
Cell-Cycle–Dependent Chromatin Dynamics at Replication Origins


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Yeast Culture and Cell-Cycle Time Courses
2.2. Chromatin Digestion with MNase and Sequencing Library Preparation
2.3. Flow Cytometry
2.4. Sequencing Data Processing and Analysis
2.5. Quantification of Nucleosome Occupancy
2.6. Quantification of Small Fragment Occupancy
2.7. Quantification of Nucleosome Disorganization by Shannon Entropy
3. Results
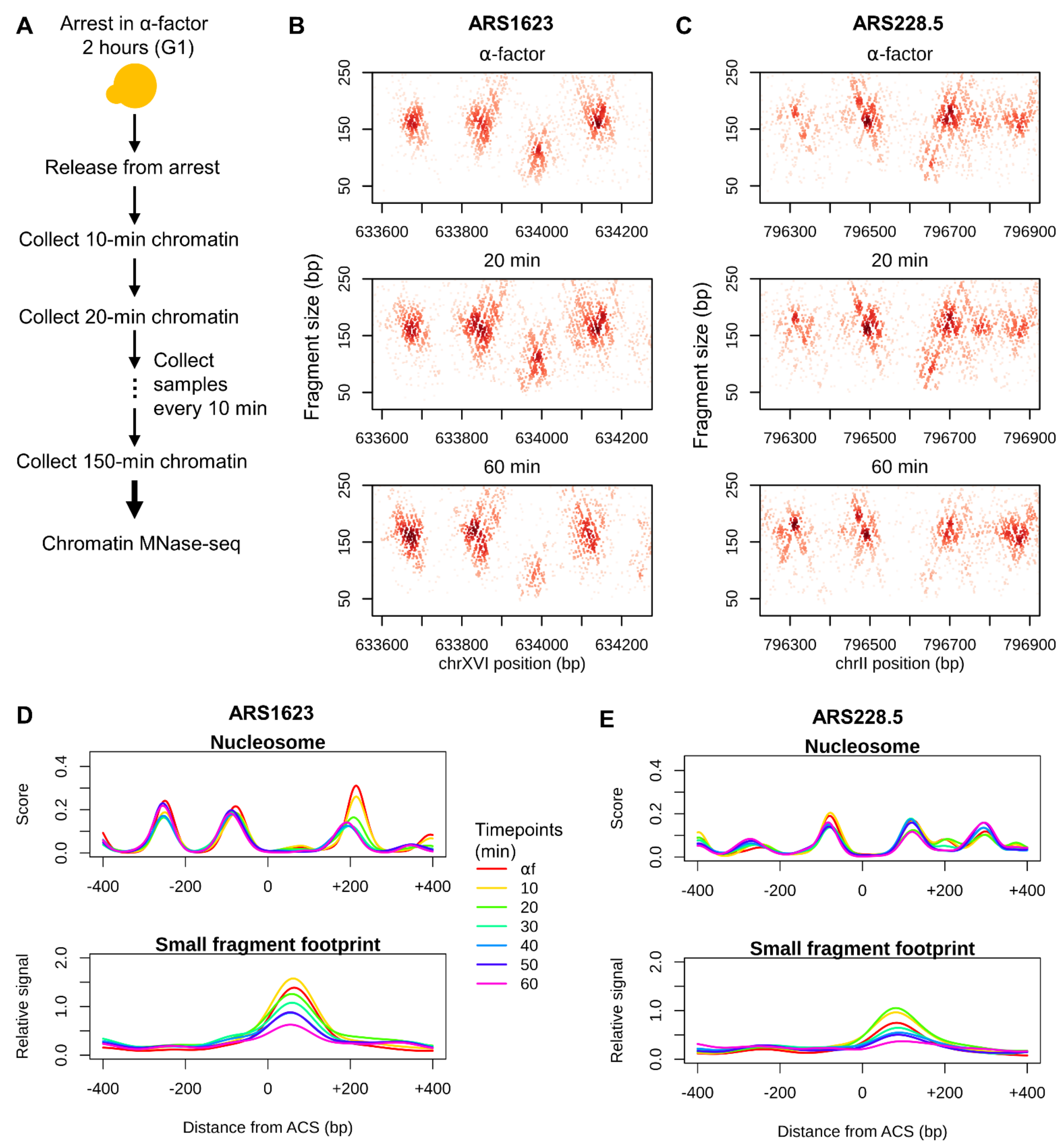
3.1. Chromatin Occupancy Profiling of Replication Origins Throughout the Cell Cycle
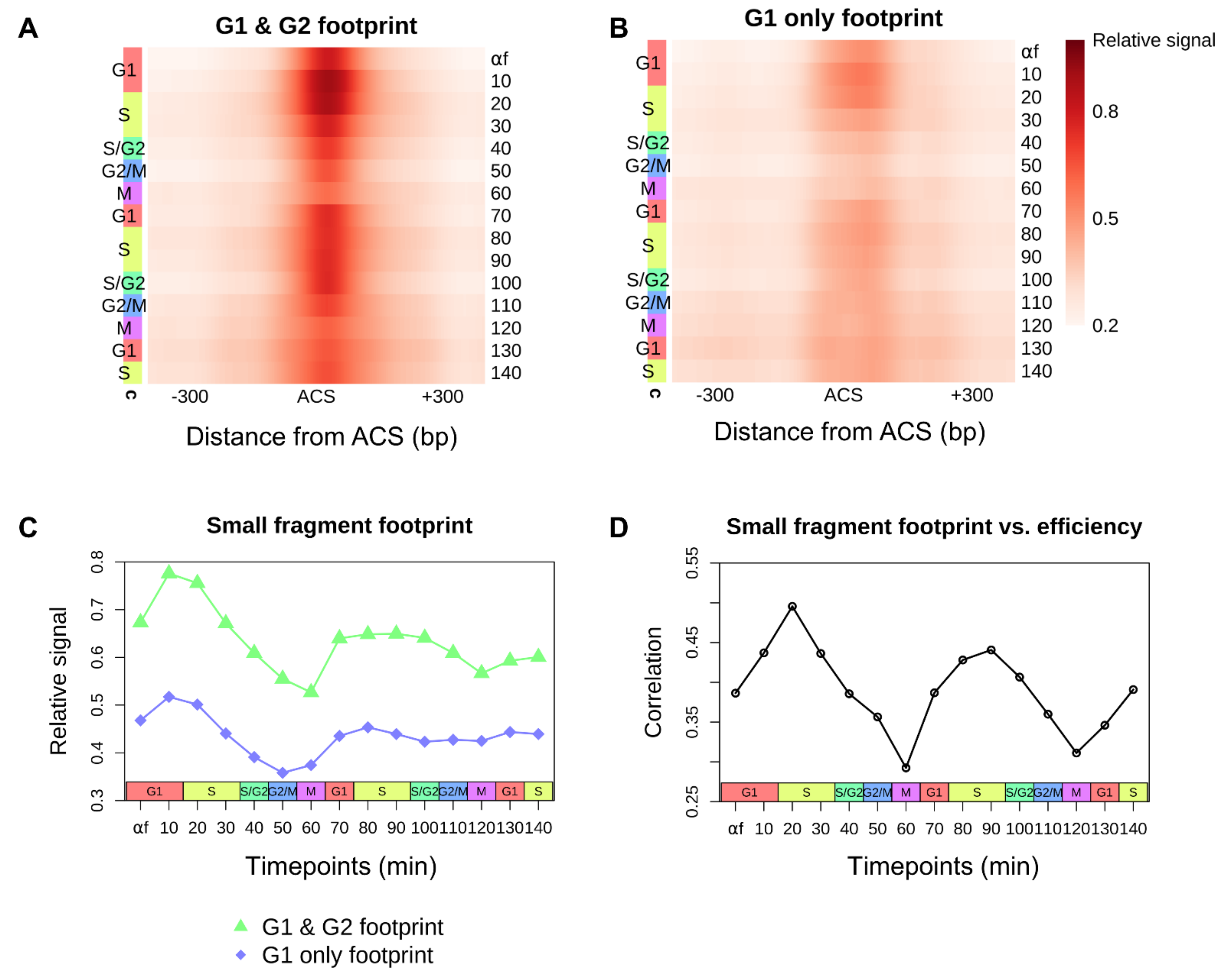
3.2. Cell-Cycle–Dependent Changes in Replication Initiation Factor Occupancy at Replication Origins
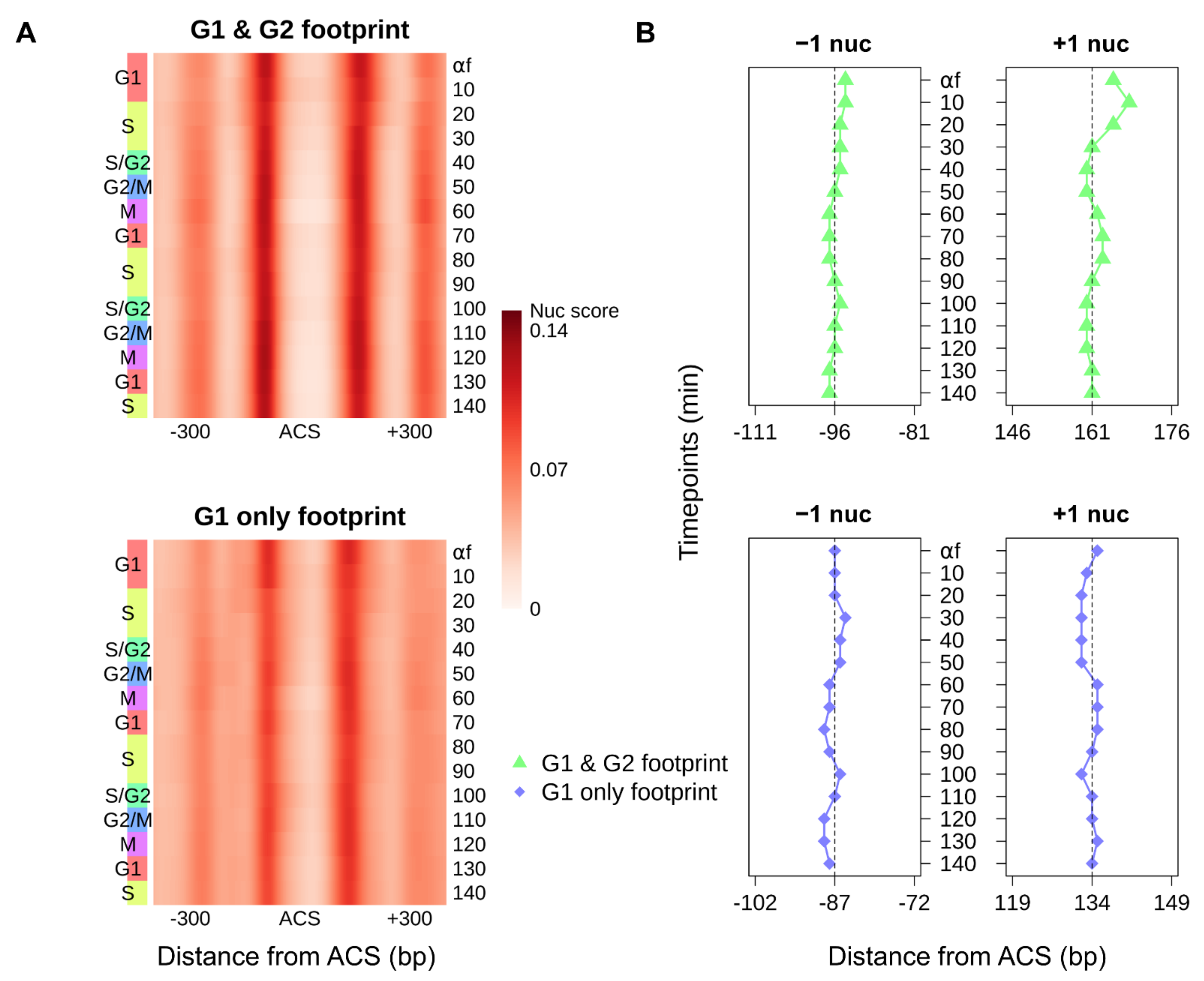
3.3. Cell-Cycle–Regulated Nucleosome Occupancy Dynamics Around Replication Origins
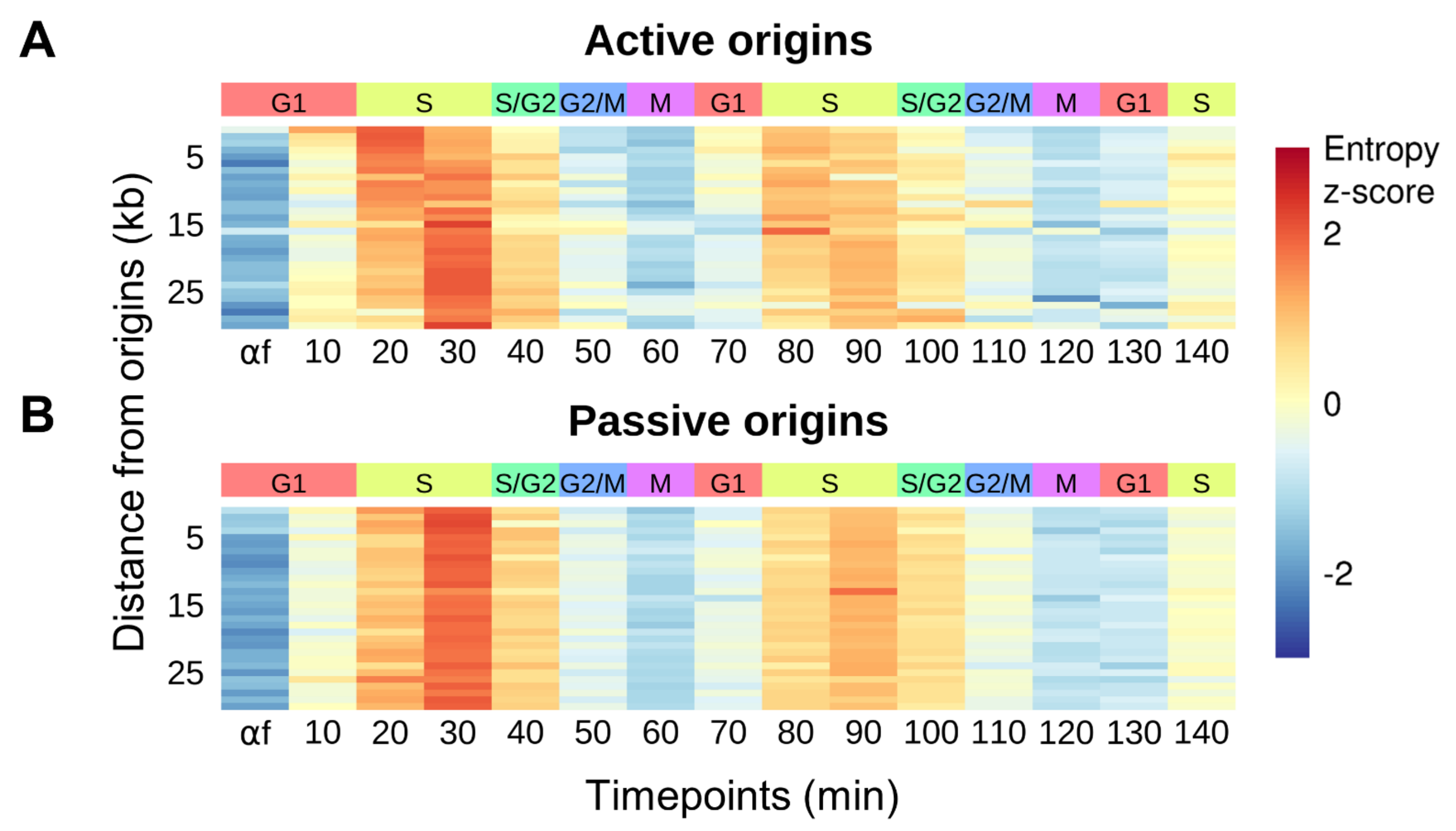
3.4. Replication-Coupled Nucleosome Disorganization
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lin, Y.C.; Prasanth, S.G. Replication initiation: Implications in genome integrity. DNA Repair 2021, 103, 103131. [Google Scholar] [CrossRef]
- McGuffee, S.R.; Smith, D.J.; Whitehouse, I. Quantitative, Genome-Wide Analysis of Eukaryotic Replication Initiation and Termination. Mol. Cell 2013, 50, 123–135. [Google Scholar] [CrossRef] [Green Version]
- Aparicio, O.M. Location, location, location: It’s all in the timing for replication origins. Genes Dev. 2013, 27, 117–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wyrick, J.J.; Aparicio, J.G.; Chen, T.; Barnett, J.D.; Jennings, E.G.; Young, R.A.; Bell, S.P.; Aparicio, O.M. Genome-Wide Distribution of ORC and MCM Proteins in S. cerevisiae: High-Resolution Mapping of Replication Origins. Science 2001, 294, 2357–2360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aparicio, O.M.; Weinstein, D.M.; Bell, S.P. Components and Dynamics of DNA Replication Complexes in S. cerevisiae: Redistribution of MCM Proteins and Cdc45p during S Phase. Cell 1997, 91, 59–69. [Google Scholar] [CrossRef] [Green Version]
- Retkute, R.; Nieduszynski, C.A.; de Moura, A. Dynamics of DNA Replication in Yeast. Phys. Rev. Lett. 2011, 107, 068103. [Google Scholar] [CrossRef] [Green Version]
- Müller, C.A.; Hawkins, M.; Retkute, R.; Malla, S.; Wilson, R.; Blythe, M.J.; Nakato, R.; Komata, M.; Shirahige, K.; de Moura, A.P.S.; et al. The dynamics of genome replication using deep sequencing. Nucleic Acids Res. 2014, 42, e3. [Google Scholar] [CrossRef] [Green Version]
- Eaton, M.L.; Galani, K.; Kang, S.; Bell, S.P.; MacAlpine, D.M. Conserved nucleosome positioning defines replication origins. Genes Dev. 2010, 24, 748–753. [Google Scholar] [CrossRef] [Green Version]
- Berbenetz, N.M.; Nislow, C.; Brown, G.W. Diversity of Eukaryotic DNA Replication Origins Revealed by Genome-Wide Analysis of Chromatin Structure. PLoS Genet. 2010, 6, e1001092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vogelauer, M.; Rubbi, L.; Lucas, I.; Brewer, B.J.; Grunstein, M. Histone Acetylation Regulates the Time of Replication Origin Firing. Mol. Cell 2002, 10, 1223–1233. [Google Scholar] [CrossRef]
- Yoshida, K.; Bacal, J.; Desmarais, D.; Padioleau, I.; Tsaponina, O.; Chabes, A.; Pantesco, V.; Dubois, E.; Parrinello, H.; Skrzypczak, M.; et al. The Histone Deacetylases Sir2 and Rpd3 Act on Ribosomal DNA to Control the Replication Program in Budding Yeast. Mol. Cell 2014, 54, 691–697. [Google Scholar] [CrossRef] [Green Version]
- Bell, S.P.; Stillman, B. ATP-dependent recognition of eukaryotic origins of DNA replication by a multiprotein complex. Nature 1992, 357, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Diffley, J.F.X.; Cocker, J.H.; Dowell, S.J.; Rowley, A. Two steps in the assembly of complexes at yeast replication origins in vivo. Cell 1994, 78, 303–316. [Google Scholar] [CrossRef]
- Bell, S.P.; Kaguni, J.M. Helicase Loading at Chromosomal Origins of Replication. CSH Perspect. Biol. 2013, 5, a010124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, S.; Araki, H. Helicase Activation and Establishment of Replication Forks at Chromosomal Origins of Replication. CSH Perspect. Biol. 2013, 5, a010371. [Google Scholar] [CrossRef] [Green Version]
- Mantiero, D.; MacKenzie, A.; Donaldson, A.; Zegerman, P. Limiting replication initiation factors execute the temporal programme of origin firing in budding yeast. EMBO J. 2011, 30, 4805–4814. [Google Scholar] [CrossRef] [Green Version]
- Lynch, K.L.; Alvino, G.M.; Kwan, E.X.; Brewer, B.J.; Raghuraman, M.K. The effects of manipulating levels of replication initiation factors on origin firing efficiency in yeast. PLoS Genet. 2019, 15, e1008430. [Google Scholar] [CrossRef]
- Xu, W.; Aparicio, J.G.; Aparicio, O.M.; Tavaré, S. Genome-wide mapping of ORC and Mcm2p binding sites on tiling arrays and identification of essential ARS consensus sequences in S. cerevisiae. BMC Genom. 2006, 7, 276. [Google Scholar] [CrossRef] [Green Version]
- MacAlpine, D.M.; Almouzni, G. Chromatin and DNA Replication. CSH Perspect. Biol. 2013, 5, a010207. [Google Scholar] [CrossRef]
- Belsky, J.A.; MacAlpine, H.K.; Lubelsky, Y.; Hartemink, A.J.; MacAlpine, D.M. Genome-wide chromatin footprinting reveals changes in replication origin architecture induced by pre-RC assembly. Genes Dev. 2015, 29, 212–224. [Google Scholar] [CrossRef] [Green Version]
- Lubelsky, Y.; Sasaki, T.; Kuipers, M.A.; Lucas, I.; Le Beau, M.M.; Carignon, S.; Debatisse, M.; Prinz, J.A.; Dennis, J.H.; Gilbert, D.M. Pre-replication complex proteins assemble at regions of low nucleosome occupancy within the Chinese hamster dihydrofolate reductase initiation zone. Nucleic Acids Res. 2011, 39, 3141–3155. [Google Scholar] [CrossRef] [Green Version]
- Cayrou, C.; Coulombe, P.; Puy, A.; Rialle, S.; Kaplan, N.; Segal, E.; Méchali, M. New insights into replication origin characteristics in metazoans. Cell Cycle 2012, 11, 658–667. [Google Scholar] [CrossRef] [Green Version]
- Cayrou, C.; Ballester, B.; Peiffer, I.; Fenouil, R.; Coulombe, P.; Andrau, J.C.; van Helden, J.; Méchali, M. The chromatin environment shapes DNA replication origin organization and defines origin classes. Genome Res. 2015, 25, 1873–1885. [Google Scholar] [CrossRef] [Green Version]
- Raghuraman, M.K.; Winzeler, E.A.; Collingwood, D.; Hunt, S.; Wodicka, L.; Conway, A.; Lockhart, D.J.; Davis, R.W.; Brewer, B.J.; Fangman, W.L. Replication Dynamics of the Yeast Genome. Science 2001, 294, 115–121. [Google Scholar] [CrossRef] [Green Version]
- Hoggard, T.; Shor, E.; Müller, C.A.; Nieduszynski, C.A.; Fox, C.A. A Link between ORC-Origin Binding Mechanisms and Origin Activation Time Revealed in Budding Yeast. PLoS Genet. 2013, 9, e1003798. [Google Scholar] [CrossRef] [PubMed]
- Das, S.P.; Borrman, T.; Liu, V.W.T.; Yang, S.C.H.; Bechhoefer, J.; Rhind, N. Replication timing is regulated by the number of MCMs loaded at origins. Genome Res. 2015, 25, 1886–1892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dukaj, L.; Rhind, N. The capacity of origins to load MCM establishes replication timing patterns. PLoS Genet. 2021, 17, e1009467. [Google Scholar] [CrossRef]
- Speck, C.; Chen, Z.; Li, H.; Stillman, B. ATPase-dependent cooperative binding of ORC and Cdc6 to origin DNA. Nat. Struct. Mol. Biol. 2005, 12, 965–971. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, R.A.; MacAlpine, H.K.; MacAlpine, D.M. Disruption of origin chromatin structure by helicase activation in the absence of DNA replication. Genes Dev. 2021, 35, 1339–1355. [Google Scholar] [CrossRef]
- Henikoff, J.G.; Belsky, J.A.; Krassovsky, K.; MacAlpine, D.M.; Henikoff, S. Epigenome characterization at single base-pair resolution. Proc. Natl. Acad. Sci. USA 2011, 108, 18318–18323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, r25. [Google Scholar] [CrossRef] [Green Version]
- Brogaard, K.R.; Xi, L.Q.; Wang, J.P.; Widom, J. A Chemical Approach to Mapping Nucleosomes at Base Pair Resolution in Yeast. Methods Enzymol. 2012, 513, 315–334. [Google Scholar] [CrossRef] [Green Version]
- Tran, T.Q.; MacAlpine, H.K.; Tripuraneni, V.; Mitra, S.; MacAlpine, D.M.; Hartemink, A.J. Linking the dynamics of chromatin occupancy and transcription with predictive models. Genome Res. 2021, 31, 1035–1046. [Google Scholar] [CrossRef] [PubMed]
- MacIsaac, K.D.; Wang, T.; Gordon, D.B.; Gifford, D.K.; Stormo, G.D.; Fraenkel, E. An improved map of conserved regulatory sites for Saccharomyces cerevisiae. BMC Bioinform. 2006, 7, 113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kent, N.A.; Adams, S.; Moorhouse, A.; Paszkiewicz, K. Chromatin particle spectrum analysis: A method for comparative chromatin structure analysis using paired-end mode next-generation DNA sequencing. Nucleic Acids Res. 2011, 39, e26. [Google Scholar] [CrossRef]
- Hawkins, M.; Retkute, R.; Müller, C.A.; Saner, N.; Tanaka, T.U.; de Moura, A.P.S.; Nieduszynski, C.A. High-Resolution Replication Profiles Define the Stochastic Nature of Genome Replication Initiation and Termination. Cell Rep. 2013, 5, 1132–1141. [Google Scholar] [CrossRef] [Green Version]
- Müller, P.; Park, S.; Shor, E.; Huebert, D.J.; Warren, C.L.; Ansari, A.Z.; Weinreich, M.; Eaton, M.L.; MacAlpine, D.M.; Fox, C.A. The conserved bromo-adjacent homology domain of yeast Orc1 functions in the selection of DNA replication origins within chromatin. Genes Dev. 2010, 24, 1418–1433. [Google Scholar] [CrossRef] [Green Version]
- Simpson, R.T. Nucleosome positioning can affect the function of a cis-acting DNA element in vivo. Nat. Cell Biol. 1990, 343, 387–389. [Google Scholar] [CrossRef]
- Lipford, J.R.; Bell, S.P. Nucleosomes Positioned by ORC Facilitate the Initiation of DNA Replication. Mol. Cell 2001, 7, 21–30. [Google Scholar] [CrossRef]
- Azmi, I.F.; Watanabe, S.; Maloney, M.F.; Kang, S.; Belsky, J.A.; MacAlpine, D.M.; Peterson, C.L.; Bell, S.P. Nucleosomes influence multiple steps during replication initiation. eLife 2017, 6, e22512. [Google Scholar] [CrossRef] [Green Version]
- Ferguson, B.M.; Fangman, W.L. A position effect on the time of replication origin activation in yeast. Cell 1992, 68, 333–339. [Google Scholar] [CrossRef]
- Knott, S.R.; Viggiani, C.J.; Tavaré, S.; Aparicio, O.M. Genome-wide replication profiles indicate an expansive role for Rpd3L in regulating replication initiation timing or efficiency, and reveal genomic loci of Rpd3 function in Saccharomyces cerevisiae. Genes Dev. 2009, 23, 1077–1090. [Google Scholar] [CrossRef] [Green Version]
- Coster, G.; Diffley, J.F.X. Bidirectional eukaryotic DNA replication is established by quasi-symmetrical helicase loading. Science 2017, 357, 314–318. [Google Scholar] [CrossRef] [Green Version]
- Miller, T.C.R.; Locke, J.; Greiwe, J.F.; Diffley, J.F.X.; Costa, A. Mechanism of head-to-head MCM double-hexamer formation revealed by cryo-EM. Nature 2019, 575, 704–710. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.B.; Zhou, H.; Katzmann, D.; Hochstrasser, M.; Atanasova, E.; Zhang, Z.G. Rtt106p is a histone chaperone involved in heterochromatin-mediated silencing. Proc. Natl. Acad. Sci. USA 2005, 102, 13410–13415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaufman, P.D.; Kobayashi, R.; Stillman, B. Ultraviolet radiation sensitivity and reduction of telomeric silencing in Saccharomyces cerevisiae cells lacking chromatin assembly factor-I. Genes Dev. 1997, 11, 345–357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meijsing, S.H.; Ehrenhofer-Murray, A.E. The silencing complex SAS-I links histone acetylation to the assembly of repressed chromatin by CAF-I and Asf1 in Saccharomyces cerevisiae. Genes Dev. 2001, 15, 3169–3182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Escobar, T.M.; Oksuz, O.; Saldaña-Meyer, R.; Descostes, N.; Bonasio, R.; Reinberg, D. Active and Repressed Chromatin Domains Exhibit Distinct Nucleosome Segregation during DNA Replication. Cell 2019, 179, 953–963. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Y.; Hartemink, A.J.; MacAlpine, D.M. Cell-Cycle–Dependent Chromatin Dynamics at Replication Origins. Genes 2021, 12, 1998. https://doi.org/10.3390/genes12121998
Li Y, Hartemink AJ, MacAlpine DM. Cell-Cycle–Dependent Chromatin Dynamics at Replication Origins. Genes. 2021; 12(12):1998. https://doi.org/10.3390/genes12121998
Chicago/Turabian StyleLi, Yulong, Alexander J. Hartemink, and David M. MacAlpine. 2021. "Cell-Cycle–Dependent Chromatin Dynamics at Replication Origins" Genes 12, no. 12: 1998. https://doi.org/10.3390/genes12121998
APA StyleLi, Y., Hartemink, A. J., & MacAlpine, D. M. (2021). Cell-Cycle–Dependent Chromatin Dynamics at Replication Origins. Genes, 12(12), 1998. https://doi.org/10.3390/genes12121998