
InpactorDB: A Classified Lineage-Level Plant LTR Retrotransposon Reference Library for Free-Alignment Methods Based on Machine Learning

 ,
,  ,
,  ,
,  and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Databases and LTR-RT Classification Processes
2.2. Statistical Analysis
2.3. Post-Processing and Generalization Tests through Deep Neural Networks
3. Results
3.1. Analysis of Significant Differences
3.2. Post-Processing and Classification Using Deep Neural Networks
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Li, S.-F.; Su, T.; Cheng, G.-Q.; Wang, B.-X.; Li, X.; Deng, C.-L.; Gao, W.-J. Chromosome Evolution in Connection with Repetitive Sequences and Epigenetics in Plants. Genes 2017, 8, 290. [Google Scholar] [CrossRef] [PubMed]
- Keidar, D.; Doron, C.; Kashkush, K. Genome-wide analysis of a recently active retrotransposon, Au SINE, in wheat: Content, distribution within subgenomes and chromosomes, and gene associations. Plant Cell Rep. 2018, 37, 193–208. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.-S. The genomes and transposable elements in plants: Are they friends or foes? Genes Genom. 2017, 39, 359–370. [Google Scholar] [CrossRef]
- De Castro Nunes, R.; Orozco-Arias, S.; Crouzillat, D.; Mueller, L.A.; Strickler, S.R.; Descombes, P.; Fournier, C.; Moine, D.; de Kochko, A.; Yuyama, P.M.; et al. Structure and Distribution of Centromeric Retrotransposons at Diploid and Allotetraploid Coffea Centromeric and Pericentromeric Regions. Front. Plant Sci. 2018, 9. [Google Scholar] [CrossRef]
- Orozco-Arias, S.; Isaza, G.; Guyot, R. Retrotransposons in Plant Genomes: Structure, Identification, and Classification through Bioinformatics and Machine Learning. Int. J. Mol. Sci. 2019, 20, 3837. [Google Scholar] [CrossRef]
- Todorovska, E. Retrotransposons and their role in plant—Genome evolution. Biotechnol. Biotechnol. Equip. 2014, 21, 294–305. [Google Scholar] [CrossRef]
- Wessler, S.R.; Bureau, T.E.; White, S.E. LTR-retrotransposons and MITEs: Important players in the evolution of plant genomes. Curr. Opin. Genet. Dev. 1995, 5, 814–821. [Google Scholar] [CrossRef]
- Casacuberta, J.M.; Santiago, N. Plant LTR-retrotransposons and MITEs: Control of transposition and impact on the evolution of plant genes and genomes. Gene 2003, 311, 1–11. [Google Scholar] [CrossRef]
- Galindo-González, L.; Mhiri, C.; Deyholos, M.K.; Grandbastien, M.-A. LTR-retrotransposons in plants: Engines of evolution. Gene 2017, 626, 14–25. [Google Scholar] [CrossRef]
- Fan, F.; Wen, X.; Ding, G.; Cui, B. Isolation, identification, and characterization of genomic LTR retrotransposon sequences from masson pine (Pinus massoniana). Tree Genet. Genomes 2013, 9, 1237–1246. [Google Scholar] [CrossRef]
- Schulman, A.H. Hitching a Ride: Nonautonomous Retrotransposons and Parasitism as a Lifestyle. In Plant Transposable Elements; Grandbastien, M.-A., Casacuberta, J.M., Eds.; Springer: Berlin/Heidelberg, Germany, 2012; pp. 71–88. [Google Scholar]
- Alzohairy, A.M.; Sabir, J.S.M.; Gyulai, G.; Younis, R.A.A.; Jansen, R.K.; Bahieldin, A. Environmental stress activation of plant long-terminal repeat retrotransposons. Funct. Plant Biol. 2014, 41, 557–567. [Google Scholar] [CrossRef]
- Serrato-Capuchina, A.; Matute, D.R. The role of transposable elements in speciation. Genes 2018, 9, 254. [Google Scholar] [CrossRef] [PubMed]
- Kidwell, M.G.; Kidwell, J.F.; Sved, J.A. Hybrid dysgenesis in Drosophila melanogaster: A syndrome of aberrant traits including mutation, sterility and male recombination. Genetics 1977, 86, 813–833. [Google Scholar]
- Zhang, Q.-J.; Gao, L.-Z. Rapid and Recent Evolution of LTR Retrotransposons Drives Rice Genome Evolution During the Speciation of AA- Genome Oryza Species. G3 Genes Genomes Genet. 2017, 7, 1875–1885. [Google Scholar] [CrossRef] [PubMed]
- Wicker, T.; Sabot, F.; Hua-Van, A.; Bennetzen, J.L.; Capy, P.; Chalhoub, B.; Flavell, A.; Leroy, P.; Morgante, M.; Panaud, O.; et al. A unified classification system for eukaryotic transposable elements. Nat. Rev. Genet. 2007, 8, 973–982. [Google Scholar] [CrossRef] [PubMed]
- Chaparro, C.; Gayraud, T.; de Souza, R.F.; Domingues, D.S.; Akaffou, S.; Laforga Vanzela, A.L.; de Kochko, A.; Rigoreau, M.; Crouzillat, D.; Hamon, S.; et al. Terminal-repeat retrotransposons with GAG domain in plant genomes: A new testimony on the complex world of transposable elements. Genome Biol. Evol. 2015, 7, 493–504. [Google Scholar] [CrossRef]
- Orozco-Arias, S.; Isaza, G.; Guyot, R.; Tabares-Soto, R. A systematic review of the application of machine learning in the detection and classi fi cation of transposable elements. PeerJ 2019, 7, 18311. [Google Scholar] [CrossRef]
- Grandbastien, M.-A.A. LTR retrotransposons, handy hitchhikers of plant regulation and stress response. Biochim. Biophys. Acta Gene Regul. Mech. 2015, 1849, 403–416. [Google Scholar] [CrossRef]
- Gao, D.; Jimenez-Lopez, J.C.; Iwata, A.; Gill, N.; Jackson, S.A. Functional and structural divergence of an unusual LTR retrotransposon family in plants. PLoS ONE 2012, 7, e48595. [Google Scholar] [CrossRef]
- Rahman, A.Y.A.; Usharraj, A.O.; Misra, B.B.; Thottathil, G.P.; Jayasekaran, K.; Feng, Y.; Hou, S.; Ong, S.Y.; Ng, F.L.; Lee, L.S.; et al. Draft genome sequence of the rubber tree Hevea brasiliensis. BMC Genom. 2013, 14, 75. [Google Scholar] [CrossRef]
- Kumar, A.; Bennetzen, J.L. Plant retrotransposons. Annu. Rev. Genet. 1999, 33, 479–532. [Google Scholar] [CrossRef] [PubMed]
- Servant, G.; Deininger, P.L. Insertion of retrotransposons at chromosome ends: Adaptive response to chromosome maintenance. Front. Genet. 2016, 6, 358. [Google Scholar] [CrossRef] [PubMed]
- Gao, D.; Chen, J.; Chen, M.; Meyers, B.C.; Jackson, S. A highly conserved, small LTR retrotransposon that preferentially targets genes in grass genomes. PLoS ONE 2012, 7, e32010. [Google Scholar] [CrossRef] [PubMed]
- Orozco-Arias, S.; Tabares-Soto, R.; Ceballos, D.; Guyot, R. Parallel Programming in Biological Sciences, Taking Advantage of Supercomputing in Genomics. In Advances in Computing; Solano, A., Ordoñez, H., Eds.; Springer: Zurich, Switzerland, 2017; Volume 735, pp. 627–643. [Google Scholar]
- Arkhipova, I.R. Using bioinformatic and phylogenetic approaches to classify transposable elements and understand their complex evolutionary histories. Mob. DNA 2017, 8, 19. [Google Scholar] [CrossRef]
- Neumann, P.; Novák, P.; Hoštáková, N.; MacAs, J. Systematic survey of plant LTR-retrotransposons elucidates phylogenetic relationships of their polyprotein domains and provides a reference for element classification. Mob. DNA 2019, 10, 1–17. [Google Scholar] [CrossRef]
- Llorens, C.; Muñoz-Pomer, A.; Bernad, L.; Botella, H.; Moya, A. Network dynamics of eukaryotic LTR retroelements beyond phylogenetic trees. Biol. Direct 2009, 4, 41. [Google Scholar] [CrossRef]
- Llorens, C.; Futami, R.; Covelli, L.; Domínguez-Escribá, L.; Viu, J.M.; Tamarit, D.; Aguilar-Rodríguez, J.; Vicente-Ripolles, M.; Fuster, G.; Bernet, G.P.; et al. The Gypsy Database (GyDB) of mobile genetic elements: Release 2.0. Nucleic Acids Res. 2011, 39, D70–D74. [Google Scholar] [CrossRef]
- Palazzo, A.; Lorusso, P.; Miskey, C.; Walisko, O.; Gerbino, A.; Marobbio, C.M.T.; Ivics, Z.; Marsano, R.M. Transcriptionally promiscuous “blurry” promoters in Tc1/mariner transposons allow transcription in distantly related genomes. Mob. DNA 2019, 10, 13. [Google Scholar] [CrossRef]
- Smit, A.F.A.; Hubley, R.; Green, P. RepeatMasker. 1996. Available online: http://www.repeatmasker.org/ (accessed on 25 January 2021).
- Piegu, B.; Guyot, R.; Picault, N.; Roulin, A.; Sanyal, A.; Kim, H.; Collura, K.; Brar, D.S.; Jackson, S.; Wing, R.A.; et al. Doubling genome size without polyploidization: Dynamics of retrotransposition-driven genomic expansions in Oryza australiensis, a wild relative of rice. Genome Res. 2006, 21, 1262–1269. [Google Scholar] [CrossRef]
- Ammiraju, J.S.S.; Zuccolo, A.; Yu, Y.; Song, X.; Piegu, B.; Chevalier, F.; Walling, J.G.; Ma, J.; Talag, J.; Brar, D.S.; et al. Evolutionary dynamics of an ancient retrotransposon family provides insights into evolution of genome size in the genus Oryza. Plant J. 2007, 52, 342–351. [Google Scholar] [CrossRef]
- Ming, R.; VanBuren, R.; Wai, C.M.; Tang, H.; Schatz, M.C.; Bowers, J.E.; Lyons, E.; Wang, M.-L.; Chen, J.; Biggers, E.; et al. The pineapple genome and the evolution of CAM photosynthesis. Nat. Genet. 2015, 47, 1435–1442. [Google Scholar] [CrossRef] [PubMed]
- Stritt, C.; Wyler, M.; Gimmi, E.L.; Pippel, M.; Roulin, A.C. Diversity, dynamics and effects of long terminal repeat retrotransposons in the model grass Brachypodium distachyon. New Phytol. 2020, 227, 1736–1748. [Google Scholar] [CrossRef] [PubMed]
- Ma, B.; Kuang, L.; Xin, Y.; He, N. New Insights into Long Terminal Repeat Retrotransposons in Mulberry Species. Genes 2019, 10, 285. [Google Scholar] [CrossRef] [PubMed]
- Domingues, D.S.; Cruz, G.M.Q.; Metcalfe, C.J.; Nogueira, F.T.S.; Vicentini, R.; Alves, C.; Van Sluys, M.-A. Analysis of plant LTR-retrotransposons at the fine-scale family level reveals individual molecular patterns. BMC Genom. 2012, 13, 137. [Google Scholar] [CrossRef] [PubMed]
- Ou, S.; Chen, J.; Jiang, N. Assessing genome assembly quality using the LTR Assembly Index (LAI). Nucleic Acids Res. 2018, 46, 1–11. [Google Scholar] [CrossRef]
- Orozco-Arias, S.; Piña, J.S.; Tabares-Soto, R.; Castillo-Ossa, L.F. Measuring performance metrics of machine learning algorithms for detecting and classifying transposable elements. Processes 2020, 8, 638. [Google Scholar] [CrossRef]
- Mustafin, R.N.; Khusnutdinova, E.K. The Role of Transposons in Epigenetic Regulation of Ontogenesis. Russ. J. Dev. Biol. 2018, 49, 61–78. [Google Scholar] [CrossRef]
- Loureiro, T.; Camacho, R.; Vieira, J.; Fonseca, N.A. Boosting the Detection of Transposable Elements Using Machine Learning. In 7th International Conference on Practical Applications of Computational Biology & Bioinformatics; Springer: Berlin/Heidelberg, Germany, 2013; pp. 85–91. [Google Scholar]
- Loureiro, T.; Camacho, R.; Vieira, J.; Fonseca, N.A. Improving the performance of Transposable Elements detection tools. J. Integr. Bioinform. 2013, 10, 231. [Google Scholar] [CrossRef][Green Version]
- Santos, B.Z.; Cerri, R.; Lu, R.W. A New Machine Learning Dataset for Hierarchical Classification of Transposable Elements. In Proceedings of the XIII Encontro Nacional de Inteligência Artificial, Recife, Brazil, 9–12 October 2016; pp. 9–12. [Google Scholar]
- Cornut, G.; Choisne, N.; Alaux, M.; Alfama-Depauw, F.; Jamilloux, V.; Maumus, F.; Letellier, T.; Luyten, I.; Pommier, C.; Adam-Blondon, A.-F.; et al. RepetDB: A unified resource for transposable element references. Mob. DNA 2019, 10, 6. [Google Scholar]
- Schietgat, L.; Vens, C.; Cerri, R.; Fischer, C.N.; Costa, E.; Ramon, J.; Carareto, C.M.A.; Blockeel, H. A machine learning based framework to identify and classify long terminal repeat retrotransposons. PLoS Comput. Biol. 2018, 14, e1006097. [Google Scholar] [CrossRef]
- Nakano, F.K.; Mastelini, S.M.; Barbon, S.; Cerri, R. Improving Hierarchical Classification of Transposable Elements using Deep Neural Networks. In Proceedings of the International Joint Conference on Neural Networks, Rio de Janeiro, Brazil, 8–13 July 2018. [Google Scholar]
- Da Cruz, M.H.P.; Domingues, D.S.; Saito, P.T.M.; Paschoal, A.R.; Bugatti, P.H. TERL: Classification of Transposable Elements by Convolutional Neural Networks. bioRxiv 2020. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Bombarely, A.; Li, S. DeepTE: A computational method for de novo classification of transposons with convolutional neural network. Bioinformatics 2020. [Google Scholar] [CrossRef] [PubMed]
- Jurka, J.; Kapitonov, V.V.; Pavlicek, A.; Klonowski, P.; Kohany, O.; Walichiewicz, J. Repbase Update, a database of eukaryotic repetitive elements. Cytogenet. Genome Res. 2005, 110, 462–467. [Google Scholar] [CrossRef]
- Spannagl, M.; Bader, K.; Pfeifer, M.; Nussbaumer, T.; Mayer, K.F.X. PGSB/MIPS Plant Genome Information Resources and Concepts for the Analysis of Complex Grass Genomes. In Plant Bioinformatics; Springer: Berlin/Heidelberg, Germany, 2016; pp. 165–186. [Google Scholar]
- Spannagl, M.; Nussbaumer, T.; Bader, K.C.; Martis, M.M.; Seidel, M.; Kugler, K.G.; Gundlach, H.; Mayer, K.F.X. PGSB PlantsDB: Updates to the database framework for comparative plant genome research. Nucleic Acids Res. 2015, 44, D1141–D1147. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, E.M.; McDonald, J.F. LTR STRUC: A novel search and identification program for LTR retrotransposons. Bioinformatics 2003, 19, 362–367. [Google Scholar] [CrossRef]
- Ou, S.; Su, W.; Liao, Y.; Chougule, K.; Agda, J.R.A.; Hellinga, A.J.; Lugo, C.S.B.; Elliott, T.A.; Ware, D.; Peterson, T.; et al. Benchmarking transposable element annotation methods for creation of a streamlined, comprehensive pipeline. Genome Biol. 2019, 20, 275. [Google Scholar] [CrossRef]
- Xu, Z.; Wang, H. LTR-FINDER: An efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res. 2007, 35, 265–268. [Google Scholar] [CrossRef]
- Ellinghaus, D.; Kurtz, S.; Willhoeft, U. LTRharvest, an efficient and flexible software for de novo detection of LTR retrotransposons. BMC Bioinform. 2008, 14. [Google Scholar] [CrossRef]
- Ou, S.; Jiang, N. LTR_retriever: A highly accurate and sensitive program for identification of long terminal-repeat retrotransposons. Plant Physiol. 2017, 176. [Google Scholar] [CrossRef]
- Orozco-Arias, S.; Liu, J.; Id, R.T.; Ceballos, D.; Silva, D.; Id, D.; Ming, R.; Guyot, R. Inpactor, Integrated and Parallel Analyzer and Classifier of LTR Retrotransposons and Its Application for Pineapple LTR Retrotransposons Diversity and Dynamics. Biology 2018, 7, 32. [Google Scholar] [CrossRef]
- Arango-López, J.; Orozco-Arias, S.; Salazar, J.A.; Guyot, R. Application of Data Mining Algorithms to Classify Biological Data: The Coffea canephora Genome Case. In Advances in Computing; Springer: Berlin/Heidelberg, Germany, 2017; Volume 735, pp. 156–170. [Google Scholar]
- Altschup, S.F.; Gish, W.; Pennsylvania, T.; Park, U. Basic Local Alignment Search Tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Miele, V.; Penel, S.; Duret, L. Ultra-fast sequence clustering from similarity networks with SiLiX. BMC Bioinform. 2011, 12, 116. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Capella-Gutiérrez, S.; Silla-Martínez, J.M.; Gabaldón, T. trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef] [PubMed]
- Rice, P.; Longden, I.; Bleasby, A. EMBOSS: The European molecular biology open software suite. TIG 2000, 16, 276–277. [Google Scholar] [CrossRef]
- Xu, Z.; Pu, X.; Gao, R.; Demurtas, O.C.; Fleck, S.J.; Richter, M.; He, C.; Ji, A.; Sun, W.; Kong, J.; et al. Tandem gene duplications drive divergent evolution of caffeine and crocin biosynthetic pathways in plants. BMC Biol. 2020, 18, 1–14. [Google Scholar] [CrossRef]
- Iorizzo, M.; Ellison, S.; Senalik, D.; Zeng, P.; Satapoomin, P.; Huang, J.; Bowman, M.; Iovene, M.; Sanseverino, W.; Cavagnaro, P.; et al. A high-quality carrot genome assembly provides new insights into carotenoid accumulation and asterid genome evolution. Nat. Genet. 2016, 48, 657–666. [Google Scholar] [CrossRef]
- Zhang, R.; Wang, Y.-H.; Jin, J.-J.; Stull, G.W.; Bruneau, A.; Cardoso, D.; De Queiroz, L.P.; Moore, M.J.; Zhang, S.-D.; Chen, S.-Y.; et al. Exploration of plastid phylogenomic conflict yields new insights into the deep relationships of Leguminosae. Syst. Biol. 2020, 69, 613–622. [Google Scholar] [CrossRef]
- Li, Q.; Zhang, N.; Zhang, L.; Ma, H. Differential evolution of members of the rhomboid gene family with conservative and divergent patterns. New Phytol. 2015, 206, 368–380. [Google Scholar] [CrossRef]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-learn: Machine Learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Abadi, M.; Barham, P.; Chen, J.; Chen, Z.; Davis, A.; Dean, J.; Devin, M.; Ghemawat, S.; Irving, G.; Isard, M.; et al. Tensorflow: A System for Large-Scale Machine Learning. In Proceedings of the 12th USENIX Symposium on Operating Systems Design and Implementation (OSDI 16), Savannah, GA, USA, 2–4 November 2016; pp. 265–283. [Google Scholar]
- Bonchev, G.N. Useful parasites: The evolutionary biology and biotechnology applications of transposable elements. J. Genet. 2016, 95, 1039–1052. [Google Scholar] [CrossRef] [PubMed]
- Cossu, R.M.; Buti, M.; Giordani, T.; Natali, L.; Cavallini, A. A computational study of the dynamics of LTR retrotransposons in the Populus trichocarpa genome. Tree Genet. Genomes 2012, 8, 61–75. [Google Scholar] [CrossRef]
- Bento, M.; Tomás, D.; Viegas, W.; Silva, M. Retrotransposons represent the most labile fraction for genomic rearrangements in polyploid plant species. Cytogenet. Genome Res. 2013, 140, 286–294. [Google Scholar] [CrossRef] [PubMed]
- Vicient, C.M.; Casacuberta, J.M. Impact of transposable elements on polyploid plant genomes. Ann. Bot. 2017, 120, 195–207. [Google Scholar] [CrossRef] [PubMed]
- Paz, R.C.; Kozaczek, M.E.; Rosli, H.G.; Andino, N.P.; Sanchez-Puerta, M.V.; Cristina Paz, R.; Eliana Kozaczek, M.; Guillermo Rosli, H.; Pilar Andino, N.; Virginia Sanchez-Puerta, M. Diversity, distribution and dynamics of full-length Copia and Gypsy LTR retroelements in Solanum lycopersicum. Genetica 2017, 145, 417–430. [Google Scholar] [CrossRef]
- Gao, D.; Li, Y.; Kim, K.D.; Abernathy, B.; Jackson, S.A. Landscape and evolutionary dynamics of terminal repeat retrotransposons in miniature in plant genomes. Genome Biol. 2016, 17, 7. [Google Scholar] [CrossRef]
- Tang, X.; Datema, E.; Guzman, M.O.; de Boer, J.M.; van Eck, H.J.; Bachem, C.W.B.; Visser, R.G.F.; de Jong, H. Chromosomal organizations of major repeat families on potato (Solanum tuberosum) and further exploring in its sequenced genome. Mol. Genet. Genom. 2014, 289, 1307–1319. [Google Scholar] [CrossRef]
- Gao, D.; Abernathy, B.; Rohksar, D.; Schmutz, J.; Jackson, S.A. Annotation and sequence diversity of transposable elements in common bean (Phaseolus vulgaris). Front. Plant Sci. 2014, 5, 339. [Google Scholar] [CrossRef]
- Gao, D.; Jiang, N.; Wing, R.A.; Jiang, J.; Jackson, S.A. Transposons play an important role in the evolution and diversification of centromeres among closely related species. Front. Plant Sci. 2015, 6, 216. [Google Scholar] [CrossRef]
- Jiang, S.-Y.; Ramachandran, S. Genome-wide survey and comparative analysis of LTR retrotransposons and their captured genes in rice and sorghum. PLoS ONE 2013, 8, e71118. [Google Scholar] [CrossRef]
- Rawal, K.; Ramaswamy, R. Genome-wide analysis of mobile genetic element insertion sites. Nucleic Acids Res. 2011, 39, 6864–6878. [Google Scholar] [CrossRef] [PubMed]
- Hermann, D.; Egue, F.; Tastard, E.; Nguyen, D.-H.; Casse, N.; Caruso, A.; Hiard, S.; Marchand, J.; Chenais, B.; Morant-Manceau, A.; et al. An introduction to the vast world of transposable elements—What about the diatoms? Diatom Res. 2014, 29, 91–104. [Google Scholar] [CrossRef]
- Wicker, T.; Matthews, D.E.; Keller, B. TREP: A database for Triticeae repetitive elements. Trends Plant Sci. 2002, 7, 561. [Google Scholar] [CrossRef]
- Du, J.; Grant, D.; Tian, Z.; Nelson, R.T.; Zhu, L.; Shoemaker, R.C.; Ma, J. SoyTEdb: A comprehensive database of transposable elements in the soybean genome. BMC Genom. 2010, 11, 113. [Google Scholar] [CrossRef]
- Arensburger, P.; Piégu, B.; Bigot, Y. The future of transposable element annotation and their classification in the light of functional genomics—What we can learn from the fables of Jean de la Fontaine? Mob. Genet. Elements 2016, 6, e1256852. [Google Scholar] [CrossRef]
- Kersey, P.J. Plant genome sequences: Past, present, future. Curr. Opin. Plant Biol. 2019, 48, 1–8. [Google Scholar] [CrossRef]
- Rishishwar, L.; Wang, L.; Clayton, E.A.; Mariño-Ramírez, L.; McDonald, J.F.; Jordan, I.K. Population and clinical genetics of human transposable elements in the (post) genomic era. Mob. Genet. Elements 2017, 7, 1–20. [Google Scholar] [CrossRef]
- Chen, W.; Feng, P.M.; Deng, E.Z.; Lin, H.; Chou, K.C. iTIS-PseTNC: A sequence-based predictor for identifying translation initiation site in human genes using pseudo trinucleotide composition. Anal. Biochem. 2014, 462, 76–83. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
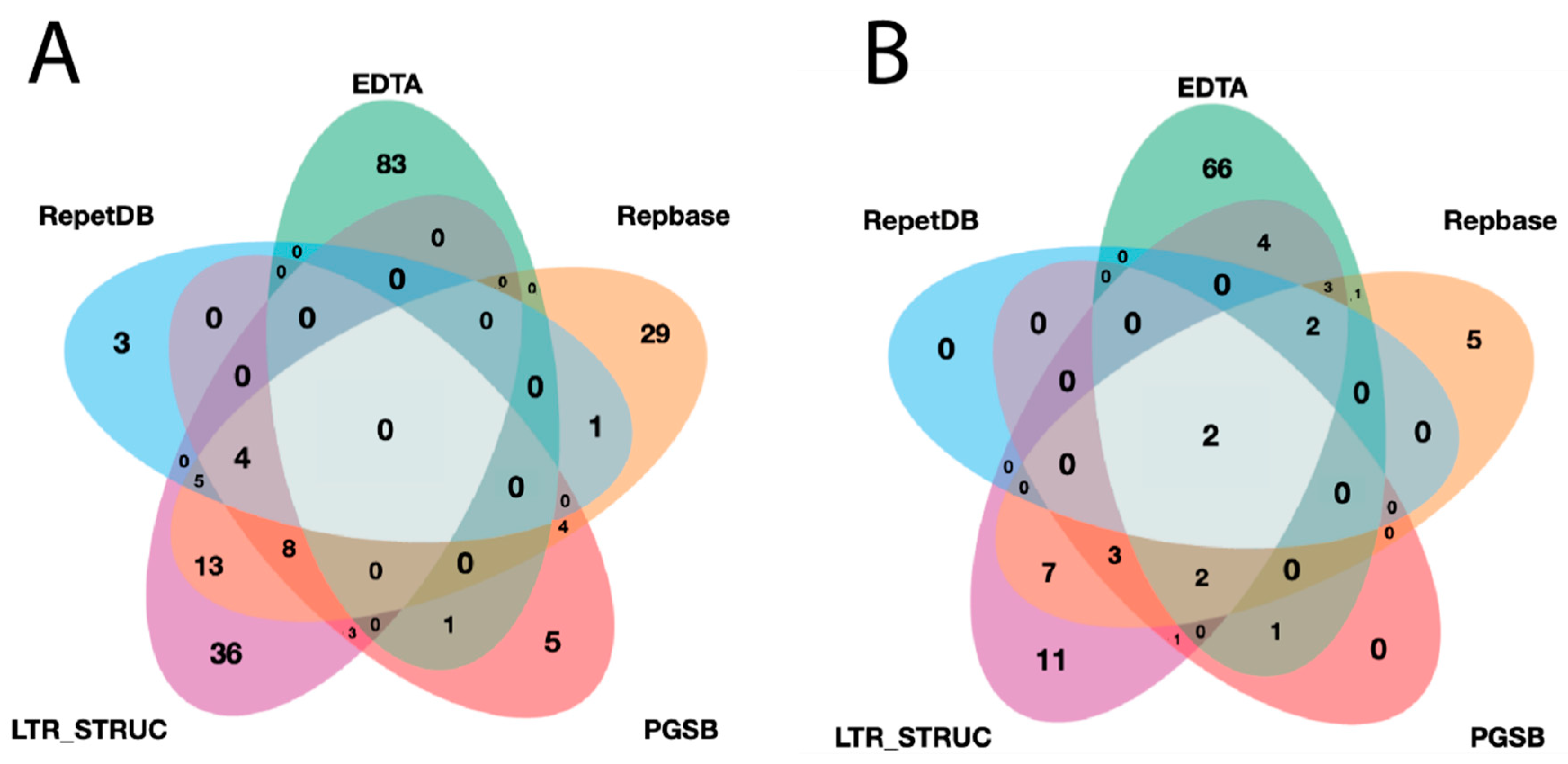{kind=link}
{kind=link}
{kind=link}
| Name | Observations |
|---|---|
| Repbase | Curated consensus sequences. |
| PGSB | Curated individual genomic sequences. |
| RepetDB | Non-curated consensus sequences. |
| LTR_STRUC | Non-curated individual genomic sequences. |
| Consensus | Union between Repbase and RepetDB. |
| Genomics | Union between PGSB and LTR_STRUC. |
| Curated | Union between Repbase and PGSB. |
| Non-curated | Union between RepetDB and LTR_STRUC. |
| All | Union between Repbase, PGSB, RepetDB, and LTR_STRUC. |
| Curated | Consensus | PGSB | RepetDB | |
|---|---|---|---|---|
| Curated | ||||
| Consensus | 1 | |||
| PGSB | 1 | 0.188 | ||
| RepetDB | 1 | 0.162 | 1 |
| Superfamilies | Lineages | Number of Sequences (Redundant) | Number Sequences (Non-Redundant) |
|---|---|---|---|
| Copia | ALE/RETROFIT | 19,888 | 12,026 |
| Copia | ANGELA | 6889 | 1458 |
| Copia | BIANCA | 2872 | 1827 |
| Copia | IKEROS | 149 | 84 |
| Copia | IVANA | 88 | 68 |
| Copia | ORYCO | 6135 | 3468 |
| Copia | SIRE | 10,892 | 3130 |
| Copia | TORK/TAR | 11,460 | 6161 |
| Total Copia | 58,373 | 28,222 | |
| Gypsy | ATHILA | 6611 | 3499 |
| Gypsy | CRM | 4811 | 2134 |
| Gypsy | DEL/TEKAY | 18,330 | 10,383 |
| Gypsy | GALADRIEL | 1715 | 549 |
| Gypsy | REINA | 6387 | 4531 |
| Gypsy | TAT | 34,212 | 17,923 |
| Total Gypsy | 72,066 | 39,019 |
| Superfamilies | Lineages/Families | Precision | Recall | F1-Score | Support |
|---|---|---|---|---|---|
| Copia | ALE/RETROFIT | 0.99 | 0.99 | 0.99 | 1220 |
| Copia | ANGELA | 0.96 | 0.98 | 0.97 | 145 |
| Copia | BIANCA | 0.99 | 0.99 | 0.99 | 166 |
| Copia | IKEROS | 0.67 | 0.57 | 0.62 | 7 |
| Copia | IVANA/ORYCO | 0.95 | 0.97 | 0.96 | 319 |
| Copia | TORK/TAR | 0.98 | 0.95 | 0.96 | 575 |
| Copia | SIRE | 0.99 | 0.98 | 0.99 | 325 |
| Gypsy | CRM | 0.98 | 0.97 | 0.97 | 201 |
| Gypsy | GALADRIEL | 1.00 | 0.93 | 0.96 | 58 |
| Gypsy | REINA | 0.99 | 1.00 | 0.99 | 497 |
| Gypsy | TEKAY/DEL | 0.99 | 0.99 | 0.99 | 1059 |
| Gypsy | ATHILA | 0.97 | 0.98 | 0.97 | 372 |
| Gypsy | TAT | 0.99 | 0.99 | 0.99 | 1787 |
| Superfamilies | Lineages/Families | Precision | Recall | F1-Score | Support |
|---|---|---|---|---|---|
| Copia | ALE/RETROFIT | 0.97 | 0.99 | 0.98 | 1220 |
| Copia | ANGELA | 0.96 | 0.94 | 0.95 | 145 |
| Copia | BIANCA | 1.00 | 0.95 | 0.98 | 166 |
| Copia | IKEROS | 1.00 | 0.43 | 0.60 | 7 |
| Copia | IVANA/ORYCO | 0.96 | 0.93 | 0.95 | 319 |
| Copia | TORK/TAR | 0.94 | 0.94 | 0.94 | 575 |
| Copia | SIRE | 0.99 | 0.97 | 0.98 | 325 |
| Gypsy | CRM | 0.97 | 0.92 | 0.94 | 201 |
| Gypsy | GALADRIEL | 1.00 | 0.74 | 0.85 | 58 |
| Gypsy | REINA | 0.98 | 0.99 | 0.98 | 497 |
| Gypsy | TEKAY/DEL | 0.98 | 0.98 | 0.98 | 1059 |
| Gypsy | ATHILA | 0.97 | 0.99 | 0.98 | 372 |
| Gypsy | TAT | 0.99 | 1.00 | 0.99 | 1787 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Orozco-Arias, S.; Jaimes, P.A.; Candamil, M.S.; Jiménez-Varón, C.F.; Tabares-Soto, R.; Isaza, G.; Guyot, R. InpactorDB: A Classified Lineage-Level Plant LTR Retrotransposon Reference Library for Free-Alignment Methods Based on Machine Learning. Genes 2021, 12, 190. https://doi.org/10.3390/genes12020190
Orozco-Arias S, Jaimes PA, Candamil MS, Jiménez-Varón CF, Tabares-Soto R, Isaza G, Guyot R. InpactorDB: A Classified Lineage-Level Plant LTR Retrotransposon Reference Library for Free-Alignment Methods Based on Machine Learning. Genes. 2021; 12(2):190. https://doi.org/10.3390/genes12020190
Chicago/Turabian StyleOrozco-Arias, Simon, Paula A. Jaimes, Mariana S. Candamil, Cristian Felipe Jiménez-Varón, Reinel Tabares-Soto, Gustavo Isaza, and Romain Guyot. 2021. "InpactorDB: A Classified Lineage-Level Plant LTR Retrotransposon Reference Library for Free-Alignment Methods Based on Machine Learning" Genes 12, no. 2: 190. https://doi.org/10.3390/genes12020190
APA StyleOrozco-Arias, S., Jaimes, P. A., Candamil, M. S., Jiménez-Varón, C. F., Tabares-Soto, R., Isaza, G., & Guyot, R. (2021). InpactorDB: A Classified Lineage-Level Plant LTR Retrotransposon Reference Library for Free-Alignment Methods Based on Machine Learning. Genes, 12(2), 190. https://doi.org/10.3390/genes12020190