cui, J.; Li, F.; Shi, Z.-L. Origin and evolution of pathogenic coronaviruses. Nat. Rev. Microbiol. 2019, 17, 181–192. [Google Scholar] [CrossRef]
- Khailany, R.A.; Safdar, M.; Ozaslan, M. Genomic characterization of a novel SARS-CoV-2. Gene Rep. 2020, 19, 100682. [Google Scholar] [CrossRef]
- Huang, Y.; Yang, C.; Xu, X.; Xu, W.; Liu, S. Structural and functional properties of SARS-CoV-2 Spike protein: Potential antivirus drug development for COVID-19. Acta Pharmacol. Sin. 2020, 41, 1141–1149. [Google Scholar] [CrossRef]
- Preliminary genomic characterisation of an emergent SARS-CoV-2 lineage in the UK defined by a novel set of Spike mutations—SARS-CoV-2 coronavirus/nCoV-2019 Genomic Epidemiology—Virological. Available online: https://virological.org/t/preliminary-genomic-characterisation-of-an-emergent-sars-cov-2-lineage-in-the-uk-defined-by-a-novel-set-of-Spike-mutations/563 (accessed on 21 December 2020).
- Korber, B.; Fischer, W.M.; Gnanakaran, S.; Yoon, H.; Theiler, J.; Abfalterer, W.; Hengartner, N.; Giorgi, E.E.; Bhattacharya, T.; Foley, B.; et al. Tracking Changes in SARS-CoV-2 Spike: Evidence that D614G Increases Infectivity of the COVID-19 Virus. Cell 2020, 182, 812.e19–827.e19. [Google Scholar] [CrossRef]
- Grubaugh, N.D.; Hanage, W.P.; Rasmussen, A.L. Making Sense of Mutation: What D614G Means for the COVID-19 Pandemic Remains Unclear. Cell 2020, 182, 794–795. [Google Scholar] [CrossRef]
- Baric, R.S. Emergence of a Highly Fit SARS-CoV-2 Variant. N. Engl. J. Med. 2020. [Google Scholar] [CrossRef]
- Quick, J. ARTIC Coronavirus Method Development Community 1 more workspace. 2020. [Google Scholar] [CrossRef]
- artic-ncov2019/primer_schemes/nCoV-2019/V3 at Master artic-network/artic-ncov2019 Publisher: GitHub (https://github.com/). Available online: https://github.com/artic-network/artic-ncov2019/tree/master/primer_schemes/nCoV-2019/V3 (accessed on 15 December 2020).
- Artic Network. Available online: https://artic.network/ncov-2019/ncov2019-bioinformatics-sop.html (accessed on 15 December 2020).
- Matsuyama, S.; Nao, N.; Shirato, K.; Kawase, M.; Saito, S.; Takayama, I.; Nagata, N.; Sekizuka, T.; Katoh, H.; Kato, F.; et al. Enhanced isolation of SARS-CoV-2 by TMPRSS2—Expressing cells. Proc. Natl. Acad. Sci. USA 2020, 117, 7001–7003. [Google Scholar] [CrossRef]
- Jacobson, M.P.; Pincus, D.L.; Rapp, C.S.; Day, T.J.F.; Honig, B.; Shaw, D.E.; Friesner, R.A. A Hierarchical Approach to All-Atom Protein Loop Prediction. Proteins Struct. Funct. Genet. 2004, 55, 351–367. [Google Scholar] [CrossRef] [PubMed]
- Yan, R.; Zhang, Y.; Li, Y.; Xia, L.; Guo, Y.; Zhou, Q. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science 2020, 367, 1444–1448. [Google Scholar] [CrossRef] [PubMed]
- Madhavi Sastry, G.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided. Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Bowers, K.J.; Chow, E.; Xu, H.; Dror, R.O.; Eastwood, M.P.; Gregersen, B.A.; Klepeis, J.L.; Kolossvary, I.; Moraes, M.A.; Sacerdoti, F.D.; et al. Scalable Algorithms for Molecular Dynamics Simulations on Commodity Clusters. In Proceedings of the 2006 ACM/IEEE Conference on Supercomputing; Association for Computing Machinery, New York, NY, USA, 11–17 November 2006; p. 84–es. [Google Scholar]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Harder, E.; Damm, W.; Maple, J.; Wu, C.; Reboul, M.; Xiang, J.Y.; Wang, L.; Lupyan, D.; Dahlgren, M.K.; Knight, J.L.; et al. OPLS3: A Force Field Providing Broad Coverage of Drug-like Small Molecules and Proteins. J. Chem. Theory Comput. 2016, 12, 281–296. [Google Scholar] [CrossRef] [PubMed]
- Nosé, S. A molecular dynamics method for simulations in the canonical ensemble. Mol. Phys. 1984, 52, 255–268. [Google Scholar] [CrossRef]
- Hoover, W.G. Canonical dynamics: Equilibrium phase-space distributions. Phys. Rev. A 1985, 31, 1695–1697. [Google Scholar] [CrossRef]
- Martyna, G.J.; Tobias, D.J.; Klein, M.L. Constant pressure molecular dynamics algorithms. J. Chem. Phys. 1994, 101, 4177–4189. [Google Scholar] [CrossRef]
- Frey, B.J.; Dueck, D. Clustering by Passing Messages between Data Points. Science 2007, 315, 972LP–976LP. [Google Scholar] [CrossRef]
- PyMOL|pymol.org. Available online: https://pymol.org/2/ (accessed on 15 December 2020).
- Artimo, P.; Jonnalagedda, M.; Arnold, K.; Baratin, D.; Csardi, G.; De Castro, E.; Duvaud, S.; Flegel, V.; Fortier, A.; Gasteiger, E.; et al. ExPASy: SIB bioinformatics resource portal. Nucleic Acids Res. 2012, 40, W597. [Google Scholar] [CrossRef]
- Bateman, A. UniProt: A worldwide hub of protein knowledge. Nucleic Acids Res. 2019, 47, D506–D515. [Google Scholar] [CrossRef]
- Starr, T.N.; Greaney, A.J.; Hilton, S.K.; Ellis, D.; Crawford, K.H.D.; Dingens, A.S.; Navarro, M.J.; Bowen, J.E.; Tortorici, M.A.; Walls, A.C.; et al. Deep Mutational Scanning of SARS-CoV-2 Receptor Binding Domain Reveals Constraints on Folding and ACE2 Binding. Cell 2020, 182, 1295–1310. [Google Scholar] [CrossRef] [PubMed]
- Lo, S.W.; Jamrozy, D. Genomics and epidemiological surveillance. Nat. Rev. Microbiol. 2020, 18, 478. [Google Scholar] [CrossRef]
- Moore, S.C.; Penrice-Randal, R.; Alruwaili, M.; Randle, N.; Armstrong, S.; Hartley, C.; Haldenby, S.; Dong, X.; Alrezaihi, A.; Almsaud, M.; et al. Amplicon-Based Detection and Sequencing of SARS-CoV-2 in Nasopharyngeal Swabs from Patients With COVID-19 and Identification of Deletions in the Viral Genome That Encode Proteins Involved in Interferon Antagonism. Viruses 2020, 12, 1164. [Google Scholar] [CrossRef] [PubMed]
- Wu, A.; Peng, Y.; Huang, B.; Ding, X.; Wang, X.; Niu, P.; Meng, J.; Zhu, Z.; Zhang, Z.; Wang, J.; et al. Genome Composition and Divergence of the Novel Coronavirus (2019-nCoV) Originating in China. Cell Host Microbe 2020, 27, 325–328. [Google Scholar] [CrossRef]
- Siu, K.-L.; Yuen, K.-S.; Castano-Rodriguez, C.; Ye, Z.-W.; Yeung, M.-L.; Fung, S.-Y.; Yuan, S.; Chan, C.-P.; Yuen, K.-Y.; Enjuanes, L.; et al. Severe acute respiratory syndrome Coronavirus ORF3a protein activates the NLRP3 inflammasome by promoting TRAF3-dependent ubiquitination of ASC. FASEB J. 2019, 33, 8865–8877. [Google Scholar] [CrossRef]
- Kanzawa, N.; Nishigaki, K.; Hayashi, T.; Ishii, Y.; Furukawa, S.; Niiro, A.; Yasui, F.; Kohara, M.; Morita, K.; Matsushima, K.; et al. Augmentation of chemokine production by severe acute respiratory syndrome coronavirus 3a/X1 and 7a/X4 proteins through NF-κB activation. FEBS Lett. 2006, 580, 6807–6812. [Google Scholar] [CrossRef]
- Young, B.E.; Fong, S.-W.; Chan, Y.-H.; Mak, T.-M.; Ang, L.W.; Anderson, D.E.; Lee, C.Y.-P.; Amrun, S.N.; Lee, B.; Goh, Y.S.; et al. Effects of a major deletion in the SARS-CoV-2 genome on the severity of infection and the inflammatory response: An observational cohort study. Lancet 2020, 396, 603–611. [Google Scholar] [CrossRef]
- Su, Y.C.F.; Anderson, D.E.; Young, B.E.; Linster, M.; Zhu, F.; Jayakumar, J.; Zhuang, Y.; Kalimuddin, S.; Low, J.G.H.; Tan, C.W.; et al. Discovery and Genomic Characterization of a 382-Nucleotide Deletion in ORF7b and ORF8 during the Early Evolution of SARS-CoV-2. MBio 2020, 11, e01610-20. [Google Scholar] [CrossRef]
- Islam, M.R.; Hoque, M.N.; Rahman, M.S.; Alam, A.S.M.R.U.; Akther, M.; Puspo, J.A.; Akter, S.; Sultana, M.; Crandall, K.A.; Hossain, M.A. Genome-wide analysis of SARS-CoV-2 virus strains circulating worldwide implicates heterogeneity. Sci. Rep. 2020, 10, 14004. [Google Scholar] [CrossRef]
- McCarthy, K.R.; Rennick, L.J.; Nambulli, S.; Robinson-McCarthy, L.R.; Bain, W.G.; Haidar, G.; Duprex, W.P. Natural deletions in the SARS-CoV-2 Spike glycoprotein drive antibody escape. bioRxiv 2020. [Google Scholar] [CrossRef]
- Hou, Y.J.; Chiba, S.; Halfmann, P.; Ehre, C.; Kuroda, M.; Dinnon, K.H.; Leist, S.R.; Schäfer, A.; Nakajima, N.; Takahashi, K.; et al. SARS-CoV-2 D614G variant exhibits efficient replication ex vivo and transmission in vivo. Science 2020, eabe8499. [Google Scholar] [CrossRef] [PubMed]
- He, J.F.; Peng, G.W.; Min, J.; Yu, D.W.; Liang, W.J.; Zhang, S.Y.; Xu, R.H.; Zheng, H.Y.; Wu, X.W.; Xu, J.; et al. Molecular Evolution of the SARS Coronavirus, during the Course of the SARS Epidemic in China. Science 2004, 303, 1666–1669. [Google Scholar] [CrossRef]
- Urbanowicz, R.A.; McClure, C.P.; Sakuntabhai, A.; Sall, A.A.; Kobinger, G.; Müller, M.A.; Holmes, E.C.; Rey, F.A.; Simon-Loriere, E.; Ball, J.K. Human Adaptation of Ebola Virus during the West African Outbreak. Cell 2016, 167, 1079–1087. [Google Scholar] [CrossRef]
- Thi Nhu Thao, T.; Labroussaa, F.; Ebert, N.; V’kovski, P.; Stalder, H.; Portmann, J.; Kelly, J.; Steiner, S.; Holwerda, M.; Kratzel, A.; et al. Rapid reconstruction of SARS-CoV-2 using a synthetic genomics platform. Nature 2020, 582, 561–565. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Date | PCR Diagnostic Result | Symptoms and Test |
|---|---|---|
| 18 March 2020 | n/a | Tiredness, headache, fever (38.3 °C) |
| 19 March 2020 | n/a | Loss of taste and smell, sore throat, fever |
| 20 March 2020 | Positive | Tiredness, limb weakness, subfebrility (37.5 °C), Charlson Comorbidity index (CCI): 1 |
| 22 March 2020 | n/a | severe numbness in the limbs and tongue |
| 30 March 2020 | Positive | asymptomatic |
| 6 April 2020 | Positive | asymptomatic |
| 16 April 2020 | Negative | asymptomatic |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kemenesi, G.; Tóth, G.E.; Bajusz, D.; Keserű, G.M.; Terhes, G.; Burián, K.; Zeghbib, S.; Somogyi, B.A.; Jakab, F. Effect of An 84-bp Deletion of the Receptor-Binding Domain on the ACE2 Binding Affinity of the SARS-CoV-2 Spike Protein: An In Silico Analysis. Genes 2021, 12, 194. https://doi.org/10.3390/genes12020194
Kemenesi G, Tóth GE, Bajusz D, Keserű GM, Terhes G, Burián K, Zeghbib S, Somogyi BA, Jakab F. Effect of An 84-bp Deletion of the Receptor-Binding Domain on the ACE2 Binding Affinity of the SARS-CoV-2 Spike Protein: An In Silico Analysis. Genes. 2021; 12(2):194. https://doi.org/10.3390/genes12020194
Chicago/Turabian StyleKemenesi, Gábor, Gábor Endre Tóth, Dávid Bajusz, György M. Keserű, Gabriella Terhes, Katalin Burián, Safia Zeghbib, Balázs A. Somogyi, and Ferenc Jakab. 2021. "Effect of An 84-bp Deletion of the Receptor-Binding Domain on the ACE2 Binding Affinity of the SARS-CoV-2 Spike Protein: An In Silico Analysis" Genes 12, no. 2: 194. https://doi.org/10.3390/genes12020194
APA StyleKemenesi, G., Tóth, G. E., Bajusz, D., Keserű, G. M., Terhes, G., Burián, K., Zeghbib, S., Somogyi, B. A., & Jakab, F. (2021). Effect of An 84-bp Deletion of the Receptor-Binding Domain on the ACE2 Binding Affinity of the SARS-CoV-2 Spike Protein: An In Silico Analysis. Genes, 12(2), 194. https://doi.org/10.3390/genes12020194