Long-Term Waterlogging as Factor Contributing to Hypoxia Stress Tolerance Enhancement in Cucumber: Comparative Transcriptome Analysis of Waterlogging Sensitive and Tolerant Accessions



Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Material and Experiment Conditions
2.2. Stress Treatment
2.3. Measurement of Morphological Parameters
2.4. RNA Extraction for RNA-Seq and qRT-PCR
2.5. RNA Library Construction and Illumina Sequencing
2.6. Data Filtering and Quality Control
2.7. Mapping to the Reference Genome
2.8. Transcriptome de novo Assembly
2.9. Differential Expression Estimation
2.10. Functional Analysis
2.11. qRT-PCR Assay
2.12. Statistical Analysis
3. Results
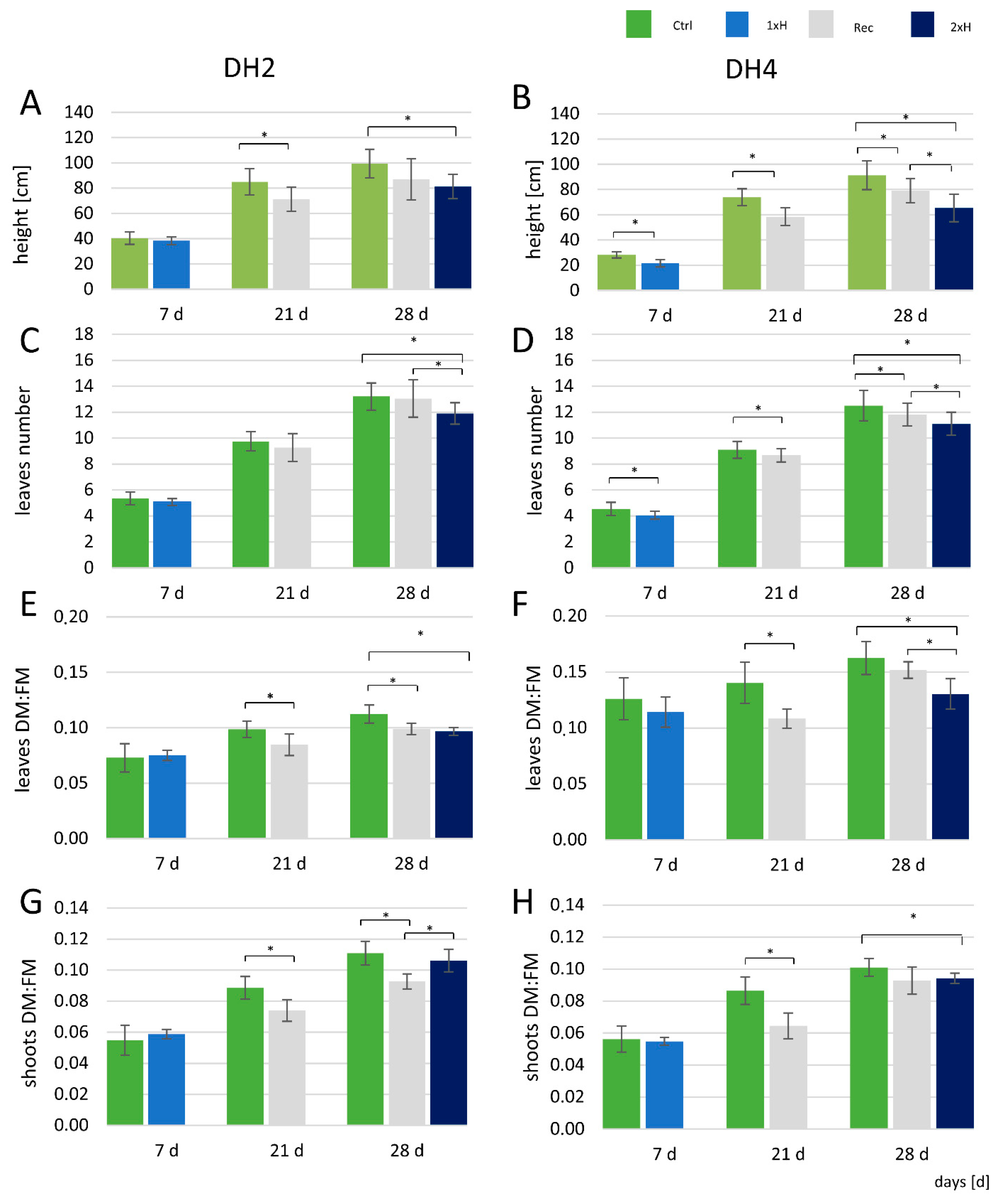
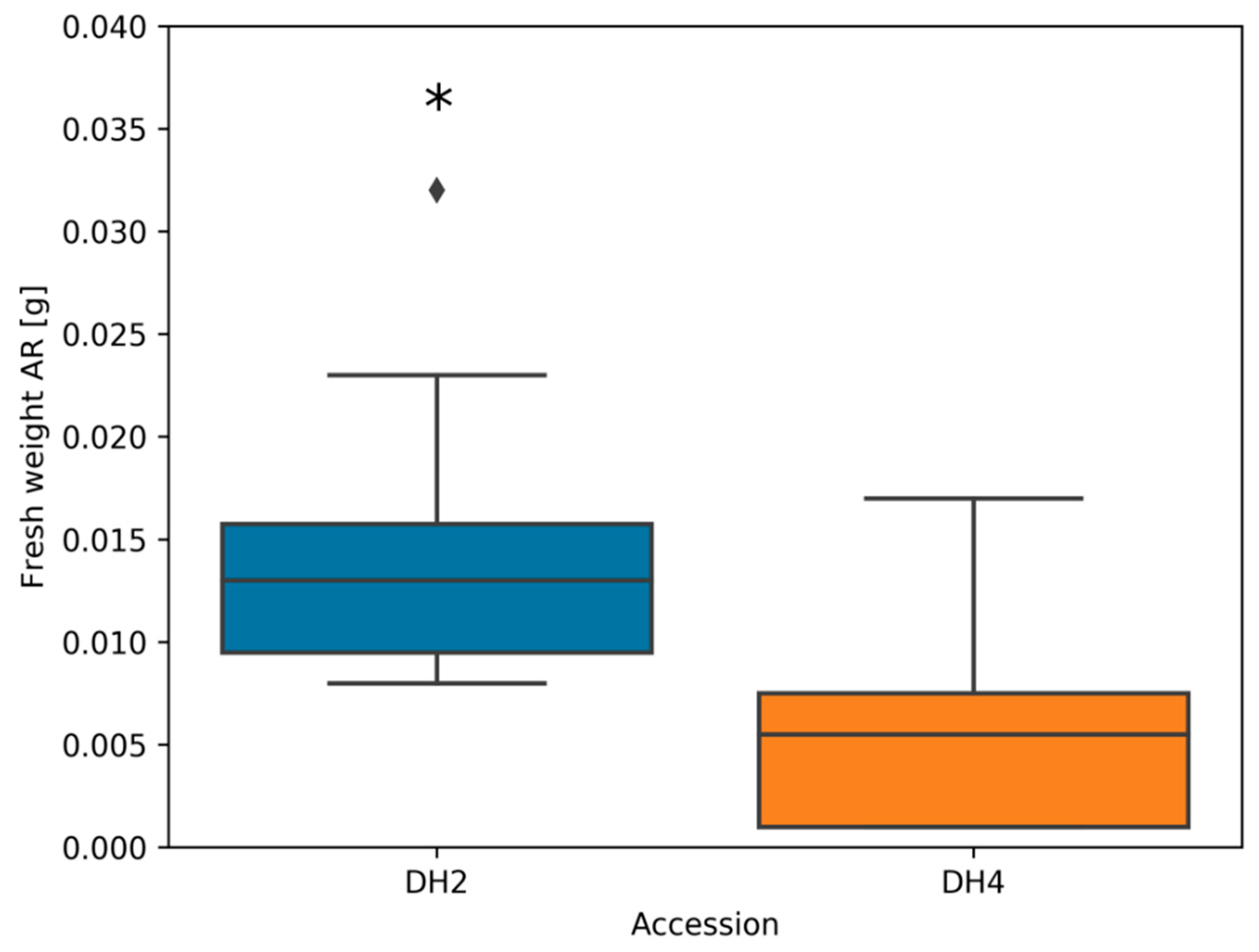
3.1. Plant Morphological Traits
3.2. RNA Sequencing and Transcriptome Assembly
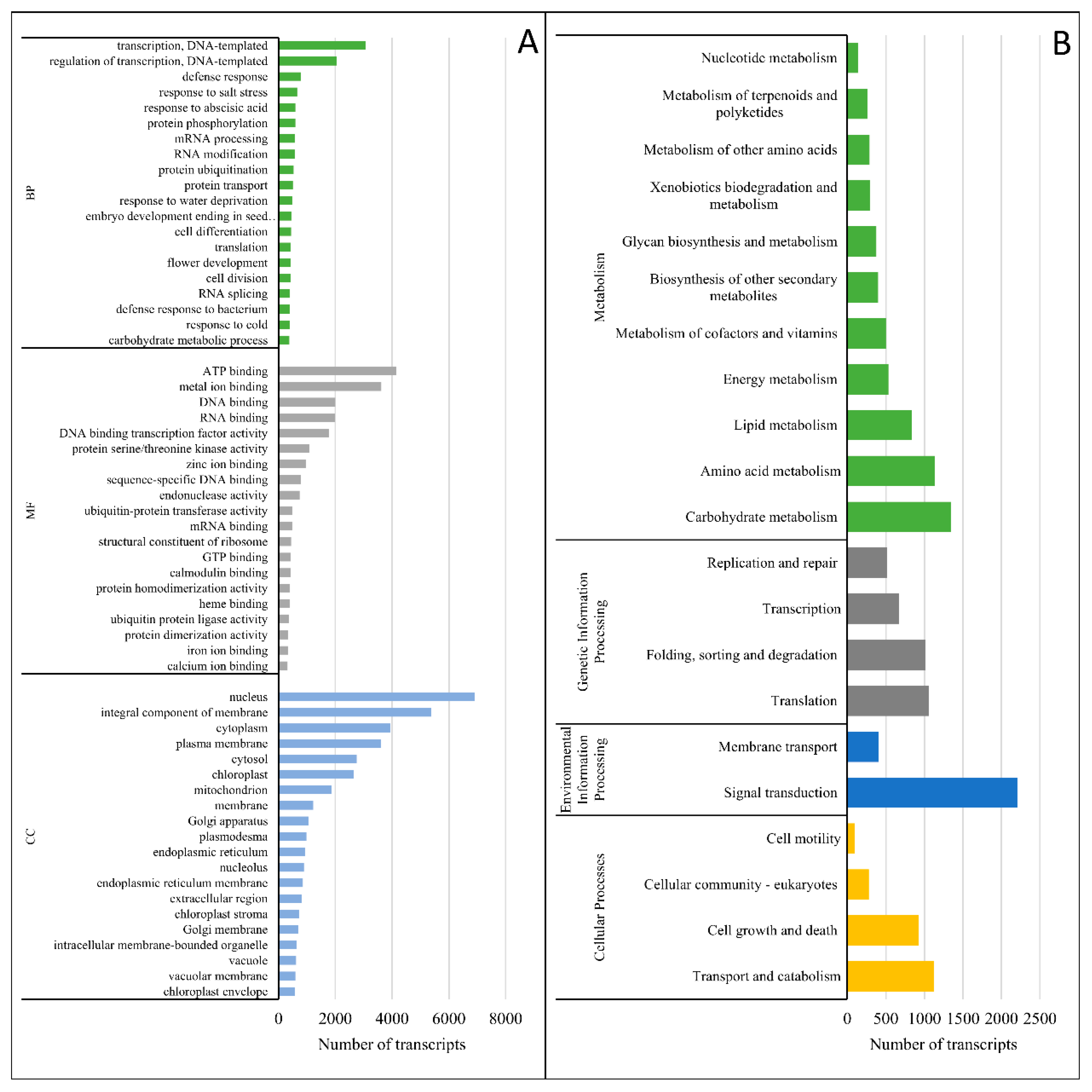
3.3. Transcriptome Characterization and Functional Annotation
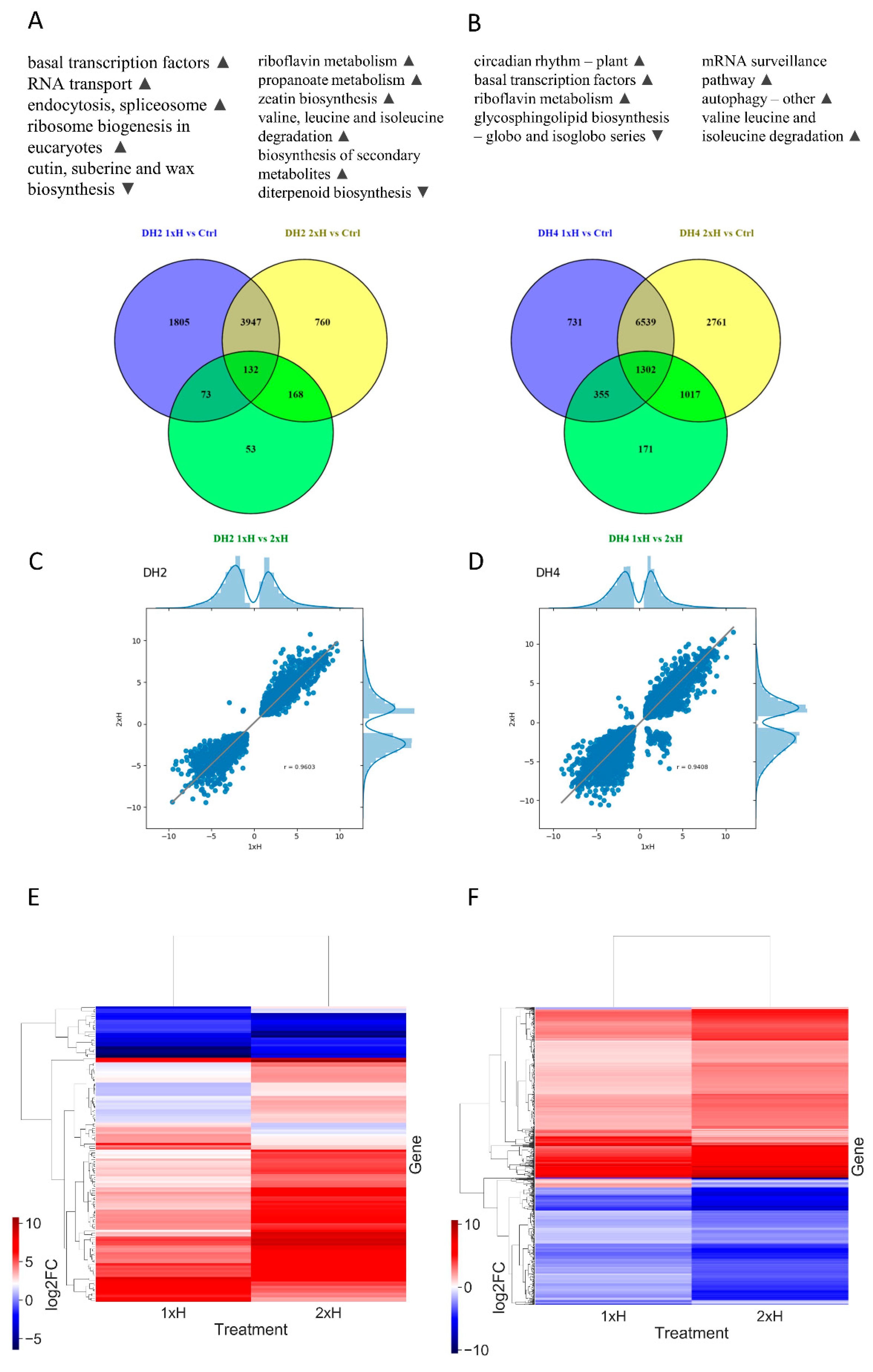
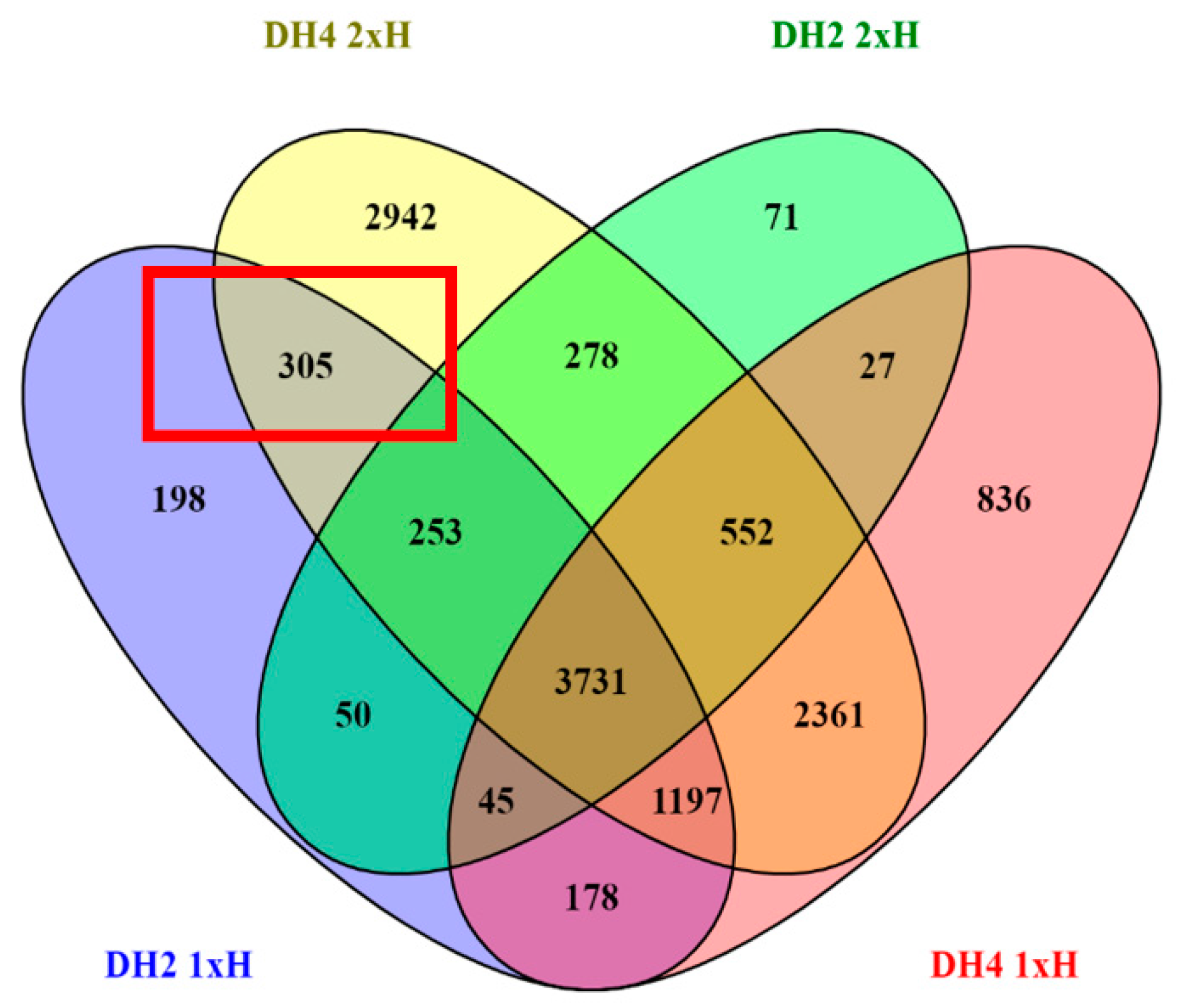
3.4. Identification of Differentially Expressed Genes (DEGs)
3.5. Differential Response to Waterlogging of Primed and Non-Primed WL-T and WL-S Cucumbers
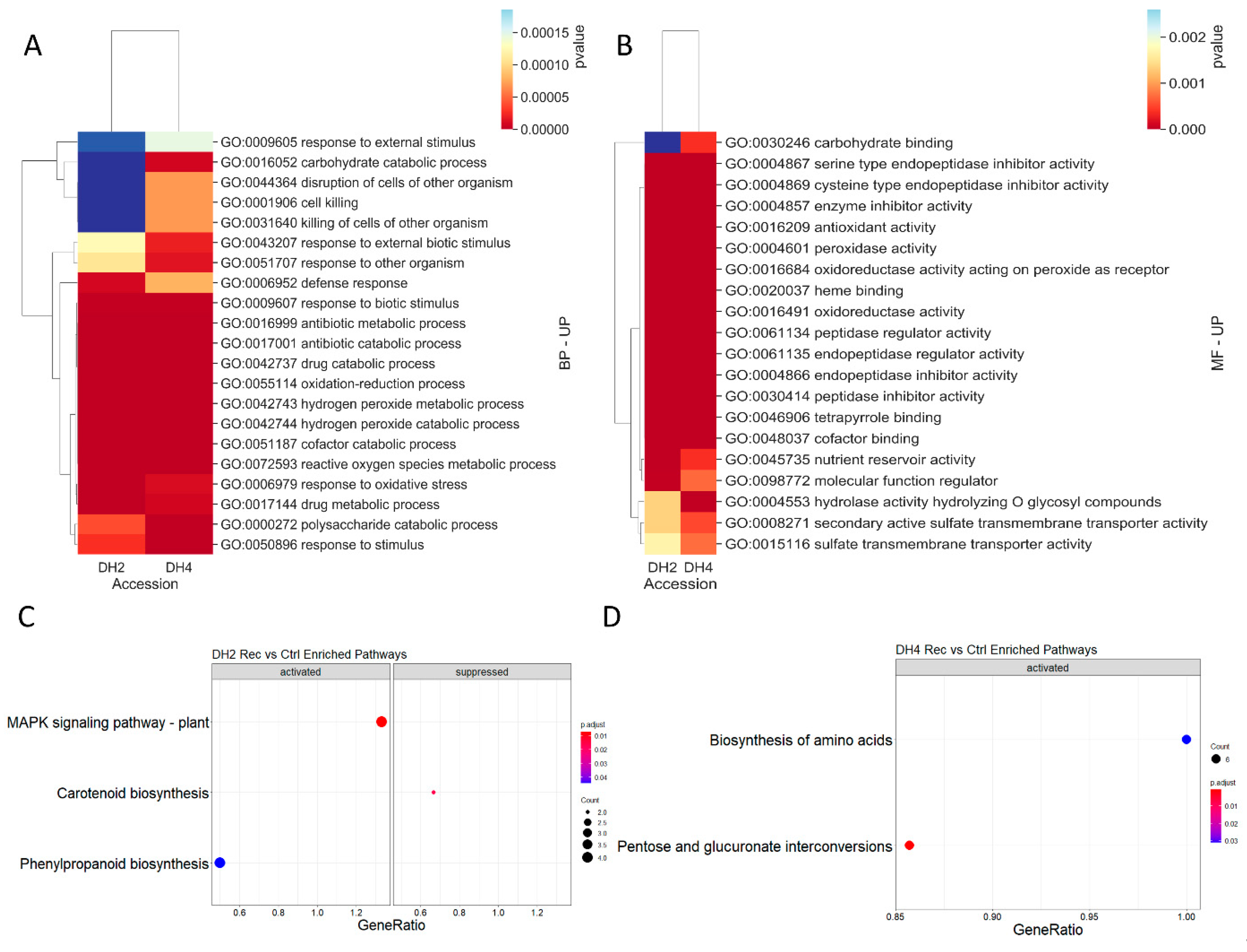
3.6. DEGs Determined in WL-T DH2 and WL-S DH4 Cucumbers in Recovery
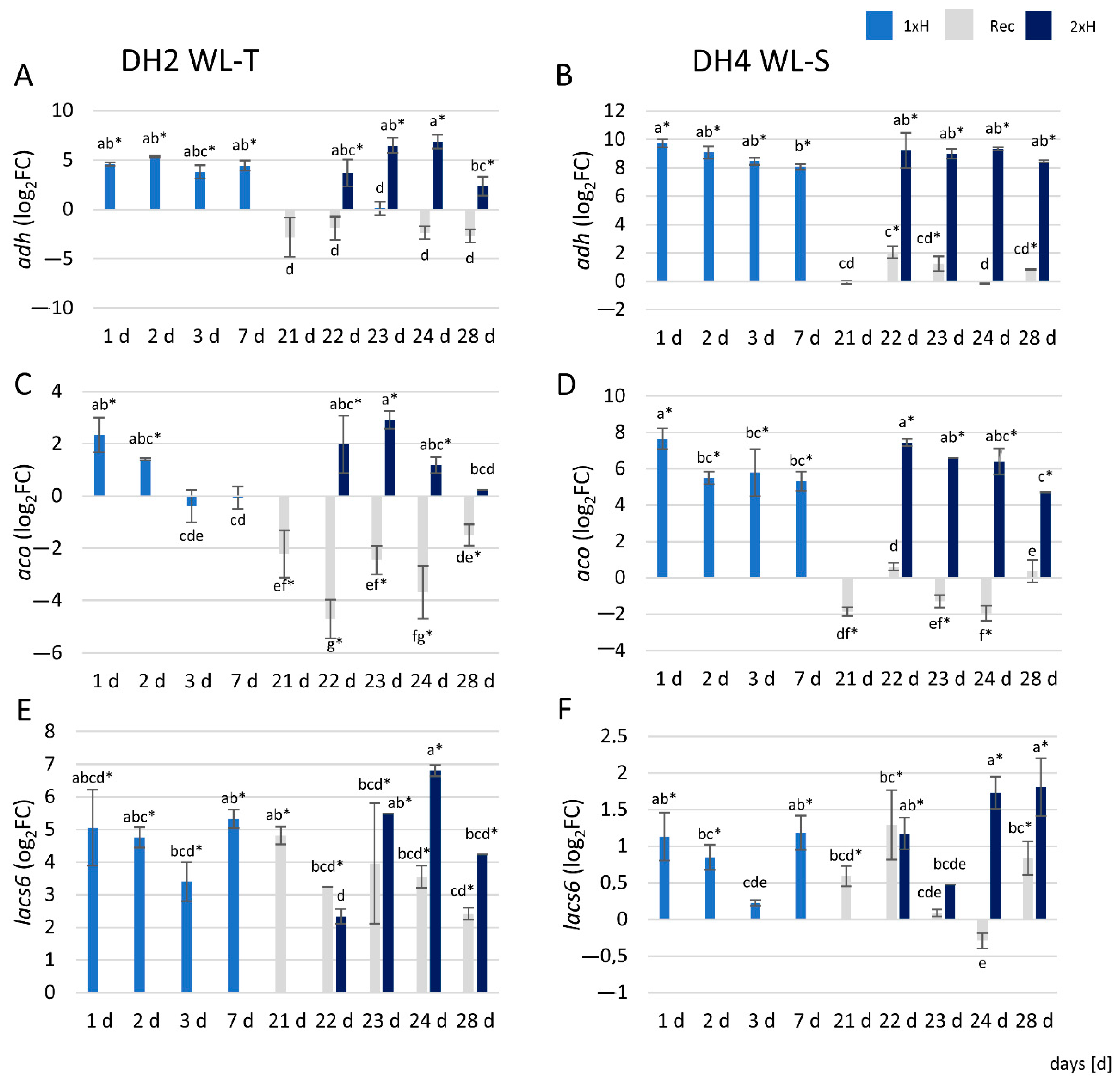
3.7. qRT-PCR Analysis
4. Discussion
4.1. Morphological Changes in Response to Waterlogging Stress
4.2. Changes at the Transcriptomic Level
4.3. Effects of Waterlogging Stress Priming on the WL-T and WL-S Cucumber Accessions
4.4. Genes Potentially Involved in Tolerance to Long-Term Waterlogging in Cucumber
4.5. DEGs Determined in WL-S and WL-T Cucumbers in Recovery
4.6. qRT-PCR Assay Revealed Differences Throughout Waterlogging Stress Duration
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Arduini, I.; Kokubun, M.; Licausi, F. Editorial: Crop Response to Waterlogging. Front. Plant Sci. 2019, 10, 1578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manik, S.M.N.; Pengilley, G.; Dean, G.; Field, B.; Shabala, S.; Zhou, M. Soil and Crop Management Practices to Minimize the Impact of Waterlogging on Crop Productivity. Front. Plant Sci. 2019, 10, 140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, L.V.; Le, T.M.; Ta, P.D.V.; Tran, G.H.; Rumanzi, M.S.; Tang, H.T.; Nguyen, L.V. Variation in root growth responses of sweet potato to hypoxia and waterlogging. Vegetos 2020, 33, 367–375. [Google Scholar] [CrossRef]
- Zou, X.; Jiang, Y.; Liu, L.; Zhang, Z.; Zheng, Y. Identification of transcriptome induced in roots of maize seedlings at the late stage of waterlogging. BMC Plant Biol. 2010, 10, 189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghobadi, M.E.; Ghobadi, M.; Zebarjadi, A. Effect of waterlogging at different growth stages on some morphological traits of wheat varieties. Int. J. Biometeorol. 2017, 61, 635–645. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Li, B.; Lu, X.; Yuan, L.; Yang, Y.; Yuan, Y.; Du, J.; Guo, S. The effect of exogenous calcium on mitochondria, respiratory metabolism enzymes and ion transport in cucumber roots under hypoxia. Sci. Rep. 2015, 5, 11391. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.; Ji, J.; Xu, Q.; Qi, X.; Chen, X. Inheritance and quantitative trail loci mapping of adventitious root numbers in cucumber seedlings under waterlogging conditions. Mol. Genet. Genom. 2017, 292, 353–364. [Google Scholar] [CrossRef]
- Barickman, T.C.; Simpson, C.R.; Sams, C.E. Waterlogging causes early modification in the physiological performance, carotenoids, chlorophylls, proline, and soluble sugars of cucumber plants. Plants 2019, 8, 160. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Sun, J.; Yang, Y.; Guo, S.; Glick, B.R. Identification of hypoxic-responsive proteins in cucumber roots using a proteomic approach. Plant Physiol. Biochem. 2012, 51, 74–80. [Google Scholar] [CrossRef]
- Kołton, A.; Kęska, K.; Czernicka, M. Selection of Tomato and Cucumber Accessions for Waterlogging Sensitivity through Morpho-Physiological Assessment at an Early Vegetative Stage. Agronomy 2020, 10, 1490. [Google Scholar] [CrossRef]
- Van Veen, H.; Vashisht, D.; Akman, M.; Girke, T.; Mustroph, A.; Reinen, E.; Hartman, S.; Kooiker, M.; van Tienderen, P.; Schranz, M.E.; et al. Transcriptomes of eight Arabidopsis thaliana accessions reveal core conserved, genotype-and organ-specific responses to flooding stress. Plant Physiol. 2016, 172, 668–689. [Google Scholar] [CrossRef] [PubMed]
- Garg, R.; Verma, M.; Agrawal, S.; Shankar, R.; Majee, M.; Jain, M. Deep transcriptome sequencing of wild halophyte rice, porteresiacoarctata, provides novel insights into the salinity and submergence tolerance factors. DNA Res. 2013, 21, 69–84. [Google Scholar] [CrossRef] [PubMed]
- Zou, X.-L.; Zeng, L.; Lu, G.-Y.; Cheng, Y.; Xu, J.-S.; Zhang, X.-K. Comparison of transcriptomes undergoing waterlogging at the seedling stage between tolerant and sensitive varieties of Brassica napus L. J. Integr. Agric. 2015, 14, 1723–1734. [Google Scholar] [CrossRef]
- Lin, Y.; Li, W.; Zhang, Y.; Xia, C.; Liu, Y.; Wang, C.; Xu, R.; Zhang, L. Identification of Genes/Proteins Related to Submergence Tolerance by Transcriptome and Proteome Analyses in Soybean. Sci. Rep. 2019, 9, 14688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Zhong, Y.; Bai, D.; Abid, M.; Zhang, Y.; Lin, M.; Qi, X.; Hu, C.; Fang, J. Comparative Transcriptome Analysis of Two Contrasting Kiwifruit (Actinidia) Genotypes under Waterlogging Stress. Preprints 2020, 2020070395. [Google Scholar] [CrossRef]
- Dossa, K.; You, J.; Wang, L.; Zhang, Y.; Li, D.; Zhou, R.; Yu, J.; Wei, X.; Zhu, X.; Jiang, S.; et al. Transcriptomic profiling of sesame during waterlogging and recovery. Sci. Data 2019, 6, 204. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.; Chen, M.; Ji, J.; Xu, Q.; Qi, X.; Chen, X. Comparative RNA-seq based transcriptome profiling of waterlogging response in cucumber hypocotyls reveals novel insights into the de novo adventitious root primordia initiation. BMC Plant Biol. 2017, 17, 129. [Google Scholar] [CrossRef] [Green Version]
- Crisp, P.A.; Ganguly, D.R.; Eichten, S.R.; Borevitz, J.O.; Pogson, B.J. Reconsidering plant memory: Intersections between stress recovery, RNA turnover, and epigenetics. Sci. Adv. 2016, 2, e1501340. [Google Scholar] [CrossRef] [Green Version]
- Yeung, E.; van Veen, H.; Vashisht, D.; Paiva, A.L.S.; Hummel, M.; Rankenberg, T.; Steffens, B.; Steffen-Heins, A.; Sauter, M.; de Vries, M.; et al. A stress recovery signaling network for enhanced flooding tolerance in Arabidopsis thaliana. Proc. Natl. Acad. Sci. USA 2018, 115, E6085–E6094. [Google Scholar] [CrossRef] [Green Version]
- Qi, X.H.; Li, T.; Xu, Q.; Chen, X.H. Modulation of chlorophyll contents and anti-oxidant systems in two cucumber varieties under waterlogging stress and subsequent drainage. J. Hortic. Sci. Biotechnol. 2011, 86, 337–342. [Google Scholar] [CrossRef]
- Lämke, J.; Bäurle, I. Epigenetic and chromatin-based mechanisms in environmental stress adaptation and stress memory in plants. Genome Biol. 2017, 18, 124. [Google Scholar] [CrossRef] [PubMed]
- Alves de Freitas Guedes, F.; Menezes-Silva, P.E.; DaMatta, F.M.; Alves-Ferreira, M. Using transcriptomics to assess plant stress memory. Theor. Exp. Plant Physiol. 2019, 31, 47–58. [Google Scholar] [CrossRef]
- Conrath, U.; Beckers, G.J.; Langenbach, C.J.; Jaskiewicz, M.R. Priming for enhanced defense. Annu. Rev. Phytopathol. 2015, 53, 97–119. [Google Scholar] [CrossRef] [PubMed]
- Trewavas, T. Plant Intelligence: An Overview. BioScience 2016, 66, 542–551. [Google Scholar] [CrossRef] [Green Version]
- Hossain, M.A.; Bhattacharjee, S.; Armin, S.-M.; Qian, P.; Xin, W.; Li, H.-Y.; Burritt, D.J.; Fujita, M.; Tran, L.-S.P. Hydrogen peroxide priming modulates abiotic oxidative stress tolerance: Insights from ROS detoxification and scavenging. Front. Plant Sci. 2015, 6, 420. [Google Scholar] [CrossRef] [Green Version]
- Zhang, N.; Zhao, B.; Zhang, H.J.; Weeda, S.; Yang, C.; Yang, Z.C.; Ren, S.; Guo, Y.D. Melatonin promotes water-stress tolerance, lateral root formation, and seed germination in cucumber (Cucumis sativus L.). J. Pineal Res. 2013, 54, 15–23. [Google Scholar] [CrossRef]
- Zhang, N.; Zhang, H.J.; Zhao, B.; Sun, Q.Q.; Cao, Y.Y.; Li, R.; Wu, X.X.; Weeda, S.; Li, L.; Ren, S.; et al. The RNA-seq approach to discriminate gene expression profiles in response to melatonin on cucumber lateral root formation. J. Pineal Res. 2014, 56, 39–50. [Google Scholar] [CrossRef]
- Wang, C.-J.; Yang, W.; Wang, C.; Gu, C.; Niu, D.-D.; Liu, H.-X.; Wang, Y.-P.; Guo, J.H. Induction of Drought Tolerance in Cucumber Plants by a Consortium of Three Plant Growth-Promoting Rhizobacterium Strains. PLoS ONE 2012, 7, e52565. [Google Scholar] [CrossRef] [Green Version]
- Lal, S.K.; Kumar, S.; Sheri, V.; Mehta, S.; Varakumar, P.; Ram, B.; Borphukan, B.; James, D.; Fartyal, D.; Reddy, M.K. Seed Priming: An Emerging Technology to Impart Abiotic Stress Tolerance in Crop Plants. In Advances in Seed Priming; Rakshit, A., Singh, H., Eds.; Springer: Singapore, 2018; pp. 41–50. [Google Scholar]
- Sanyal, R.P.; Misra, H.S.; Saini, A. Heat-stress priming and alternative splicing-linked memory. J. Exp. Bot. 2018, 69, 2431–2434. [Google Scholar] [CrossRef] [Green Version]
- Turgut-Kara, N.; Arikan, B.; Celik, H. Epigenetic memory and priming in plants. Genetica 2020, 148, 47–54. [Google Scholar] [CrossRef]
- Ashraf, M.A.; Rasheed, R.; Hussain, I.; Iqbal, M.; Riaz, M.; Arif, M.S. Chemical priming for multiple stress tolerance. In Priming and Pretreatment of Seeds and Seedlings; Hasanuzzaman, M., Fotopoulos, V., Eds.; Springer: Singapore, 2019; pp. 385–415. [Google Scholar]
- Tan, X.; Zwiazek, J.J. Stable expression of aquaporins and hypoxia-responsive genes in adventitious roots are linked to maintaining hydraulic conductance in tobacco (Nicotiana tabacum) exposed to root hypoxia. PLoS ONE 2019, 14, e0212059. [Google Scholar] [CrossRef] [PubMed]
- Dobin, A.; Gingeras, T.R. Mapping RNA-seq Reads with STAR. Curr. Protoc. Bioinform. 2015, 51, 11–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, S.; Li, R.; Zhang, Z.; Li, L.; Gu, X.; Fan, W.; Lucas, W.L.; Wang, X.; Xie, B.; Ni, P.; et al. The genome of the cucumber, Cucumis sativus L. Nat. Genet. 2009, 41, 1275–1281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seqreads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haas, B.J.; Papanicolaou, A.; Yassour, M.; Grabherr, M.; Blood, P.D.; Bowden, J.; Couger, M.B.; Eccles, D.; Li, B.; Lieber, M.; et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 2013, 8, 1494–1512. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [Green Version]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. Edger: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Das, S.; Mykles, D.L. A Comparison of Resources for the Annotation of a De Novo Assembled Transcriptome in the Molting Gland (Y-Organ) of the Blackback Land Crab, Gecarcinus lateralis. Integr. Comp. Biol. 2016, 56, 1103–1112. [Google Scholar] [CrossRef]
- Boeckmann, B.; Bairoch, A.; Apweiler, R.; Blatter, M.C.; Estreicher, A.; Gasteiger, E.; Martin, M.J.; Michoud, K.; O’Donovan, C.; Phan, I.; et al. The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Res. 2003, 31, 365–370. [Google Scholar] [CrossRef]
- Finn, R.D.; Clements, J.; Eddy, S.R. HMMER web server: Interactive sequence similarity searching. Nucleic Acids Res. 2011, 39, 29–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finn, R.D.; Bateman, A.; Clements, J.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Heger, A.; Hetherington, K.; Holm, L.; Mistry, J.; et al. Pfam: The protein families database. Nucleic Acids Res. 2014, 42, 222–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petersen, T.; Brunak, S.; von Heijne, G.; Nielsen, H. SignalP 4.0: Discriminating signal peptides from transmembrane regions. Nat. Methods 2011, 8, 785–786. [Google Scholar] [CrossRef] [PubMed]
- Powell, S.; Forslund, K.; Szklarczyk, D.; Trachana, K.; Roth, A.; Huerta-Cepas, J.; Gabaldon, T.; Rattei, T.; Creevey, C.; Kuhn, M.; et al. eggNOG v4.0: Nested orthology inference across 3686 organisms. Nucleic Acids Res. 2014, 42, D231–D239. [Google Scholar] [CrossRef] [PubMed]
- Ashburner, M.; Ball, C.; Blake, J.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanehisa, M.; Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Alexa, A.; Rahnenfuhrer, J. TopGo: Enrichment Analysis for Gene Ontology. R Package Version 2.38.1. 2019. Available online: https://bioconductor.riken.jp/packages/3.3/bioc/html/topGO.html (accessed on 27 November 2020).
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Pradhan, B.; Sarkar, M.; Kundagrami, S. Alcohol Dehydrogenase (ADH) Enzyme is a Potent Biochemical Marker for Submergence Tolerance in Rice (Oryza sativa L.) During Seedling Stage of Growth. Int. J. Curr. Res. 2019, 11, 16–21. [Google Scholar] [CrossRef]
- Qi, X.H.; Xu, X.W.; Lin, X.J.; Zhang, W.J.; Chen, X.H. Identification of differentially expressed genes in cucumber (Cucumis sativus L.) root under waterlogging stress by digital gene expression profile. Genomics 2012, 99, 160–168. [Google Scholar] [CrossRef] [Green Version]
- Xie, L.; Tan, W.; Yang, Y.; Tan, Y.; Zhou, Y.; Zhou, D.; Xiao, S.; Chen, Q. Long-chain acyl-CoA synthetase LACS2 contributes to submergence tolerance by modulating cuticle permeability in Arabidopsis. Plants 2020, 9, 262. [Google Scholar] [CrossRef] [Green Version]
- Bustin, S.A.; Benes, V.; Garson, J.A.; Hellemans, J.; Huggett, J.; Kubista, M.; Mueller, R.; Nolan, T.; Pfaffl, M.W.; Shipley, G.L.; et al. The MIQE Guidelines: Minimum Information for Publication of Quantitative Real-Time PCR Experiments. Clin. Chem. 2009, 55, 611–622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hellemans, J.; Mortier, G.; De Paepe, A.; Speleman, F.; Vandesompele, J. qBase relative quantification framework and software for management and automated analysis of real-time quantitative PCR data. Genome Biol. 2007, 8, R19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luan, H.; Guo, B.; Pan, Y.; Lv, C.; Shen, H.; Xu, R. Morpho-anatomical and physiological responses to waterlogging stress in different barley (Hordeum vulgare L.) genotypes. Plant Growth Regul. 2018, 85, 399–409. [Google Scholar] [CrossRef]
- Pan, R.; He, D.; Xu, L.; Zhou, M.; Li, C.; Wu, C.; Xu, Y.; Zhang, W. Proteomic analysis reveals response of differential wheat (Triticum aestivum L.) genotypes to oxygen deficiency stress. BMC Genom. 2019, 20, 60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sauter, M.; Steffens, B. Root responses to flooding. Curr. Opin. Plant Biol. 2013, 16, 282–286. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Ji, J.; Ma, X.; Xu, Q.; Qi, X.; Chen, X. Comparative Proteomic Analysis Provides Insight into the Key Proteins Involved in Cucumber (Cucumis sativus L.) Adventitious Root Emergence under Waterlogging Stress. Front. Plant Sci. 2016, 7, 1515. [Google Scholar] [CrossRef] [Green Version]
- Qi, X.H.; Chen, R.F.; Xu, Q.; Chen, X.H. Preliminary analysis of submergence tolerance of cucumber at seedling stage. China Veg. 2011, 4, 23–28. [Google Scholar]
- Vidoz, M.L.; Loreti, E.; Mensuali, A.; Alpi, A.; Perata, P. Hormonal interplay during adventitious root formation in flooded tomato plants. Plant J. 2010, 63, 551–562. [Google Scholar] [CrossRef]
- Butsayawarapat, P.; Juntawong, P.; Khamsuk, O.; Somta, P. Comparative transcriptome analysis of waterlogging-sensitive and tolerant zombi pea (Vigna vexillata) reveals energy conservation and root plasticity controlling waterlogging tolerance. Plants 2019, 8, 264. [Google Scholar] [CrossRef] [Green Version]
- Kreuzwieser, J.; Hauberg, J.; Howell, K.A.; Carroll, A.; Rennenberg, H.; Millar, A.H.; Whelan, J. Differential response of gray poplar leaves and roots underpins stress adaptation during hypoxia. Plant Physiol. 2009, 149, 461–473. [Google Scholar] [CrossRef] [Green Version]
- Do Amaral, M.N.; Arge, L.W.P.; Auler, P.A.; Rosatto, T.; Milech, C.; Magalhães, A.M., Jr.; Braga, E.J.B. Long-term transcriptional memory in rice plants submitted to salt shock. Planta 2020, 251, 111. [Google Scholar] [CrossRef] [PubMed]
- Bittner, A.; van Buer, J.; Baier, M. Cold priming uncouples light- and cold-regulation of gene expression in Arabidopsis thaliana. BMC Plant Biol. 2020, 20, 281. [Google Scholar] [CrossRef] [PubMed]
- Borrego-Benjumea, A.; Carter, A.; Tucker, J.R.; Yao, Z.; Xu, W.; Badea, A. Genome-wide analysis of gene expression provides new insights into waterlogging responses in barley (Hordeum vulgare L.). Plants 2020, 9, 240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phukan, U.J.; Jeena, G.S.; Tripathi, V.; Shukla, R.K. MaRAP 2-4, a waterlogging-responsive ERF from Mentha, regulates bidirectional sugar transporter AtSWEET 10 to modulate stress response in Arabidopsis. Plant Biotechnol. J. 2018, 16, 221–233. [Google Scholar] [CrossRef] [Green Version]
- Du, X.M.; Ni, X.L.; Ren, X.L.; Xin, G.L.; Jia, G.L.; Liu, H.D.; Liu, W.-Z. De novo transcriptomic analysis to identify differentially expressed genes during the process of aerenchyma formation in Typha angustifolia leaves. Gene 2018, 662, 66–75. [Google Scholar] [CrossRef]
- Nishiuchi, S.; Yamauchi, T.; Takahashi, H.; Kotula, L.; Nakazano, M. Mechanisms for coping with submergence and waterlogging in rice. Rice 2012, 5, 2. [Google Scholar] [CrossRef] [Green Version]
- Krishnan, H.B.; Oehrle, N.W.; Alaswad, A.A.; Stevens, W.G.; Maria John, K.M.; Luthria, D.L.; Natarajan, S.S. Biochemical and anatomical investigation of Sesbania herbacea (Mill.) McVaugh nodules grown under flooded and non-flooded conditions. Int. J. Mol. Sci. 2019, 20, 1824. [Google Scholar] [CrossRef] [Green Version]
- Loreti, E.; Perata, P. The Many Facets of Hypoxia in Plants. Plants 2020, 9, 745. [Google Scholar] [CrossRef]
- Nanjo, Y.; Maruyama, K.; Yasue, H.; Yamaguchi-Shinozaki, K.; Shinozaki, K.; Komatsu, S. Transcriptional responses to flooding stress in roots including hypocotyl of soybean seedlings. Plant Mol. Biol. 2011, 77, 129–144. [Google Scholar] [CrossRef]
- Souza, S.C.; Mazzafera, P.; Sodek, L. Flooding of the root system in soybean: Biochemical and molecular aspects of N metabolism in the nodule during stress and recovery. Amino Acids 2016, 48, 1285–1295. [Google Scholar] [CrossRef]
- Zhang, X.; Lin, H.; Ming, H.; Hu, X.; Hu, L. γ-aminobutyric acid mediates nicotine biosynthesis in tobacco under flooding stress. Plant Divers. 2016, 38, 53–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yiu, J.; Tseng, M.; Liu, C. Exogenous catechin increases antioxidant enzyme activity and promotes flooding tolerance in tomato (Solanum lycopersicum L.). Plant Soil 2011, 344, 213–225. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Sui, X.; Guo, J.; Wang, Z.; Cheng, J.; Ma, S.; Li, X.; Zhang, Z. Antisense suppression of cucumber (Cucumis sativus L.) sucrose synthase 3 (CsSUS3) reduces hypoxic stress tolerance. Plant Cell Environ. 2014, 37, 795–810. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Sakata, K.; Komatsu, S. An Integrated Approach of Proteomics and Computational Genetic Modification Effectiveness Analysis to Uncover the Mechanisms of Flood Tolerance in Soybeans. Int. J. Mol. Sci. 2018, 19, 1301. [Google Scholar] [CrossRef] [Green Version]
- Arora, K.; Panda, K.; Mittal, S.; Mallikarjuna, M.G.; Rao, A.R.; Dash, P.K.; Thirunavukkarasu, N. RNAseq revealed the important gene pathways controlling adaptive mechanisms under waterlogged stress in maize. Sci. Rep. 2017, 7, 10950. [Google Scholar] [CrossRef] [Green Version]
- Khatoon, A.; Rehman, S.; Salavati, A.; Komatsu, S. A comparative proteomics analysis in roots of soybean to compatible symbiotic bacteria under flooding stress. Amino Acids 2012, 43, 2513–2525. [Google Scholar] [CrossRef]
- Marowa, P.; Ding, A.; Kong, Y. Expansins: Roles in plant growth and potential applications in crop improvement. Plant Cell Rep. 2016, 35, 949–965. [Google Scholar] [CrossRef] [Green Version]
- Guo, W.; Zhao, J.; Li, X.; Qin, L.; Yan, X.; Liao, H. A soybean β-expansin gene GmEXPB2 intrinsically involved in root system architecture responses to abiotic stresses. Plant J. 2011, 66, 541–552. [Google Scholar] [CrossRef]
- Chen, Y.; Han, Y.; Kong, X.; Kang, H.; Ren, Y.; Wang, W. Ectopic expression of wheat expansin gene TaEXPA2 improved the salt tolerance of transgenic tobacco by regulating Na+/K+ and antioxidant competence. Physiol. Plant. 2017, 159, 161–177. [Google Scholar] [CrossRef]
- Lee, Y.; Kende, H. Expression of α-expansin and expansin-like genes in deepwater rice. Plant Physiol. 2002, 130, 1396–1405. [Google Scholar] [CrossRef] [Green Version]
- Arora, K.; Panda, K.K.; Mittal, S.; Mallikarjuna, M.G.; Thirunavukkarasu, N. In silico characterization and functional validation of cell wall modification genes imparting waterlogging tolerance in maize. Bioinform. Biol. Insights 2017, 11, 1177932217747277. [Google Scholar] [CrossRef] [PubMed]
- Lü, P.; Kang, M.; Jiang, X.; Dai, F.; Gao, J.; Zhang, C. RhEXPA4, a rose expansin gene, modulates leaf growth and confers drought and salt tolerance to Arabidopsis. Planta 2013, 237, 1547–1559. [Google Scholar] [CrossRef] [PubMed]
- Sasidharan, R.; Bailey-Serres, J.; Ashikari, M.; Atwell, B.J.; Colmer, T.D.; Fagerstedt, K.; Fukao, T.; Geigenberger, P.; Hebelstrup, K.H.; Hill, R.D.; et al. Community recommendations on terminology and procedures used in flooding and low oxygen stress research. New Phytol. 2017, 214, 1403–1407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pang, J.; Zhou, M.; Mendham, N.; Shabala, S. comparison of growth and physiological responses to waterlogging and subsequent recovery in six barley genotypes. Aust. J. Agric. Res. 2004, 55, 895–906. [Google Scholar] [CrossRef]
- Blokhina, O.B.; Virolainen, E.; Fagerstedt, K.V. Antioxidants, oxidative damage and oxygen deprivation stress: A review. Ann. Bot. 2003, 91, 179–194. [Google Scholar] [CrossRef] [Green Version]
- Le Gall, H.; Philippe, F.; Domon, J.M.; Gillet, F.; Pelloux, J.; Rayon, C. Cell Wall Metabolism in Response to Abiotic Stress. Plants 2015, 4, 112–166. [Google Scholar] [CrossRef]
- Garcia, M.J.; Garcia-Mateo, M.J.; Lucena, C.; Romera, F.J.; Rojas, C.L.; Alcantara, E.; Perez-Vicente, R. Hypoxia and bicarbonate could limit the expression of iron acquisition genes in Strategy I plants by affecting ethylene synthesis and signaling in different ways. Physiol. Plant 2013, 150, 95–106. [Google Scholar] [CrossRef]
- Tsai, K.J.; Chou, S.J.; Shih, M.C. Ethylene plays an essential role in the recovery of Arabidopsis during post-anaerobiosis reoxygenation. Plant Cell Environ. 2014, 37, 2391–2405. [Google Scholar] [CrossRef]
- Winkel-Shirley, B. Biosynthesis of flavonoids and effects of stress. Curr. Opin. Plant Biol. 2002, 5, 218–223. [Google Scholar] [CrossRef]
- André, C.M.; Schafleitner, R.; Legay, S.; Lefèvre, I.; Aliaga, C.A.A.; Nomberto, G.; Hoffmann, L.; Hausman, J.F.; Larondelle, Y.; Evers, D. Gene expression changes related to the production of phenolic compounds in potato tubers grown under drought stress. Phytochemistry 2009, 70, 1107–1116. [Google Scholar] [CrossRef]
- Lenka, S.K.; Katiyar, A.; Chinnusamy, V.; Bansal, K.C. Comparative analysis of drought-responsive transcriptome in Indica rice genotypes with contrasting drought tolerance. Plant Biotechnol. J. 2011, 9, 315–327. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Li, X.; Liu, Y.; Cao, B. Regulation of flavanone 3-hydroxylase gene involved in the flavonoid biosynthesis pathway in response to UV-B radiation and drought stress in the desert plant, Reaumuria soongorica. Plant Physiol. Biochem. 2013, 73, 161–167. [Google Scholar] [CrossRef] [PubMed]
- Miro, B.; Ismail, A.M. Tolerance of anaerobic conditions caused by flooding during germination and early growth in rice (Oryza sativa L.). Front. Plant Sci. 2013, 4, 269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arismendi, M.J.; Almada, R.; Pimentel, P.; Bastias, A.; Salvatierra, A.; Rojas, P.; Hinrichsen, P.; Pinto, M.; Di Genova, A.; Travisany, D.; et al. Transcriptome sequencing of Prunus sp. rootstocks roots to identify candidate genes involved in the response to root hypoxia. Tree Genet. Genomes 2015, 11, 11. [Google Scholar] [CrossRef]
- Ismond, K.P.; Dolferus, R.; de Pauw, M.; Dennis, E.S.; Good, A.G. Enhanced low oxygen survival in Arabidopsis through increased metabolic flux in the fermentative pathway. Plant Physiol. 2003, 132, 1292–1303. [Google Scholar] [CrossRef] [Green Version]
- Hwang, J.H.; Lee, M.O.; Choy, Y.H.; Ha-Lee, Y.M.; Hong, C.B.; Dong-Hee, L. Expression profile analysis of hypoxia responses in Arabidopsis roots and shoots. J. Plant Biol. 2011, 54, 373–383. [Google Scholar] [CrossRef]
- Dat, J.F.; Capelli, N.; Folzer, H.; Bourgeade, P.; Badot, P.M. Sensing and signalling during plant flooding. Plant Physiol. Biochem. 2004, 42, 273–282. [Google Scholar] [CrossRef]
- Yang, S.F.; Hoffman, N.E. Ethylene biosynthesis and its regulation in higher plants. Annu. Rev. Plant Physiol. 1984, 35, 155–189. [Google Scholar] [CrossRef]
- Shockey, J.M.; Fulda, M.S.; Browse, J.A. Arabidopsis Contains Nine Long-Chain Acyl-Coenzyme A Synthetase Gene That Participate in Fatty Acid and Glycerolipid Metabolism. Plant Physiol. 2002, 129, 1710–1722. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, N.; Koussevitzky, S.; Mittler, R.; Miller, G. ROS and redox signalling in the response of plants to abiotic stress. Plant Cell Environ. 2011, 35, 259–270. [Google Scholar] [CrossRef]
- Xie, L.J.; Yu, L.J.; Chen, Q.F.; Wang, F.Z.; Huang, L.; Xia, F.N.; Zhu, T.R.; Wu, J.X.; Yin, J.; Liao, B.; et al. Arabidopsis acyl-CoA-binding protein ACBP3 participates in plant response to hypoxia by modulating very-long-chain fatty acid metabolism. Plant J. 2015, 81, 53–67. [Google Scholar] [CrossRef] [PubMed]
- Schnurr, J.; Shockey, J.; Browse, J. The Acyl-CoA Synthetase Encoded by LACS2 Is Essential for Normal Cuticle Development in Arabidopsis. Plant Cell 2004, 16, 629–642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewandowska, M.; Keyl, A.; Feussner, I. Wax biosynthesis upon danger: Its regulation upon abiotic and biotic stress. New Phytol. 2020, 227, 698–713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hilker, M.; Schmülling, T. Stress priming, memory, and signalling in plants. Plant Cell Environ. 2019, 42, 753–761. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Trascripts | |
|---|---|
| Total numer (bp) | 35,712 |
| Total length (bp) | 79,877,390 |
| N50 length (bp) | 2711 |
| Minimum length (bp) | 162 |
| Maximum length (bp) | 20,773 |
| Average length (bp) | 2236 |
| Regulation | WL-T DH2 | WL-S DH4 | ||||||
|---|---|---|---|---|---|---|---|---|
| 1xH vs. Ctrl | Rec vs. Ctrl | 2xH vs. Ctrl | 1xH vs. 2xH | 1xH vs. Ctrl | Rec vs. Ctrl | 2xH vs. Ctrl | 1xH vs. 2xH | |
 | 2584 (493) * | 355 (66) | 2310 (474) | 78 (13) | 4211 (948) | 892 (164) | 5453 (1120) | 1649 (355) |
 | 3373 (560) | 299 (51) | 2697 (473) | 348 (71) | 4716 (809) | 985 (249) | 6166 (1112) | 1196 (238) |
| Total | 5957 (1053) | 654 (117) | 5007 (947) | 426 (84) | 8927 (1757) | 1,877 (413) | 11,619 (2232) | 2845 (593) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kęska, K.; Szcześniak, M.W.; Makałowska, I.; Czernicka, M. Long-Term Waterlogging as Factor Contributing to Hypoxia Stress Tolerance Enhancement in Cucumber: Comparative Transcriptome Analysis of Waterlogging Sensitive and Tolerant Accessions. Genes 2021, 12, 189. https://doi.org/10.3390/genes12020189
Kęska K, Szcześniak MW, Makałowska I, Czernicka M. Long-Term Waterlogging as Factor Contributing to Hypoxia Stress Tolerance Enhancement in Cucumber: Comparative Transcriptome Analysis of Waterlogging Sensitive and Tolerant Accessions. Genes. 2021; 12(2):189. https://doi.org/10.3390/genes12020189
Chicago/Turabian StyleKęska, Kinga, Michał Wojciech Szcześniak, Izabela Makałowska, and Małgorzata Czernicka. 2021. "Long-Term Waterlogging as Factor Contributing to Hypoxia Stress Tolerance Enhancement in Cucumber: Comparative Transcriptome Analysis of Waterlogging Sensitive and Tolerant Accessions" Genes 12, no. 2: 189. https://doi.org/10.3390/genes12020189